
Abstract
Over 50 loci associated with colorectal cancer (CRC) have been uncovered by genome-wide association studies (GWAS). Identifying additional loci has the potential to help elucidate aspects of the underlying biological processes leading to better understanding of the pathogenesis of the disease. We re-evaluated a GWAS by excluding controls that have family history of CRC or personal history of colorectal polyps, as we hypothesized that their inclusion reduces power to detect associations. This is supported empirically and through simulations. Two-phase GWAS analysis was performed in a total of 16,517 cases and 14,487 controls. We identified rs17094983, a SNP associated with risk of CRC [p = 2.5 × 10−10; odds ratio estimated by re-including all controls (OR) = 0.87, 95 % confidence interval (CI) 0.83–0.91; minor allele frequency (MAF) = 13 %]. Results were replicated in samples of African descent (1894 cases and 4703 controls; p = 0.01; OR = 0.86, 95 % CI 0.77–0.97; MAF = 16 %). Gene expression data in 195 colon adenocarcinomas and 59 normal colon tissues from two different studies revealed that this locus has genotypes that are associated with RTN1 (Reticulon 1) expression (p = 0.001), a protein-coding gene involved in survival and proliferation of cancer cells which is highly expressed in normal colon tissues but has significantly reduced expression in tumor cells (p = 1.3 × 10−8).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genome-wide association studies (GWAS) have been successful at identifying germline common variations associated with the risk of developing colorectal cancer (CRC). Success of the genome-wide design has been driven mainly by large international collaborative efforts to pool resources and samples to produce large datasets of tens of thousands of cases and controls, to help identify genetic risk factors that only had moderate associated risks. Over 50 genetic risk variants have been identified thus far (Al-Tassan et al. 2015; Broderick et al. 2007; Cui et al. 2011; Dunlop et al. 2012; Houlston et al. 2008, 2010; Jaeger et al. 2008; Jia et al. 2013; Peters et al. 2012, 2013; Schmit et al. 2014; Schumacher et al. 2015; Tenesa et al. 2008; Tomlinson et al. 2007, 2008, 2011; Wang et al. 2014; Whiffin et al. 2014; Zanke et al. 2007; Zhang et al. 2014), with odds ratio typically in the range 1.10–1.25 and minor allele frequencies typically no less than ~10 % (partly by design of genotyping arrays). Once the low-hanging fruits have been picked, the design becomes more challenging since the discovery of additional variants with smaller effect or lower allelic frequency may require increasing the sample size by an order of magnitude. Although not as informative from a public health perspective, these additional, undiscovered variants still have the potential to help elucidate parts of the pathobiology.
The American Cancer Society and the US Multi-Society Task Force on Colorectal Cancer recommend early detection testing starting at 40 years of age for those with a family history of CRC, given their higher risk of developing tumors (Read and Kodner 1999; Levin et al. 2008; Lieberman et al. 2012). The lifetime increase in risk in those with a family history of CRC is about twofold (Slattery et al. 2003), partly due to shared genes and/or shared environment with the affected relative (Lichtenstein et al. 2000). Because they share the genome and the genetic risk background of their affected relative, the inclusion of controls with a family history of CRC may reduce the power to detect a genetic association with the disease in a case–control study. By excluding these controls from the study, we show that power can be increased even if the sample size is reduced. Moreover, we argue with empirical evidence that excluding controls that were diagnosed with colorectal (CR) polyps (potential precursors of tumors), when such a diagnostic is available, may also lead to an increase in power. This allows for a re-evaluation of GWAS without the need to increase the sample size or genotype additional samples.
Materials and methods
Sample description and genotyping
The cases and controls included in the present GWAS consist of a subset of samples that were collected across multiple study centers, within the Genetics and Epidemiology of Colorectal Cancer Consortium/Colon Cancer Family Registries (GECCO/CCFR) (Peters et al. 2013). As a result of simulation-based power calculations and empirical observations, we attempted to increase the power to detect an association by excluding controls with a positive family history and controls that were diagnosed with CR polyps. Status of CR polyps was self-reported from answering questions such as “has a doctor ever told you that you had polyps in your large bowel or colon or rectum?”. Table 1 describes the sample sizes of each study, before and after exclusion of controls and the genotyping platform used in each. Replication of initial results from GECCO/CCFR was attempted in samples from 6 studies from the Colorectal Cancer Transdisciplinary Study (CORECT) (Wang et al. 2014) (Table 1). Genome-wide significant results were then analyzed in samples of African ancestry [1894 cases (49.6 % females; mean age 67.9) and 4703 controls (35.2 % females; mean age 61.6)] and of Japanese ancestry [2627 cases (42.1 % females; mean age 65.3) and 3797 controls (45 % females; mean age 64.7)] to evaluate trans-ethnic effects of the SNPs. These samples were genotyped using Illumina 1 M-Duo, 660 W-Quad or Omni 2.5 M depending on the center (see Wang et al. 2014 for details).
Statistical power comparison
To confirm that the exclusion of controls with a positive family history of CRC would not lead to a reduction, but rather an increase in power, we performed a simulation study. We simulated the segregation of a susceptibility SNP in nuclear families. Sibship size followed a Poisson distribution with mean 3.5 sibs. One susceptibility SNP was simulated with varying allele frequency and relative risk (with risk alleles acting multiplicatively on the risk). The segregation of alleles in the nuclear families and the simulation of the disease state of all family members were performed using SLINK (Schäffer et al. 2011). Lifetime risk of the simulated disease was fixed at 5 % (Siegel et al. 2014). 11,800 cases and 14,300 controls (the approximate sample size of all samples in GECCO/CCFR) were randomly selected among all affected and unaffected individuals, respectively. Once an individual was selected, all other members of the nuclear family became ineligible to enter the case–control sample. Having a family history of the simulated disease was defined as having at least one first-degree affected relative (sib or parent). For each combination of allele frequency and effect size, 400 replicates were assessed for association between the simulated SNP and the disease status using a simple allelic Chi-square test (–assoc command in PLINK; Purcell et al. 2007), before and after exclusion of the controls with a positive family history. Power was estimated from the proportion of replicates reaching significance p < 5 × 10−8.
Genome-wide association analysis
Imputation to HapMap2 Release 24 was performed using MACH for all studies, with the exception of OFCCR, which was imputed to HapMap Release 22 using BEAGLE. Log-additive models were fit and adjusted for age, sex, center, batch effect (in the ASTERISK study), smoking status (in the PHS study), and the first 3 principal components on study level (using HapMap-imputed data). Replication was attempted in CORECT for the SNPs with meta-analysis p < 10−5 in GECCO/CCFR.
RNA expression studies
Two sample sets were used to assess the association between a SNP and expression of genes within a 2 Mbp window centered at the SNP position. Both studies evaluated gene expression in colon adenocarcinomas and normal colon tissues.
The first study (TCGA) consists of data from 155 colon adenocarcinomas and 19 normal colon tissues (from a total of 162 distinct donors: 12 matched tumor and normal adjacent pairs are included) from The Cancer Genome Atlas (TCGA; downloaded from CG Hub: https://cghub.ucsc.edu/). These samples have gene expression data derived from an Agilent 244 K Custom Gene Expression Array and genotypes derived from Affymetrix Genome-Wide Human SNP 6.0 Array. We used Level 3 expression data, which consists of normalized signals and expression calls per gene, per sample. Genotype data were obtained under approved access. We compared the genotype calls between tissues of the same donors. A patient was excluded if he or she presented discordant homozygous genotype calls at >1 % of homozygous markers (heterozygous genotypes were ignored because of the potential for loss of heterozygosity in tumors). The SNP data was analyzed with the –homozyg command in PLINK to identify regions with loss of heterozygosity (LOH); gene expression values in samples displaying LOH in the gene interval were ignored in analyses.
The second study (CCFR) consists of data from 40 tumors and 40 paired adjacent normal tissues from 40 participants enrolled in CCFR, with gene expression data derived from the Affymetrix GeneChip Human Exon 1.0 ST Array and genotype data derived from Affymetrix Genome-Wide Human SNP 6.0 Array. This set of tumor/normal samples has been used in an eQTL (expression quantitative trait loci) study of previously published GWAS loci for CRC (Loo et al. 2012).
Differential expression was assessed using a non-parametric Wilcoxon rank-sum test when comparing two factors, or a Kruskal–Wallis rank-sum test when comparing three factors.
Results
Controls with a family history or CR polyps potentially reduce power to detect association
As a proof of concept that power may be reduced when including controls with a positive family history of CRC in a case–control study, we evaluated a genetic risk score in GECCO by counting the number of risk alleles that an individual possessed across 36 SNPs identified by GWAS, after pruning those in LD (Al-Tassan et al. 2015; Broderick et al. 2007; Cui et al. 2011; Dunlop et al. 2012; Houlston et al. 2008, 2010; Jaeger et al. 2008; Jia et al. 2013; Peters et al. 2012, 2013; Schumacher et al. 2015; Tenesa et al. 2008; Tomlinson et al. 2007, 2008, 2011; Wang et al. 2014; Whiffin et al. 2014; Zanke et al. 2007; Zhang et al. 2014). The distribution of this genetic risk score was stratified by disease status and family history. Figure 1 shows that controls with a family history of CRC have genetic risk scores that are intermediate between that of cases and family-history-negative controls, indicating that controls with a family history share some genetic risk with their affected first-degree relatives.

Count of risk alleles. Boxplot representation of the total count of risk alleles in cases and controls, stratified on family history (FH)
Simulation-based power calculations support the strategy of excluding controls with a family history of CRC: across a wide spectrum of allele frequencies and relative risks, Supplementary Table S2 indicates a gain in statistical power even though the number of controls is reduced by over 20 %. This motivated exclusion of controls with a positive family history.
Family history is a feature that can easily be simulated, through specification of penetrances (including phenocopies), segregation of alleles or shared environmental variables, and ascertainment. For other traits or features—such as diagnosis of CR polyps in controls, it can be hypothesized that power may be reduced from inclusion of samples that display them. However, these traits may not be straightforward to incorporate in an assessment of power; interpretation would only be as good as the underlying model linking the trait (say, presence of CR polyps) to the likelihood of developing the disease. For these traits, stratifying the risk score, as was done for family history, can provide insights. Similar to family history-based stratification, Supplementary Figure S1 shows that controls that were previously diagnosed with CR polyps have a genetic risk score intermediate to that of cases and other controls. Because the diagnosis of CR polyps is correlated with family history of CRC, Supplementary Figure S1 only focuses on samples without a family history. Based on this empirical evidence and the results from simulations described above, we excluded from this analysis controls that have a family history and/or controls diagnosed with CR polyps.
Genome-wide association study and replication
Samples in the discovery phase of this study, which were collected across multiple study centers within GECCO/CCFR, were analyzed after exclusion of controls with a family history of CRC or diagnosis of CR polyps. Of note, among the centers that sampled both sexes, female controls were more likely to have reported a family history of CRC than males (fixed effect model: OR = 1.31; p = 0.0006) and less likely to have reported CR polyps than males (OR = 0.65; p = 2 × 10−8). Control individuals who reported family history were slightly older than those who did not (mean of 64.06 years compared to 63.49; p = 0.011, adjusted for center). In contrast, control individuals who reported polyps were substantially older than those who did not (mean of 65.9 years compared to 63.3; p < 10−8).
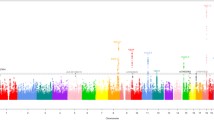
Association results between genetic variants and risk of developing CRC in the resulting samples are graphically summarized in the Manhattan plot depicted in Fig. 2. The inflation factor (λ = 1.019) is comparable to the one calculated when no controls are excluded (λ = 1.021; Fig. 2b, c).

Association results. a Manhattan plot of results in GECCO/CCFR. Controls with family history and/or polyps are excluded from the analysis. Each dot represents a SNP plotted on the x axis relative to its position in the genome, whose level of significance is represented on the y axis. Green dots represent SNPs in LD with SNPs identified in published GWAS for CRC. Replication in CORECT was attempted for SNPs with p < 10−5 (blue horizontal line). The red horizontal line indicates p = 5 × 10−8; b quantile–quantile plot of p values in (a), on the negative log scale. λ is the inflation factor (the ratio of observed to expected median); c quantile–quantile plot of p values when no controls are excluded from the analysis
Replication was attempted in samples from CORECT for SNPs that reached significance at p < 10−5 in the discovery phase. Supplementary Table S3 shows results for these SNPs in both phases of the study after pruning for linkage disequilibrium (LD) (reporting the most significant SNP among SNPs with r 2 > 0.5).
One SNP, rs17094983, reached genome-wide significance in the meta-analysis of all studies combined (p = 2.5 × 10−10) with no evidence of heterogeneity across centers (p het = 0.97) (Supplementary Figure S2). The minor allele of the SNP has a frequency of 13 % and is inversely associated with risk; the odds ratio (estimated by re-including the controls with FH or CR polyps, to eliminate the effect of the selection bias) is OR = 0.87 (95 % confidence interval 0.83–0.91; p = 4.7 × 10−9) compared to OR = 0.85 when these controls are excluded (Supplementary Figure S2). To evaluate trans-ethnic associations for that SNP, we first note that rs17094983 is monomorphic in populations of Asian ancestry according to the 1000 Genomes project, and it has thus not been observed in the samples of Japanese descent; this also has been reported elsewhere (Peters et al. 2013). In samples of African descent, the SNP replicated (p = 0.01) with a minor allele frequency of 16 % and a consistent effect size (OR = 0.86, 95 % confidence interval 0.77–0.97).
Genes and transcripts in the region surrounding rs17094983 are illustrated in Fig. 3.

UCSC browser representation of the 14q23.1 locus. Window is centered at rs17094983 ± 2Mbp. Top track indicates position of SNPs in LD with rs17094983 (r 2 > 0.05) along with r 2 values
Study of expression quantitative trait loci
In the 2Mbp window centered on rs17094983, The Cancer Genome Atlas (TCGA) includes expression data on 11 transcripts: ACTR10, ARID4A, JKAMP (C14orf100), C14orf37, DAAM1, DACT1, GPR135, KIAA0586, PSMA3, RTN1 and TIMM9. Figure 4 and Supplementary Figures S3–S12 show expression values of these genes in normal colon tissues and tumors as well as expression values in tumors stratified by genotypes at 3 SNPs in high LD with rs17094983 (which is not part of the Affymetrix 6.0 array available from TCGA): rs17094971 (r 2 = 0.81 with rs17094983, calculated from the EUR samples of the 1000 Genomes Project), rs1432096 (r 2 = 0.80) and rs710005 (r 2 = 0.54). RTN1 (Fig. 4) displays lower expression in tumors than in normal tissue and is the transcript that shows the most differential expression in the region (p = 1.3 × 10−8; based on a non-parametric Wilcoxon test). Notably, of the transcripts targeted by the expression array, RTN1 is among the genes with the highest average expression across normal colon tissues: only 13 % of transcripts in the genome have expression values higher than that of RTN1. In tumors, eQTL analyses reveal that RTN1 shows differential expression between genotypes of both rs1432096 (p = 0.022; based on a non-parametric Kruskal–Wallis test) and rs710005 (p = 0.0013), the latter being statistically significant even after accounting for the 33 eQTL combinations (SNP-transcript expression) that we tested [false discovery rate (FDR) = 4.2 % for rs710005]. It is, however, the SNP with the weakest LD with rs17094983. Expression values for the heterozygous genotypes are elevated compared to values for the common homozygous genotypes (homozygous for the apparent “risk” allele); this direction of association is consistent with the minor allele being inversely associated with risk, as normal tissue shows higher expression of RTN1. The number of normal tissues (n = 15) is too small to draw meaningful conclusions from eQTL analyses. No other transcript is associated (after accounting for multiple testing) with any of these SNPs (Supplementary Figures S3–S12).

Expression of RTN1 in TCGA. a Boxplot representation of the expression of RTN1 in normal colon tissues and tumors. Significance calculated from Wilcoxon test. b–d Boxplot representations of the expression of RTN1 in tumors as a function of b rs17094971; c rs1432096; d rs710005. Significance calculated from Kruskal–Wallis tests
We sought to replicate RTN1 expression association results from TCGA using data from 40 normal colon tissues and 40 matched tumors from CCFR. Consistent with the TCGA data, RTN1 shows significantly lower expression in tumors compared to normal tissues (p = 1.1 × 10−8) (Fig. 5a). When stratified on genotypes, RTN1 expression levels show patterns of associations that are in the same direction as seen in the TCGA data, in both normal colon tissues [p = 0.041 for rs1432096 (r 2 = 0.80 with rs17094983); Fig. 5] and tumors (p = 0.041 for rs1432096; Supplemental Figure S13), suggesting that heterozygous individuals tend to show higher expression of RTN1 than common homozygous individuals, irrespective of whether the colon cells are normal or malignant.

Expression of RTN1 in CCFR. a Boxplot representation of the expression of RTN1 in normal colon tissues and tumors. Significance calculated from Wilcoxon test. b–d Boxplot representations of the expression of RTN1 in normal tissues as a function of b rs17094971; c rs1432096; d rs710005. Significance calculated from Kruskal–Wallis tests
Discussion
We describe a strategy to re-evaluate GWAS data that may facilitate identification of additional genetic risk variants at genome-wide significance levels without necessitating an increase in sample size. By excluding controls with a family history of the disease from a case–control study (or other features that may potentially make controls more likely to possess genetic risk factors for the disease under study—such as diagnosis of CR polyps, potential precursors of tumors of the colon) power can be increased. This also has implications for study design.
We report an association between SNPs at 14q23.1 and the risk of developing CRC. rs17094983 was mentioned in a published GWAS (Peters et al. 2013) for CRC but did not reach genome-wide significance (reported p < 3 × 10−6). The present study confirms the association at genome-wide significance levels. We show that genotypes of SNPs in high LD with it are significantly associated with expression of RTN1 (Reticulon 1), a protein-coding gene highly expressed in normal colon cells whose expression is substantially reduced in colon tumor cells.
The RTN1 gene produces three transcripts, which encode for the RTN1-A, RTN1-B, and RTN1-C proteins. The expression values that we presented were derived from probes that are targeting exons present in all three transcripts; there were no probes specific to a single transcript. These proteins are members of highly conserved reticulons, which are localized in the endoplasmic reticulum (ER). Reticulons show pro-apoptotic activity via the induction of ER stress (Kuang et al. 2005; Di Sano et al. 2007). The mechanisms by which RTN1 exerts its effects are not well understood. RTN1-A has been recently described as a mediator of chronic kidney disease progression that promotes renal injury through ER stress (Fan et al. 2015). In kidney epithelial cells, RTN1-A but not RTN1-C interacts with PERK, an ER stress molecule that activates apoptotic pathway. RTN1-C is regulated by acetylation and its DNA-binding activity is required for its role as an inhibitor of histone deacetylases (HDAC) activity (Fazi et al. 2009). Inhibition of HDACs can result in hyperacetylation of proteins, which, in turn, induces apoptosis of tumor cells and sensitizes tumors to cell-death processes and to other drugs (Heerboth et al. 2014). RTN1-C overexpression sensitizes cancer cells to chemotherapeutic-induced apoptosis through p53-independent pathways (Di Sano et al. 2003). In androgen-dependent LNCaP prostate cancer cells, knock down using siRNA targeting all RTN1 transcript isoforms enabled androgen independent growth of these cells (Levina et al. 2015). Gastrointestinal stromal tumors (GISTs) with mutations in KIT or PDGFRA show frequent alterations of the 14q23.1 region, which includes the RTN1 gene (Astolfi et al. 2010). Moreover, the knockdown of RTN1 results in increased proliferation of mutation-harboring GIST cells. These studies indicate that decreased expression of RTN1 is related to survival and proliferation of cancer cells. In the present study, reduced expression of RTN1 in tumors and a further decrease in patients with risk-associated alleles are consistent with the abovementioned roles of RTN1 in cancer.
The strengths of this study are the large sample size and the increase in power to detect a genetic association, caused by the removal of controls with family history of CRC or personal history of CR polyps. By excluding controls that may share the genetic risk background of their affected relatives, we have increased the differences between cases and the remaining controls. However, the OR estimated from samples that underwent this selection bias does not readily generalize to the whole population; we thus provided an OR estimated from the complete sample set thereby making a distinction between the discovery aspects of the study and the estimation of the effect size. In the present study, genome-wide significance was observed with or without the excluded controls, due to the large sample size at hand. Excluding these controls, the p value was more than one order of magnitude smaller, consistent with higher power; for smaller studies, an order of magnitude difference might be all that is needed for additional discoveries at genome-wide significance levels.
References
Al-Tassan NA, Whiffin N, Hosking FJ, Palles C, Farrington SM, Dobbins SE, Harris R, Gorman M, Tenesa A, Meyer BF, Wakil SM, Kinnersley B, Campbell H, Martin L, Smith CG, Idziaszczyk S, Barclay E, Maughan TS, Kaplan R, Kerr R, Kerr D, Buchannan DD, Ko Win A, Hopper J, Jenkins M, Lindor NM, Newcomb PA, Gallinger S, Conti D, Schumacher F, Casey G, Dunlop MG, Tomlinson IP, Cheadle JP, Houlston RS (2015) A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Sci Rep 5:10442
Astolfi A, Nannini M, Pantaleo MA, Di Battista M, Heinrich MC, Santini D, Catena F, Corless CL, Maleddu A, Saponara M, Lolli C, Di Scioscio V, Formica S, Biasco G (2010) A molecular portrait of gastrointestinal stromal tumors: an integrative analysis of gene expression profiling and high-resolution genomic copy number. Lab Invest 90:1285–1294
Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, Lubbe S, Spain S, Sullivan K, Fielding S, Jaeger E, Vijayakrishnan J, Kemp Z, Gorman M, Chandler I, Papaemmanuil E, Penegar S, Wood W, Sellick G, Qureshi M, Teixeira A, Domingo E, Barclay E, Martin L, Sieber O, CORGI Consortium, Kerr D, Gray R, Peto J, Cazier JB, Tomlinson I, Houlston RS (2007) A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet 39:1315–1317
Cui R, Okada Y, Jang SG, Ku JL, Park JG, Kamatani Y, Hosono N, Tsunoda T, Kumar V, Tanikawa C, Kamatani N, Yamada R, Kubo M, Nakamura Y, Matsuda K (2011) Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut 60:799–805
Di Sano F, Fazi B, Citro G, Lovat PE, Cesareni G, Piacentini M (2003) Glucosylceramide synthase and its functional interaction with RTN-1C regulate chemotherapeutic-induced apoptosis in neuroepithelioma cells. Cancer Res 63:3860–3865
Di Sano F, Fazi B, Tufi R, Nardacci R, Piacentini M (2007) Reticulon-1C acts as a molecular switch between endoplasmic reticulum stress and genotoxic cell death pathway in human neuroblastoma cells. J Neurochem 102:345–353
Dunlop MG, Dobbins SE, Farrington SM, Jones AM, Palles C, Whiffin N, Tenesa A, Spain S, Broderick P, Ooi LY, Domingo E, Smillie C, Henrion M, Frampton M, Martin L, Grimes G, Gorman M, Semple C, Ma YP, Barclay E, Prendergast J, Cazier JB, Olver B, Penegar S, Lubbe S, Chander I, Carvajal-Carmona LG, Ballereau S, Lloyd A, Vijayakrishnan J, Zgaga L, Rudan I, Theodoratou E, Colorectal Tumor Gene Identification (CORGI) Consortium, Starr JM, Deary I, Kirac I, Kovacević D, Aaltonen LA, Renkonen-Sinisalo L, Mecklin JP, Matsuda K, Nakamura Y, Okada Y, Gallinger S, Duggan DJ, Conti D, Newcomb P, Hopper J, Jenkins MA, Schumacher F, Casey G, Easton D, Shah M, Pharoah P, Lindblom A, Liu T, Swedish Low-Risk Colorectal Cancer Study Group, Smith CG, West H, Cheadle JP, COIN Collaborative Group, Midgley R, Kerr DJ, Campbell H, Tomlinson IP, Houlston RS (2012) Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet 44:770–776
Fan Y, Xiao W, Li Z, Li X, Chuang PY, Jim B, Zhang W, Wei C, Wang N, Jia W, Xiong H, Lee K, He JC (2015) RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun 6:7841
Fazi B, Melino S, De Rubeis S, Bagni C, Paci M, Piacentini M, Di Sano F (2009) Acetylation of RTN-1C regulates the induction of ER stress by the inhibition of HDAC activity in neuroectodermal tumors. Oncogene 28:3814–3824
Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S (2014) Use of epigenetic drugs in disease: an overview. Genet Epigenet 6:9–19
Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E, Farrington S, Prendergast JG, Pittman AM, Theodoratou E, Smith CG, Olver B, Walther A, Barnetson RA, Churchman M, Jaeger EE, Penegar S, Barclay E, Martin L, Gorman M, Mager R, Johnstone E, Midgley R, Niittymäki I, Tuupanen S, Colley J, Idziaszczyk S, COGENT Consortium, Thomas HJ, Lucassen AM, Evans DG, CORGI Consortium, COIN Collaborative Group, COINB Collaborative Group, Maher ER, Maughan T, Dimas A, Dermitzakis E, Cazier JB, Aaltonen LA, Pharoah P, Kerr DJ, Carvajal-Carmona LG, Campbell H, Dunlop MG, Tomlinson IP (2010) Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 42:973–977
Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I, Vijayakrishnan J, Sullivan K, Penegar S, Colorectal Cancer Association Study Consortium, Carvajal-Carmona L, Howarth K, Jaeger E, Spain SL, Walther A, Barclay E, Martin L, Gorman M, Domingo E, Teixeira AS, CoRGI Consortium, Kerr D, Cazier JB, Niittymäki I, Tuupanen S, Karhu A, Aaltonen LA, Tomlinson IP, Farrington SM, Tenesa A, Prendergast JG, Barnetson RA, Cetnarskyj R, Porteous ME, Pharoah PD, Koessler T, Hampe J, Buch S, Schafmayer C, Tepel J, Schreiber S, Völzke H, Chang-Claude J, Hoffmeister M, Brenner H, Zanke BW, Montpetit A, Hudson TJ, Gallinger S, Campbell H, Dunlop MG (2008) Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet 40:1426–1435
Jaeger E, Webb E, Howarth K, Carvajal-Carmona L, Rowan A, Broderick P, Walther A, Spain S, Pittman A, Kemp Z, Sullivan K, Heinimann K, Lubbe S, Domingo E, Barclay E, Martin L, Gorman M, Chandler I, Vijayakrishnan J, Wood W, Papaemmanuil E, Penegar S, Qureshi M, CORGI Consortium, Farrington S, Tenesa A, Cazier JB, Kerr D, Gray R, Peto J, Dunlop M, Campbell H, Thomas H, Houlston R, Tomlinson I (2008) Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat Genet 40:26–28
Jia WH, Zhang B, Matsuo K, Shin A, Xiang YB, Jee SH, Kim DH, Ren Z, Cai Q, Long J, Shi J, Wen W, Yang G, Delahanty RJ, Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), Colon Cancer Family Registry (CCFR), Ji BT, Pan ZZ, Matsuda F, Gao YT, Oh JH, Ahn YO, Park EJ, Li HL, Park JW, Jo J, Jeong JY, Hosono S, Casey G, Peters U, Shu XO, Zeng YX, Zheng W (2013) Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet. 45:191–196
Kuang E, Wan Q, Li X, Xu H, Liu Q, Qi Y (2005) ER Ca2+ depletion triggers apoptotic signals for endoplasmic reticulum (ER) overload response induced by overexpressed reticulon 3 (RTN3/HAP). J Cell Physiol 204:549–559
Levin B, Lieberman DA, McFarland B, Andrews KS, Brooks D, Bond J, Dash C, Giardiello FM, Glick S, Johnson D, Johnson CD, Levin TR, Pickhardt PJ, Rex DK, Smith RA, Thorson A, Winawer SJ, American Cancer Society Colorectal Cancer Advisory Group, US Multi-Society Task Force, American College of Radiology Colon Cancer Committee (2008) Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 134:1570–1595
Levina E, Ji H, Chen M, Baig M, Oliver D, Ohouo P, Lim CU, Schools G, Carmack S, Ding Y, Broude EV, Roninson IB, Buttyan R, Shtutman M (2015) Identification of novel genes that regulate androgen receptor signaling and growth of androgen-deprived prostate cancer cells. Oncotarget 6:13088–13104
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K (2000) Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343:78–85
Lieberman DA, Rex DK, Winawer SJ, Giardiello FM, Johnson DA, Levin TR, United States Multi-Society Task Force on Colorectal Cancer (2012) Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 143:844–857
Loo LW, Cheng I, Tiirikainen M, Lum-Jones A, Seifried A, Dunklee LM, Church JM, Gryfe R, Weisenberger DJ, Haile RW, Gallinger S, Duggan DJ, Thibodeau SN, Casey G, Le Marchand L (2012) cis-Expression QTL analysis of established colorectal cancer risk variants in colon tumors and adjacent normal tissue. PLoS One 7:e30477
Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, Berndt SI, Bézieau S, Brenner H, Butterbach K, Caan BJ, Campbell PT, Carlson CS, Casey G, Chan AT, Chang-Claude J, Chanock SJ, Chen LS, Coetzee GA, Coetzee SG, Conti DV, Curtis KR, Duggan D, Edwards T, Fuchs CS, Gallinger S, Giovannucci EL, Gogarten SM, Gruber SB, Haile RW, Harrison TA, Hayes RB, Henderson BE, Hoffmeister M, Hopper JL, Hudson TJ, Hunter DJ, Jackson RD, Jee SH, Jenkins MA, Jia WH, Kolonel LN, Kooperberg C, Küry S, Lacroix AZ, Laurie CC, Laurie CA, Le Marchand L, Lemire M, Levine D, Lindor NM, Liu Y, Ma J, Makar KW, Matsuo K, Newcomb PA, Potter JD, Prentice RL, Qu C, Rohan T, Rosse SA, Schoen RE, Seminara D, Shrubsole M, Shu XO, Slattery ML, Taverna D, Thibodeau SN, Ulrich CM, White E, Xiang Y, Zanke BW, Zeng YX, Zhang B, Zheng W, Hsu L, Colon Cancer Family Registry and the Genetics and Epidemiology of Colorectal Cancer Consortium (2013) Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 144:799–807
Peters U, Hutter CM, Hsu L, Schumacher FR, Conti DV, Carlson CS, Edlund CK, Haile RW, Gallinger S, Zanke BW, Lemire M, Rangrej J, Vijayaraghavan R, Chan AT, Hazra A, Hunter DJ, Ma J, Fuchs CS, Giovannucci EL, Kraft P, Liu Y, Chen L, Jiao S, Makar KW, Taverna D, Gruber SB, Rennert G, Moreno V, Ulrich CM, Woods MO, Green RC, Parfrey PS, Prentice RL, Kooperberg C, Jackson RD, Lacroix AZ, Caan BJ, Hayes RB, Berndt SI, Chanock SJ, Schoen RE, Chang-Claude J, Hoffmeister M, Brenner H, Frank B, Bézieau S, Küry S, Slattery ML, Hopper JL, Jenkins MA, Le Marchand L, Lindor NM, Newcomb PA, Seminara D, Hudson TJ, Duggan DJ, Potter JD, Casey G (2012) Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet 131:217–234
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Read TE, Kodner IJ (1999) Colorectal cancer: risk factors and recommendations for early detection. Am Fam Physician 59:3083–3092
Schäffer AA, Lemire M, Ott J, Lathrop GM, Weeks DE (2011) Coordinated conditional simulation with SLINK and SUP of many markers linked or associated to a trait in large pedigrees. Hum Hered 71:126–134
Schmit SL, Schumacher FR, Edlund CK, Conti DV, Raskin L, Lejbkowicz F, Pinchev M, Rennert HS, Jenkins MA, Hopper JL, Buchanan DD, Lindor NM, Le Marchand L, Gallinger S, Haile RW, Newcomb PA, Huang SC, Rennert G, Casey G, Gruber SB (2014) A novel colorectal cancer risk locus at 4q32.2 identified from an international genome-wide association study. Carcinogenesis 35:2512–2519
Schumacher FR, Schmit SL, Jiao S, Edlund CK, Wang H, Zhang B, Hsu L, Huang SC, Fischer CP, Harju JF, Idos GE, Lejbkowicz F, Manion FJ, McDonnell K, McNeil CE, Melas M, Rennert HS, Shi W, Thomas DC, Van Den Berg DJ, Hutter CM, Aragaki AK, Butterbach K, Caan BJ, Carlson CS, Chanock SJ, Curtis KR, Fuchs CS, Gala M, Giocannucci EL, Gogarten SM, Hayes RB, Henderson B, Hunter DJ, Jackson RD, Kolonel LN, Kooperberg C, Kury S, LaCroix A, Laurie CC, Laurie CA, Lemire M, Levine D, Ma J, Makar KW, Qu C, Taverna D, Ulrich CM, Wu K, Kono S, West DW, Berndt SI, Bezieau S, Brenner H, Campbell PT, Chan AT, Chang-Claude J, Coetzee GA, Conti DV, Duggan D, Figueiredo JC, Fortini BK, Gallinger SJ, Gauderman WJ, Giles G, Green R, Haile R, Harrison TA, Hoffmeister M, Hopper JL, Hudson TJ, Jacobs E, Iwasaki M, Jee SH, Jenkins M, Jia WH, Joshi A, Li L, Lindor NM, Matsuo K, Moreno V, Mukherjee B, Newcomb PA, Potter JD, Raskin L, Rennert G, Rosse S, Severi G, Schoen RE, Seminara D, Shu XO, Slattery ML, Tsugane S, White E, Xiang YB, Zanke BW, Zheng W, Le Marchand L, Casey G, Gruber SB, Peters U (2015) Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun. 6:7138
Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics. CA Cancer J Clin 64:9–29
Slattery ML, Levin TR, Ma K, Goldgar D, Holubkov R, Edwards S (2003) Family history and colorectal cancer: predictors of risk. Cancer Causes Control 14:879–887
Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, Semple C, Clark AJ, Reid FJ, Smith LA, Kavoussanakis K, Koessler T, Pharoah PD, Buch S, Schafmayer C, Tepel J, Schreiber S, Völzke H, Schmidt CO, Hampe J, Chang-Claude J, Hoffmeister M, Brenner H, Wilkening S, Canzian F, Capella G, Moreno V, Deary IJ, Starr JM, Tomlinson IP, Kemp Z, Howarth K, Carvajal-Carmona L, Webb E, Broderick P, Vijayakrishnan J, Houlston RS, Rennert G, Ballinger D, Rozek L, Gruber SB, Matsuda K, Kidokoro T, Nakamura Y, Zanke BW, Greenwood CM, Rangrej J, Kustra R, Montpetit A, Hudson TJ, Gallinger S, Campbell H, Dunlop MG (2008) Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40:631–637
Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, CORGI Consortium, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier JB, Houlston R (2007) A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 39:984–988
Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, Tenesa A, Jones AM, Howarth K, Palles C, Broderick P, Jaeger EE, Farrington S, Lewis A, Prendergast JG, Pittman AM, Theodoratou E, Olver B, Walker M, Penegar S, Barclay E, Whiffin N, Martin L, Ballereau S, Lloyd A, Gorman M, Lubbe S, COGENT Consortium, CORGI Collaborators, EPICOLON Consortium, Howie B, Marchini J, Ruiz-Ponte C, Fernandez-Rozadilla C, Castells A, Carracedo A, Castellvi-Bel S, Duggan D, Conti D, Cazier JB, Campbell H, Sieber O, Lipton L, Gibbs P, Martin NG, Montgomery GW, Young J, Baird PN, Gallinger S, Newcomb P, Hopper J, Jenkins MA, Aaltonen LA, Kerr DJ, Cheadle J, Pharoah P, Casey G, Houlston RS, Dunlop MG (2011) Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet 7:e1002105
Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, Jaeger E, Fielding S, Rowan A, Vijayakrishnan J, Domingo E, Chandler I, Kemp Z, Qureshi M, Farrington SM, Tenesa A, Prendergast JG, Barnetson RA, Penegar S, Barclay E, Wood W, Martin L, Gorman M, Thomas H, Peto J, Bishop DT, Gray R, Maher ER, Lucassen A, Kerr D, Evans DG, CORGI Consortium, Schafmayer C, Buch S, Völzke H, Hampe J, Schreiber S, John U, Koessler T, Pharoah P, van Wezel T, Morreau H, Wijnen JT, Hopper JL, Southey MC, Giles GG, Severi G, Castellví-Bel S, Ruiz-Ponte C, Carracedo A, Castells A, EPICOLON Consortium, Försti A, Hemminki K, Vodicka P, Naccarati A, Lipton L, Ho JW, Cheng KK, Sham PC, Luk J, Agúndez JA, Ladero JM, de la Hoya M, Caldés T, Niittymäki I, Tuupanen S, Karhu A, Aaltonen L, Cazier JB, Campbell H, Dunlop MG, Houlston RS (2008) A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 40:623–630
Wang H, Burnett T, Kono S, Haiman CA, Iwasaki M, Wilkens LR, Loo LW, Van Den Berg D, Kolonel LN, Henderson BE, Keku TO, Sandler RS, Signorello LB, Blot WJ, Newcomb PA, Pande M, Amos CI, West DW, Bézieau S, Berndt SI, Zanke BW, Hsu L, Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), Lindor NM, Haile RW, Hopper JL, Jenkins MA, Gallinger S, Casey G, Colon Cancer Family Registry (CCFR), Stenzel SL, Schumacher FR, Peters U, Gruber SB, Colorectal Transdisciplinary Study (CORECT), Tsugane S, Stram DO, Le Marchand L (2014) Trans-ethnic genome-wide association study of colorectal cancer identifies a new susceptibility locus in VTI1A. Nat Commun 5:4613
Whiffin N, Hosking FJ, Farrington SM, Palles C, Dobbins SE, Zgaga L, Lloyd A, Kinnersley B, Gorman M, Tenesa A, Broderick P, Wang Y, Barclay E, Hayward C, Martin L, Buchanan DD, Win AK, Hopper J, Jenkins M, Lindor NM, Newcomb PA, Gallinger S, Conti D, Schumacher F, Casey G, Liu T, Swedish Low-Risk Colorectal Cancer Study Group, Campbell H, Lindblom A, Houlston RS, Tomlinson IP, Dunlop MG (2014) Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet 23:4729–4737
Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O’Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellié C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG (2007) Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39:989–994
Zhang B, Jia WH, Matsuda K, Kweon SS, Matsuo K, Xiang YB, Shin A, Jee SH, Kim DH, Cai Q, Long J, Shi J, Wen W, Yang G, Zhang Y, Li C, Li B, Guo Y, Ren Z, Ji BT, Pan ZZ, Takahashi A, Shin MH, Matsuda F, Gao YT, Oh JH, Kim S, Ahn YO, Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), Chan AT, Chang-Claude J, Slattery ML, Colorectal Transdisciplinary (CORECT) Study, Gruber SB, Schumacher FR, Stenzel SL, Colon Cancer Family Registry (CCFR), Casey G, Kim HR, Jeong JY, Park JW, Li HL, Hosono S, Cho SH, Kubo M, Shu XO, Zeng YX, Zheng W (2014) Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 46:533–542
Acknowledgments
The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
ASTERISK: We are very grateful to Dr. Bruno Buecher without whom this project would not have existed. We also thank all those who agreed to participate in this study, including the patients and the healthy control persons, as well as all the physicians, technicians and students.
CANCER PREVENTION STUDY II: CPS-II is supported by the American Cancer Society (ACS). We would like to thank the CPS-II participants and the CPS-II Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program.
DACHS: We thank all participants and cooperating clinicians, and Ute Handte-Daub, Utz Benscheid, Muhabbet Celik and Ursula Eilber for excellent technical assistance.
GECCO: The authors would like to thank all those at the GECCO Coordinating Center for helping bring together the data and people that made this project possible. The authors also acknowledge COMPASS (Comprehensive Center for the Advancement of Scientific Strategies) at the Fred Hutchinson Cancer Research Center for their work harmonizing the GECCO epidemiological data set. The authors acknowledge Dave Duggan and team members at TGEN (Translational Genomics Research Institute), the Broad Institute, and the Génome Québec Innovation Center for genotyping DNA samples of cases and controls, and for scientific input for GECCO.
HPFS, NHS and PHS: We would like to acknowledge Patrice Soule and Hardeep Ranu of the Dana Farber Harvard Cancer Center High-Throughput Polymorphism Core who assisted in the genotyping for NHS, HPFS, and PHS under the supervision of Dr. Immaculata Devivo and Dr. David Hunter, Qin (Carolyn) Guo and Amit Joshi who assisted in programming for NHS and HPFS, and Haiyan Zhang who assisted in programming for the PHS. We would like to thank the participants and staff of the Nurses’ Health Study and the Health Professionals Follow-Up Study, for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
PLCO: The authors thank Drs. Christine Berg and Philip Prorok, Division of Cancer Prevention, National Cancer Institute, the Screening Center investigators and staff or the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, Mr. Tom Riley and staff, Information Management Services, Inc., Ms. Barbara O’Brien and staff, Westat, Inc., and Drs. Bill Kopp and staff, SAIC-Frederick. Most importantly, we acknowledge the study participants for their contributions to making this study possible. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by NCI.
PMH: The authors would like to thank the study participants and staff of the Hormones and Colon Cancer study.
WHI: The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
GECCO: National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088; R01 CA059045).
ASTERISK: a Hospital Clinical Research Program (PHRC) and supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC), the Association Anne de Bretagne Génétique and the Ligue Régionale Contre le Cancer (LRCC).
CCFR: This work was supported by grant UM1 CA167551 from the National Cancer Institute and through cooperative agreements with the following CCFR centers: Australasian Colorectal Cancer Family Registry (U01 CA074778 and U01/U24 CA097735), Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (U01/U24 CA074800), Ontario Familial Colorectal Cancer Registry (U01/U24 CA074783), Seattle Colorectal Cancer Family Registry (U01/U24 CA074794), USC Consortium Colorectal Cancer Family Registry U01/U24 CA074799). The Colon CFR GWAS was supported by funding from the National Cancer Institute, National Institutes of Health (U01 CA122839 and R01 CA143237 to Graham Casey). CRC tumor and normal tissues used for the gene expression study were made available by The Jeremy Jass Memorial Pathology Bank. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the CCFR.
COLO2&3: National Institutes of Health (R01 CA60987).
CORECT: National Cancer Institute, National Institutes of Health under RFA# CA-09-002 (U19 CA148107). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in CORECT, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or CORECT.
DACHS: German Research Council (Deutsche Forschungsgemeinschaft, BR 1704/6-1, BR 1704/6-3, BR 1704/6-4 and CH 117/1-1), and the German Federal Ministry of Education and Research (01KH0404 and 01ER0814).
DALS: National Institutes of Health (R01 CA48998 to M. L. Slattery); HPFS is supported by the National Institutes of Health (P01 CA 055075, UM1 CA167552, R01 137178, R01 CA151993 and P50 CA127003), NHS by the National Institutes of Health (UM1 CA186107, R01 CA137178, P01 CA87969, R01 CA151993 and P50 CA127003,) and PHS by the National Institutes of Health (R01 CA042182).
MEC: National Institutes of Health (R37 CA54281, P01 CA033619, and R01 CA63464).
OFCCR: National Institutes of Health, through funding allocated to the Ontario Registry for Studies of Familial Colorectal Cancer (U01 CA074783); see CCFR section above. Additional funding toward genetic analyses of OFCCR includes the Ontario Research Fund, the Canadian Institutes of Health Research, and the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation.
PLCO: Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Additionally, a subset of control samples were genotyped as part of the Cancer Genetic Markers of Susceptibility (CGEMS) Prostate Cancer GWAS (Yeager, M et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007 May;39(5):645–9), Colon CGEMS pancreatic cancer scan (PanScan) (Amundadottir, L et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009 Sep;41(9):986–90, and Petersen, GM et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010 Mar;42(3):224–8), and the Lung Cancer and Smoking study (Landi MT, et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am J Hum Genet. 2009 Nov;85(5):679–91). The prostate and PanScan study datasets were accessed with appropriate approval through the dbGaP online resource (http://cgems.cancer.gov/data/) accession numbers phs000207.v1.p1 and phs000206.v3.p2, respectively, and the lung datasets were accessed from the dbGaP website (http://www.ncbi.nlm.nih.gov/gap) through accession number phs000093.v2.p2. Funding for the Lung Cancer and Smoking study was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438. For the lung study, the GENEVA Coordinating Center provided assistance with genotype cleaning and general study coordination, and the Johns Hopkins University Center for Inherited Disease Research conducted genotyping.
PMH: National Institutes of Health (R01 CA076366 to P.A. Newcomb).
VITAL: National Institutes of Health (K05 CA154337).
WHI: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C.
Additional information
M. Lemire and C. Qu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lemire, M., Qu, C., Loo, L.W.M. et al. A genome-wide association study for colorectal cancer identifies a risk locus in 14q23.1. Hum Genet 134, 1249–1262 (2015). https://doi.org/10.1007/s00439-015-1598-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-015-1598-6