
Abstract
An interplay between gene expression, mineral concentration, and beef quality traits in Bos indicus muscle has been reported previously under a network approach. However, growing evidence suggested that miRNAs not only modulate gene expression but are also involved with mineral homeostasis. To our knowledge, understanding of the miRNA–gene expression-mineral concentration relationship in mammals is still minimal. Therefore, we carried out a miRNA co-expression and multi-level miRNA–mRNA integration analyses to predict the putative drivers (miRNAs and genes) associated with muscle mineral concentration in Nelore steers. In this study, we identified calcium and iron to be the pivotal minerals associated with miRNAs and gene targets. Furthermore, we identified the miR-29 family (miR-29a, -29b, -29c, -29d-3p, and -29e) as the putative key regulators modulating mineral homeostasis. The miR-29 family targets genes involved with AMPK, insulin, mTOR, and thyroid hormone signaling pathways. Finally, we reported an interplay between miRNAs and minerals acting cooperatively to modulate co-expressed genes and signaling pathways both involved with mineral and energy homeostasis in Nelore muscle. Although we provided some evidence to understand this complex relationship, future work should determine the functional implications of minerals for miRNA levels and their feedback regulation system.\\An interplay between gene expression, mineral concentration, and beef quality traits in Bos indicus muscle has been reported previously under a network approach. However, growing evidence suggested that miRNAs not only modulate gene expression but are also involved with mineral homeostasis. To our knowledge, understanding of the miRNA–gene expression-mineral concentration relationship in mammals is still minimal. Therefore, we carried out a miRNA co-expression and multi-level miRNA–mRNA integration analyses to predict the putative drivers (miRNAs and genes) associated with muscle mineral concentration in Nelore steers. In this study, we identified calcium and iron to be the pivotal minerals associated with miRNAs and gene targets. Furthermore, we identified the miR-29 family (miR-29a, -29b, -29c, -29d-3p, and -29e) as the putative key regulators modulating mineral homeostasis. The miR-29 family targets genes involved with AMPK, insulin, mTOR, and thyroid hormone signaling pathways. Finally, we reported an interplay between miRNAs and minerals acting cooperatively to modulate co-expressed genes and signaling pathways both involved with mineral and energy homeostasis in Nelore muscle. Although we provided some evidence to understand this complex relationship, future work should determine the functional implications of minerals for miRNA levels and their feedback regulation system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Advancements have been made to understand mineral metabolism and its role in human health and animal production (Suttle 2010; Fleet et al. 2011). However, mineral deficiencies, mainly iron and zinc, are the most prevalent worldwide nutritional disorder (Ritchie and Roser 2018). Although required in small amounts in the diet, an adequate mineral supply is necessary for the body’s metabolism, which includes muscle performance and energy utilization (Garmyn et al. 2011; Tizioto et al. 2015). Macro and micro minerals have multiple roles. They are crucial for biological processes such as DNA synthesis, gene expression, cell growth and differentiation, and energy metabolism (Fleet et al. 2011; Davis et al. 2012a; Beckett et al. 2014). Mineral deficiency or overload are potentially deleterious (Suttle 2010). Therefore, tight regulation is necessary to keep mineral levels within a narrow range. Besides the intake-output imbalance and environmental factors, several transcriptional and post-transcriptional mechanisms involved in mineral homeostasis were reported (Suttle 2010).
Genome-wide association studies in cattle have suggested that muscle mineral content is under genetic control (Tizioto et al. 2015; Mateescu et al. 2017). Tizioto et al. (2015) reported candidate genes with an additive effect on muscle mineral concentration in cattle. Several differentially expressed genes underlie the complex network regulating muscle mineral deposition in Nelore steers (Diniz et al. 2019; Afonso et al. 2019). Based on co-expression analysis, we reported that genes acting in pathways related to energy and protein metabolism were associated with variation in muscle mineral concentration. Furthermore, studies have reported that minerals not only modulate gene expression but are involved with miRNA biosynthesis, which in turn regulates mineral homeostasis (El Azzouzi et al. 2013; Beckett et al. 2014; Magenta et al. 2016). MiRNAs have a role in a wide range of biological functions (Sengar et al. 2018), underpinning traits like meat tenderness (Kappeler et al. 2019) and intramuscular fat content (de Oliveira et al. 2018, Oliveira et al. 2018). Likewise, Ca, Fe, and Zn were also associated with these meat traits (Garmyn et al. 2011; Tizioto et al. 2014; Casas et al. 2014; Ahlberg et al. 2014) and modulate miRNA biosynthesis (Beckett et al. 2014; Magenta et al. 2016; Ripa et al. 2017).
Mineral metabolism should be viewed as a system, both because of their interactions among themselves, as well as for their role with the functional genome variation in different regulatory layers (Fleet et al. 2011). Supporting this holistic approach, a growing number of studies have shown the interaction between minerals and gene expression (Davis et al. 2012b; Xu et al. 2013; Beckett et al. 2014). In this scenario, a feedback loop acts as the mechanism modulating the miRNA–gene–mineral interaction (Beckett et al. 2014). Integrative genomic approaches have proven to be a fruitful tool to study these interactions (Su et al. 2014; Feng et al. 2018). However, to date, there is still a knowledge gap regarding the genetic architecture underlying muscle mineral homeostasis, as well as the miRNA–gene–mineral relationship.
Therefore, we carried out exploratory miRNA co-expression and multi-level miRNA–mRNA integration analyses to uncover the pathways and regulatory networks underlying the mineral concentration in Nelore cattle muscle. From a systems biology perspective, we identified miRNAs acting cooperatively to modulate co-expressed genes and signaling pathways, involved with mineral and energy homeostasis. Understanding this complex relationship opens up several nutrigenomic opportunities. For example, by identifying mineral-responsive miRNA and their targets, one can manage the animal’s diet to regulate the miRNA expression and, consequently, the associated phenotype.
Results
We established co-expression networks to shed light on the miRNA–trait relationship, as well as the regulatory mechanisms among miRNA, gene targets, and mineral concentration. We first identified co-expressed miRNA modules relying on the Weighted Correlation Network Analysis (WGCNA) method. Next, a linear association model was fitted to identify modules biologically associated with the phenotypes. Subsequently, we integrated these miRNAs to gene co-expression modules identified in our previous study (Diniz et al. 2019), based on module eigengene (MEs) correlation. We intersected the significant negatively correlated modules to miRNA–mRNA interactions predicted from TargetScan (Agarwal et al. 2015). Lastly, we carried out a transcription factor prediction and over-representation analysis to bring up putative regulators and biological pathways (Fig. 1).

Co-expression pipeline analysis. The main analyses steps to data processing and co-expression, and data integration are shown in white boxes. Tools applied in each analysis are shown in green boxes. Inputs and outputs are shown in lightyellow boxes. QC quality control, MM module membership. amRNA modules from (Diniz et al. 2019). bBased on the curated compendium of bovine transcription factors (TFs) from (de Souza et al. 2018)
Phenotypic and sequencing data
The heritability for mineral concentration evaluated for this Nelore population ranged from 0.29 to 0.33, as previously published (Tizioto et al. 2015). There was a strong and significant correlation among the minerals themselves ranging from 0.45 to 0.99 (Diniz et al. 2019). Descriptive statistics for the mineral concentrations are reported in Supplementary Table S1 (Diniz et al. 2019). Among the minerals, the average values for Ca and Fe were 153.95 and 46.48 mg/kg, respectively. The lowest values were for Se (0.14 mg/kg) and Mn (0.20 mg/kg).
MiRNA sequencing from 50 Longissimus thoracis (LT) samples yielded 1.4 million sequence reads per sample, on average. An average of 84.7% of the reads was mapped to Bos taurus ARS-UCD 1.2. After filtering out the lowly expressed miRNAs, we kept 343 known miRNAs used for the co-expression analysis.
Co-expression analysis and module-trait association
To identify the miRNA co-expression network, we clustered the 343 miRNAs adopting the WGCNA framework (Langfelder and Horvath 2008). We gathered 24 modules labeled by color names and module sizes ranged from 5 (miR.MEdarkgreen, miR.MEdargrey, and miR.MEred) to 34 miRNAs (miR.MEturquoise) (Supplementary Table S1). We calculated the proportion of variance explained by the MEs, which ranged from 0.29 (miR.MEgreen) to 0.67 (miR.MEdarkgreen) (Supplementary Table S1).
We fitted a linear model to associate the MEs with mineral concentration, selected the significant (p ≤ 0.05) modules, and investigated their biological relevance. Our approach identified nine miRNA modules, which were significantly (p ≤ 0.05) associated with at least one mineral, as summarized in Table 1. The linear model coefficient values for the significant associations ranged from − 0.058 to 0.071 (Supplementary Fig. S1). A positive association meant the trait measure increases with increasing “eigengene expression” or vice-versa for a negative association. We found the highest number of significantly associated traits between miR.MEcyan (positively associated with ten minerals) and miR.MEgreen (negatively associated with six minerals) modules. A positive association between minerals and miR.MEbrown (Ca, Na, and S), miR.MEmidnightblue (Ca, Fe, S, and Zn), and miR.MEgrey60 (Fe) was identified. Further, we showed a negative relationship between minerals and the miR.MElightyellow (Ca, Mg, and Na), miR.MEmagenta (Fe), miR.MEtan (Cu), and miR.MEred (Cu and Mn) modules.
Next, we selected the miRNAs with a pivotal role in the network topology and biological pathways based on the module membership (MM) criteria (Langfelder and Horvath 2008). We identified 50 miRNAs with a MM ≥ 0.7 (Table 1) that belong to 23 families through the nine associated modules (Supplementary Table S2). The main miRNA families identified were let-7 and miR-29, with six members each, followed by miR-154 and miR-199, both with four miRNAs.
MiRNA–mRNA regulatory network and identification of key transcription factors
To have an overview of the miRNA–gene–trait interactions and to better understand their regulatory relationship, we integrated miRNA and mRNA modules. We selected 15 mRNA modules associated with mineral concentration and meat quality traits (intramuscular fat content—IMF, and tenderness-WBSF7) from our previous work (Diniz et al. 2019) (Supplementary Table S3) and the nine miRNA modules reported here. We identified 48 animals with paired data (miRNA and mRNA) and calculated Pearson’s correlation among the MEs. A total of 16 pairs of miRNAs and mRNAs MEs showed significant negative correlations, which ranged from r = − 0.3 to r = − 0.68 (p ≤ 0.05) (Fig. 2). We further identified strong, positive, and significant module correlations ranging from r = 0.3 to r = 0.61. Nonetheless, it is still unclear whether the positively correlated pairs are a direct (Mamdani et al. 2015) or intermediate, such as feedback motifs, miRNA effect (Ritchie et al. 2009; Su et al. 2014). Thus, as the main direct effect of miRNAs is downregulating the mRNA abundance (Su et al. 2014), we focused on the negative correlations for further investigations.

MiRNA–mRNA module correlation. MiRNA (x-axis) and mRNA (y-axis) modules are labeled by color. The matrix is color-coded based on Pearson’s correlation (p values in the parenthesis) according to the legend. Positive and negative correlations are showed in red and green colors, respectively. Significant negative correlations (p ≤ 0.05) are highlighted with a red rectangle
We found the highest number of significant negative correlated modules between miR.MEcyan (MEgrey60, MEgreenyellow, and MEgreen), miR.MEtan (MEorange, MEsalmon, and MEyellow), and miR.MElightyellow (MElightyellow, MEsalmon, and MEyellow), followed by miR.MEmidnightblue (MEgreenyellow and MEgrey60) (Fig. 2). The miR.MEbrown, miR.MEgreen, and miR.MEred were correlated with MEgreenyellow, MEroyalblue, MEdarkturquoise, respectively. Furthermore, the miR.MEmagenta was correlated with MEroyalblue and MEdarkgrey. No significant negative correlations were identified between miR.MEgrey60 and any mRNA MEs. The network among all the trait-correlated MEs, as well as the negatively correlated mRNA–miRNA, showed that Ca, Fe, and Mg were the most associated minerals (Fig. 3).

Network of negatively correlated miRNA–mRNA modules and associated phenotypes in Nelore cattle muscle. V, hexagon, and ellipse shapes show, miRNA, mRNA, and phenotypes, respectively. Each arrow indicates the direction of regulation
To identify the miRNA–target pairs and shed light on their role in biological pathways related to mineral concentration, we applied two complementary approaches. Firstly, for the 50 miRNA hubs, we used the hoardeR package (Fischer and Sironen 2016) to build a list of predicted targets of cattle miRNA hubs from TargetScan. In total, we reported 8123 unique genes, out of 24,698 putative targets, expressed in the mRNA muscle transcriptome from the same animals (Supplementary Table S2), among the ten modules (Table 2). No correlations were observed between the miR.MEgrey60 (five hubs) with any of the mRNA modules. Due to that, miR.MEgrey60 was not considered for subsequent analyses. Then, to establish the most likely miRNA–target pairs, we intersected the predicted interactions with the significant negatively correlated miRNA–mRNA MEs (Supplementary Table S4). Putative new interactions based only on the negative ME correlation are in Supplementary Table S4.
MiRNAs targeted several genes. The miRNA–gene target interaction network, with a total of 4045 interactions among the ten mRNA modules, corresponding to 1815 unique targets, is represented in Supplementary Fig. S2 and Supplementary Table S4. On average, 41 out of 45 hub miRNAs targeted 98 genes with a maximum of 600 genes (Fig. 4, Supplementary Table S4). The miR-29 family (miR-29e, 29a, 29b, 29c, 29d-3p, in order of the number of targets) targeted the highest number of genes. At least two miRNAs targeted around 50% of the genes. We found that the genes HLF and TRAF3 were targeted by ten miRNAs each, followed by ATP2B2, DYNLL2, and YY1 with nine regulators. However, we did not find targets for the bta-miRs- 410, -411a, and -487b for miR.MEred, as well as for the bta-let-7c (miR.MEtan).

Number of targeted genes by miRNA co-expression network in Nelore muscle (ranked in descending order)
To identify enriched TFs targeted by miRNAs, 1815 reported genes were screened against the compendium of bovine transcription factors (de Souza et al. 2018). We found 131 TFs, of which at least two miRNAs targeted 58% (76/131) of the TFs (Supplementary Table S4). Among the TFs, HLF, YY1, and THRA were regulated by 10, 9, and 8 different miRNAs, respectively. We further evaluated the connectivity for these TFs and considered their MM from our previous work (Diniz et al. 2019). We identified 30 TFs with a MM higher than 0.7, which highlights their importance for the network’s topology architecture (Supplementary Table S4).
Pathway over-representation analysis
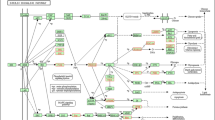
To reveal the biological pathways in which the miRNA targets acted, we carried out a pathway over-representation analysis using Cluego (Bindea et al. 2009). As we sought to point a biological relationship between miRNAs and mRNAs, we focused the analysis on those genes that overlapped between TargetScan prediction and mRNA–miRNA associated modules (Supplementary Table S4). Based on that approach, we identified 37 significant enriched KEGG pathways (pV ≤ 0.05) (Fig. 5) among the five modules (Supplementary Table S5).

Over-represented signaling pathway network of miRNA target genes in Nelore muscle co-expression. The miRNAs are colored based on their clustered module (miRMEbrown, miRMEcyan, miRMElightyellow, ad miRMEtan)
The miR.MEcyan showed the highest number of enriched pathways, such as those related to protein (e.g., mTOR signaling pathway, protein processing, ubiquitin-mediated proteolysis) and energy metabolism (insulin and thyroid hormone signaling pathways). Additional pathways were identified including ferroptosis (miR.MEbrown), TGF-beta signaling pathway, focal adhesion (miR.MElightyellow), insulin resistance (miR.MEmidnightblue), and ECM-receptor interaction (miR.MEtan). No results from miR.MEgreen, miR.MEmagenta, or miR.MEred were retrieved.
Discussion
Previously, we reported an interplay among gene expression, mineral concentration, and meat quality traits based on the gene co-expression network (Diniz et al. 2019). As part of this picture, in addition to the gene expression regulation role, growing evidence shows the cross talk between miRNA and mineral homeostasis (Xu et al. 2013; An et al. 2014; Beckett et al. 2014). However, our understanding of the miRNA–gene–mineral relationship is still minimal. Herein, we reported an interplay among miRNAs, mRNAs, and minerals in Nelore muscle for the first time. We identified mineral-associated co-expressed miRNAs along with multi-level miRNA–mRNA integration. This combined information sheds light upon regulatory networks that contribute to mineral metabolism in Nelore muscle. The results from this exploratory in silico study showed a strong relationship among several biological pathways to maintain cellular homeostasis (He and Jiang 2016).
MiRNA module association and regulatory network
Among nine co-expressed modules, we found 50 hub, out of 343 miRNAs, associated with at least one mineral (p ≤ 0.05). By integrating the associated miRNA–mRNA MEs and intersecting with the TargetScan prediction, we reduced the number of false-positive pairs when compared to predictions alone (Mamdani et al. 2015) and pointed out putative mineral–mRNA–miRNA relationships. Furthermore, by focusing the analysis on the MEs, one key advantage of our approach was that we alleviated the multiple testing problems inherent in RNA-Seq multi-level data integration (Langfelder and Horvath 2008). Our approach gathered 41 miRNAs and 1815 target genes that were inversely correlated. By definition, miRNA hubs play a pivotal role both in the network’s topology (Langfelder and Horvath 2008) and gene translation coordination within the transcriptional network (Su et al. 2014). We found genes targeted by several miRNAs (over-targeted), as well as miRNAs with multiple targets. It is worth highlighting that TFs, such as HLF, YY1, and THRA were among the over-targeted genes. Likewise, the miR-29 family, associated with the concentration of ten minerals, showed the highest number of putative targets. These results reinforce the general concept that genes are redundantly regulated by multiple miRNA interactions (Su et al. 2014), as well as by a putative combinatorial TFs co-regulation (Shalgi et al. 2007).
The interplay among Ca, Fe, and miRNA expression
Because we have a lack of knowledge regarding the miRNA–mineral relationship in cattle so far, and most mammalian genes are conserved targets of miRNAs (Friedman et al. 2008), we will draw a parallel between the results identified here and the current research in humans and animal models. Ca and Fe showed correlation with other minerals ranging from moderate to strong (Diniz et al. 2019) and were the main minerals associated with miRNA MEs. Furthermore, these minerals have been associated with gene expression and miRNA biogenesis in a bi-directional regulatory circuit (Davis and Clarke 2013; Beckett et al. 2014; Magenta et al. 2016). Thus, our discussion will focus mainly on Ca and Fe and their relationship with miRNA and gene targets.
We identified the genes PCBP1 and PCBP2 [Poly (RC) Binding Protein] were targeted by the miR-21-5p (miR.MEbrown). Additionally, the Argonaute family members, AGO1, AGO2, AGO3, and AGO4, were mutually targeted by miR-29e (miR.MEcyan). Li et al. (2012) reported that cytosolic Fe could modulate the PCBP2–AGO1 relationship, leading to decreased mature miRNA production. Some of the hub miRNAs clustered into the miR.MEcyan and miR.MEbrown were correlated with minerals and gene modules that were previously associated with miRNA biosynthesis and mineral homeostasis in humans. The miR-29 family members (-a, -b, -c, -d-3p and -e) along with miR-15a, targeted most of the genes identified, including those with known involvement in Ca, Cu, Fe, and Zn metabolism (Li et al. 2012). For instance, the transcription factors YY1 and SP1, both targets of miR-29 family, bind the human transferrin (TF) gene and modulate its expression level (Amodio et al. 2015). In addition, YY1 was targeted by miR-19a, -19b (miR.MEbrown), -30c, and 30f (miR.MEmidnightblue). Still concerning Fe metabolism, we found the transferrin receptor gene (TFRC) was targeted by four miRNAs (miR-103, -107, -15a, and 29e). In addition to other mechanisms, the level of free Fe in biological fluids is controlled by the interplay between TF and TFRC (Xu et al. 2013).
The interaction between reactive oxygen species (ROS) and hypoxia has a crucial role in miRNA biogenesis (Dengler et al. 2014; He and Jiang 2016; Magenta et al. 2016). Transition metals, such as Cu, Fe, and Zn catalyze the production of ROS (Peña and Kiselyov 2015), which were associated with hypoxia-induced miRNAs. Although Ca is not a transition metal, it has been associated with ROS and miRNA expression as well (Magenta et al. 2016). Through the MEs, we identified miRNAs hypoxia-induced, such as miR-15a, -29a (miR.MEcyan) (Gambacciani et al. 2014; Hao et al. 2014), miR-204, -214, and -199 (miR.MEmagenta) (El Azzouzi et al. 2013; Qiu et al. 2018), and miR-30c (miR.MEmidnightblue) (Gambacciani et al. 2014). We identified the TFs TFEB (transcription factor EB), HIF1A, and HIF3A (hypoxia-inducible factor) among the miR.MEtan and miR.MEcyan targets. These TFs are metal-affected and miRNA regulated (Li et al. 2006; Peña and Kiselyov 2015).
Pathway over-representation analysis
We carried out a pathway over-representation analysis to assign biological meaning to the associated miRNA–mRNA modules. Genes clustered into the module act cooperatively in the same pathway (Langfelder and Horvath 2007), which are also under the regulation of co-expressed miRNAs (Su et al. 2014). We unveiled several over-represented pathways, including those reported on our previous gene-co-expression network (apelin, insulin, mTOR, relaxin, TGF-beta signaling pathways, ECM-receptor interaction, focal adhesion, protein digestion and absorption, and ubiquitin-mediated proteolysis) (Diniz et al. 2019).
The miR.MEcyan showed over-represented KEGG signaling pathways that are central in cellular and organismal metabolism. These pathways include thyroid hormone (TH), mammalian target of rapamycin (mTOR), hypoxia-inducible factor-1 (HIF-1), and insulin. Furthermore, we identified the TGF-beta signaling pathway underlying the miR.MElightyellow. It is worth mentioning that these pathways have a pleiotropic action and are also interrelated with the AMPK pathway (5′-adenosine monophosphate-activated protein kinase) (Xu et al. 2012). Although not over-represented here, the AMPK pathway was the main one identified among the modules associated with mineral concentration in our previous work (Diniz et al. 2019). In agreement with our results, Afonso et al. (2019) reported AMPK underlying differentially expressed genes from Nelore cattle genetically divergent for Cu concentration in muscle.
By integrating the information from the literature, the clustered genes, and the KEGG database, we draw an overview of the relationship among the overmentioned pathways, miRNA, genes, and minerals. Genes from the families PIK3, EIF4EBP, and RSK were linked to the miR.MEcyan pathways. MiR-15a, -29b, 29-c, 29d-3p, and 29e targeted RPS6KA3 (members of RSK family), whereas miR-125a and -29a targeted RPS6KA1 and RPS6KB1, respectively. Nutrient and growth factors are the main sensors modulating the AMPK and mTOR pathways (Chen and Long 2018). AMPK inhibits mTOR activity by modulating the S6K and EIF4EBP genes that in turn inhibit the translation to maintain homeostasis (Xu et al. 2012). Both pathways are mineral responsive. Watson et al. (2016) reported a downregulation in mTORC1 signaling and a decreased protein synthesis as a result of iron depletion. Furthermore, calcium flux modulates AMPK pathways via CaMKK, whereas mTOR modulates intracellular Ca2+ signaling machinery under nutrient deprivation (Brini et al. 2013).
In this complex landscape, mTOR is also involved with autophagy regulation through TFEB gene expression (Roczniak-Ferguson et al. 2012). The autophagy pathway was over-represented in miR.MEbrown and miR.MEcyan and it is known to be regulated in different ways, including the TH, HIF, and p53 pathways (He and Jiang 2016). The HIF family controls the transcriptional mechanism as a response to hypoxia (Dengler et al. 2014), which may be an outcome of ROS (Magenta et al. 2016). The HIF1A and HIF3A genes are oxygen responsive and activate genes involved with iron metabolism, glucose, and glycolysis (Li et al. 2006; Dengler et al. 2014). We identified TFs related to HIFs including CREBP1, E2F4, and nuclear receptor coactivator genes (NCOA) (Dengler et al. 2014). CREBP1 and E2F4 were regulated by miR-29e and -29a, respectively. NCOA1 and NCOA2 were both targeted by miR-29e, whereas NCOA3 was targeted by miR-29b, -29c, and -29d-3p. It is essential to highlight that HIF induced genes are also under TH regulation (Otto and Fandrey 2008). Otto and Fandrey (2008) showed that triiodothyronine (T3) increased HIF1A expression as an outcome of increased hepatic leukemia factor gene (HLF), which was the most targeted gene in our analysis (10 miRNAs). Previous works have described the association between HIF genes and miRNA biogenesis (El Azzouzi et al. 2013; Magenta et al. 2016), as well as the role of metals, such as Cu and Fe, in HIF and TH pathway regulation (Li et al. 2006; Kaczmarek et al. 2009).
Iron is one of the most studied minerals due to its double role in cell metabolism. Both deficiency and overload are harmful, and iron/metal excess is closely related to ROS production (Speer et al. 2013). Among the pathways identified in the miR.MEtan, the p53 signaling pathway has a pro-oxidative activity and regulates ferroptosis (Cao and Dixon 2016). Likewise, the genes underlying the miR.MEbrown are partaking in the ferroptosis pathway. Shen et al. (2014) showed that Fe deprivation increased the p53 protein level avoiding ferroptosis. Besides p53, TFRC and IREB2 (encodes for IRP2 protein) are essential genes for ferroptosis (Cao and Dixon 2016). IREB2, a master regulator of Fe homeostasis (Cao and Dixon 2016), was targeted by the miR-29 family. Ripa et al. (2017) showed that Fe loading induces miR-29 up-regulation, whereas the downregulation of this miRNA increases the levels of TFRC, IRP2, Fe uptake, and oxidative stress.
Some of the miRNAs targeted genes that are associated with mineral homeostasis are associated with energy metabolism as well. The miR-29 family mainly targeted TFs involved with insulin and glucose metabolism, including CREB1, CRTC1, and FOXO3 (Oh et al. 2013). In addition, IGF1 and IGF2 were targeted by the let7 family (let-7a-5p, -b, -c, and -e). Family members share the seed region exhibiting a functional collaborative relationship in mRNA targets (Su et al. 2014; Oliveira et al. 2018). Massart et al. (2017) identified that miR-29a and -29c negatively regulate glucose uptake and fatty acid oxidation. Likewise, overexpression of let-7 in mice was associated with impaired glucose tolerance, decreased fat mass, and body weight (Frost and Olson 2011). Altogether, the miRNAs are an additional layer of regulation in the interrelated TH, insulin, AMPK, and mTOR signaling pathways, thus regulating energy homeostasis.
This study establishes a framework for understanding the role of minerals in gene/miRNA expression and metabolism regulation. By screening the genes, we identified several known TFs as being miRNA targets, as well as miRNAs acting cooperatively to regulate their targets. These results were supported by TargetScan prediction and negative correlation analysis. Nonetheless, in vitro and in vivo analyses should be carried out to better understand the potential for dietary modulated miRNAs and their complex relationship with gene targets. Equally important, further validation of identified miRNA–gene target interactions and hub genes in a larger cohort could support these findings.
MiRNAs showed a co-expression pattern where highly connected hubs drive gene expression. To our knowledge, this is the first exploratory study of miRNA–mRNA integration in the context of minerals in cattle. The significant associations identified among miRNAs, Ca, Fe, and their potential gene targets support the hypotheses of an intricate interplay among them. The miR-29 family plays a pivotal role in genes involved with major pathways like insulin, TH, AMPK, and mTOR, suggesting their importance in mineral metabolism, which can affect health and production. Future work should determine the functional implications of minerals for miRNA levels and their feedback regulation systems.
Methods
Animals and phenotypes
Experimental procedures involving the animals used in this study were carried out following the Institutional Animal Care and Use Committee Guidelines of the Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA—Pecuária Sudeste) (approval code CEUA 01/2013).
A population of 200 Nelore steers sired by 34 unrelated Nelore bulls was used in this study (Tizioto et al. 2015). In brief, calves were raised in grazing systems at three farms under similar diet and management (Diniz et al. 2016). After 21 months of age, the steers were moved to a feedlot at Embrapa Pecuária Sudeste (São Carlos, São Paulo, Brazil) with feed offered ad libitum twice daily. During the 90-day trial, the diet was composed of 40% corn silage and 60% of the concentrate, which contained soybean grain, soybean hull, limestone, mineral mixture, urea, and monensin (Rumensin®), on a dry matter basis.
Muscle samples were collected as a cross-section of the LT muscle (11th and 13th ribs) at slaughter for mineral measurement and RNA extraction. The samples for RNA analysis were immediately snap-frozen in liquid nitrogen and stored at – 80 °C until RNA extraction. Muscle expression profiles from 194 animals out of 200 that were reported elsewhere (Diniz et al. 2019) were used for the present analysis following the RNA-Seq quality control. The miRNA-Seq was carried out on 50 animals randomly selected from the whole population (n = 200), where the data from 48 steers were paired between both datasets.
Macro [calcium (Ca), magnesium (Mg), phosphorus (P), potassium (K), sodium (Na), and sulfur (S)] and micro minerals [copper (Cu), manganese (Mn), selenium (Se), iron (Fe), and zinc (Zn)] were measured by mass spectrometry. Briefly, muscle samples were lyophilized, and aliquots of approximately 0.1 g were digested in a closed-vessel microwave digestion system (Ethos 100, Milestone, Sorisole, Italy). The samples were digested using 2 mL of sub-boiled concentrated HNO3, 2 mL of H2O2 (30% w/w) and 6.0 mL of ultrapure water. Inductively coupled plasma-optical emission spectrometry (ICP OES; Vista Pro-CCD ICP OES1, radial view, Varian, Mulgrave, Australia) was used to measure all the minerals, except Se that was determined by ICP mass spectrometry (ICP-MS 820-MS, Varian, Mulgrave, Australia).
RNA extraction, library preparation and sequencing
The Trizol® standard protocol (Life Technologies, Carlsbad, CA, USA) was used to extract total RNA from 100 mg of frozen muscle tissue. The RNA quality and integrity were evaluated by Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) with the RNA 6000 Nano kit.
Libraries were prepared and sequenced in ESALQ’s Multiuser Laboratory in Piracicaba-SP, Brazil, as reported elsewhere (Oliveira et al. 2018). Briefly, for library preparation, 200 ng/μL of RNA from each sample was used following the TruSeq® smallRNA Sample Preparation kit (Illumina—San Diego, USA). Libraries were further quantified by quantitative PCR with the KAPA Library Quantification kit (KAPA Biosystems, Foster City, CA, USA). Only samples with an RNA integrity number (RIN) greater than or equal to 8 were sequenced. The single-end sequencing of 42 bp was carried out on MiSeq sequencer (Illumina®) using MiSeq Reagent Kit v3 (150 cycles) that generated around 1 million reads/sample.
Data quality control, miRNA identification, and expression normalization
Raw data quality control was carried out with FastQC version 0.11.2 (shorturl.at/lyIRS) (Andrews 2010) and trimmed using FASTX—Toolkit software (https://goo.gl/MueTV5) (FASTX-Toolkit 2009). Reads with Phred quality score lower than 28 and shorter than 18 nt were discarded.
The reads were mapped to the bovine reference genome Bos taurus ARS-UCD 1.2 by miRDeep2 (Friedländer et al. 2008), which uses Bowtie version 1.2.1.1 (Langmead et al. 2009), allowing one mismatch in the seed. Mapped reads were further used to identify known and novel miRNAs for B. taurus using the miRDeep2.pl module (Friedländer et al. 2008). The mature miRNA sequences of bovine and humans and the bovine hairpin structure were retrieved from the miRBase v. 22 (Kozomara and Griffiths-Jones 2014).
The raw counts generated by miRDeep2 were processed to filter out low or not expressed miRNAs applying the cpm function from edgeR version 3.24.0 (Robinson et al. 2010). MiRNA counts with less than 0.5 cpm in more than 70% of the samples were filtered out. Library normalization and data variance stabilization were carried out by the VST function implemented in DESeq2 (Anders and Huber 2010).
Principal Component Analysis (PCA) and hierarchical clustering on normalized data were performed with NOISeq version 2.26.0 (Tarazona et al. 2015). A linear model was fitted for adjusting the miRNA expression matrix for the batch effect (flow cell). Thus, the removeBatchEffect function from the R-package Limma (version 3.34.9) (Ritchie et al. 2015) was adopted.
MiRNA co-expression network analysis
A co-expression network analysis was carried out taking the expression profile of 343 miRNAs from 50 samples based on the general framework implemented in WGCNA R-package version 1.66 (Langfelder and Horvath 2008). Briefly, an adjacency matrix was calculated by raising the absolute Pearson’s correlation coefficient between the miRNAs to a power β = 9 (soft threshold) to reach a scale-free network topology index (R2 > 0.9) (Zhang and Horvath 2005).
Average linkage hierarchical clustering method was used to define the miRNA clusters using the Dynamic Tree algorithm (Langfelder et al. 2008). MiRNA modules were generated considering branch cut-off of 0.99, deepSplit = 4, and a minimum module size of 5 was chosen due to the small miRNA transcriptome (Oliveira et al. 2018). Following this, modules were detected and labeled by color. From each module, the eigengene (module eigengene—ME) was the first principal component (Langfelder and Horvath 2007) and represented a measure of miRNA expression profiles in the module.
Module-trait association and hub miRNA selection
The module–trait relationship was estimated by fitting a linear model to analyze the association between the expression profiles of the modules (MEs) and the phenotypes (Ca, Mg, P, K, Na, S, Cu, Mn, Se, Fe, and Zn), which were mean-centered and scaled. The statistical model included the fixed effect of place of birth and age at slaughter as a covariate, according to the equation:
where \(y_{ijk}\) is the expression level of the eigengene in each module (n = 24); \(\mu\) is the intercept of ME; \(C_{i}\) is the fixed effect for the place of birth (3 levels = CPPSE, IMA, NOHO); \(A_{j}\) is the covariate for the animal’s age; \(T_{k}\) is the trait observation for each animal; and \(\varepsilon_{ijk}\) is the random residual effect associated with each observation.
Putative relevant modules were taken for further analyses with p ≤ 0.05. As reported by Su et al. (2014) miRNAs exist as highly connected hub nodes within a transcriptional network and drive changes in mRNA expression. Thus, from the associated modules, the hub miRNAs were selected based on the MM ≥ 0.7 (Langfelder and Horvath 2008).
MiRNA–mRNA regulatory network and miRNA target prediction
The samples used in this study were part of a previous work that was carried out to identify co-expressed genes associated with meat quality and mineral traits in a population of 194 steers (Diniz et al. 2019). Because the miRNAs exert a pivotal role in the regulation of gene translation, the miRNA–mRNA expression data were integrated to identify the putative regulatory link between one another and their role in meat quality and mineral composition traits. In that study, 15 modules were associated with at least one trait (p ≤ 0.05), which were taken for the integrative analysis in the current study. Since multiple miRNAs can target the same gene, a module approach was adopted to alleviate the multiple test problem (Langfelder and Horvath 2008). For this, 48 animals were identified with paired expression data, and the miRNA MEs (9 modules from the current study) were correlated with mRNAs MEs (15 modules from the previous study). MiRNAs are expected to downregulate the translation level of targets (Su et al. 2014), and thereby modules with a negative correlation lower than − 0.3 and p ≤ 0.05 were selected for functional analysis.
A computational prediction method was applied based on paired miRNA–mRNA profiling to identify the potential target mRNAs of the hub miRNAs. The significant negatively correlated modules were intersected with miRNA–mRNA interactions predicted from TargetScan (Agarwal et al. 2015) to point out only those putative acting pairs. To this end, hoardeR package version 0.9.2 (Fischer and Sironen 2016) was used to search for the putative mRNA targets on the TargetScan bovine database release 7.2 (Agarwal et al. 2015). To better predict the putative targets, the genes retrieved from TargetScan were filtered by skeletal muscle expression data previously analyzed in the same samples (Diniz et al. 2019). MicroRNA family information was obtained from miRbase v. 22 (Kozomara and Griffiths-Jones 2014).
Identification of key transcription factors
The compendium of bovine transcription factors (de Souza et al. 2018) was employed to explore the gene regulatory network of the co-expressed modules, as well as the cross talk between miRNAs, transcription factors, and their targets. To detect regulatory modules, the identified miRNA–mRNA pairs were screened to reveal enriched transcription factors. The regulatory network was visualized in Cytoscape 3.7.0 (Cline et al. 2007).
Functional and pathway enrichment analyses
Based on the B. taurus genome background, KEGG pathway analysis was carried out using Cytoscape plugins: ClueGO v. 2.5.3 and CluePedia v. 1.5.3 (Bindea et al. 2009) to shed light on the biological functions over-represented in the associated module genes. KEGG pathways with pV ≤ 0.05 (group p value corrected with Bonferroni step down) were considered significantly enriched. Redundant terms were grouped based on the kappa score = 0.4 (Bindea et al. 2009). Interaction networks were constructed and visualized in Cytoscape (Cline et al. 2007).
Availability of data and material
All relevant data are within the paper and its Supporting Information files. All sequencing data is available in the European Nucleotide Archive (ENA) repository (EMBL-EBI), under accession PRJEB13188, PRJEB10898, and PRJEB19421 (https://www.ebi.ac.uk/ena/submit/sra/). All additional datasets generated and analyzed during this study are available from the corresponding author on reasonable request.
References
Afonso J, Coutinho LL, Tizioto PC, da Silva Diniz WJ, de Lima AO, Rocha MIP, Buss CE, Andrade BGN, Piaya O, da Silva JV, Lins LA, Gromboni CF, Nogueira ARA, Fortes MRS, Mourao GB, de Almeida Regitano LC (2019) Muscle transcriptome analysis reveals genes and metabolic pathways related to mineral concentration in Bos indicus. Sci Rep 9:12715. https://doi.org/10.1038/s41598-019-49089-x
Agarwal V, Bell GW, Nam JW, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4:1–38. https://doi.org/10.7554/eLife.05005
Ahlberg CM, Schiermiester LN, Howard TJ, Calkins CR, Spangler ML (2014) Genome wide association study of cholesterol and poly- and monounsaturated fatty acids, protein, and mineral content of beef from crossbred cattle. Meat Sci 98:804–814. https://doi.org/10.1016/j.meatsci.2014.07.030
Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P, Tassone P, Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P, Tassone P (2015) miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 6:12837–12861. https://doi.org/10.18632/oncotarget.3805
An JH, Ohn JH, Song JA, Yang JY, Park H, Choi HJ, Kim SW, Kim SY, Park WY, Shin CS (2014) Changes of microRNA profile and microRNA–mRNA regulatory network in bones of ovariectomized mice. J Bone Miner Res 29:644–656. https://doi.org/10.1002/jbmr.2060
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106. https://doi.org/10.1186/gb-2010-11-10-r106
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 2 Apr 2018
Beckett EL, Yates Z, Veysey M, Duesing K, Lucock M (2014) The role of vitamins and minerals in modulating the expression of microRNA. Nutr Res Rev 27:94–106. https://doi.org/10.1017/S0954422414000043
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093. https://doi.org/10.1093/bioinformatics/btp101
Brini M, Ottolini D, Calì T, Carafoli E (2013) Calcium in health and disease. In: Astrid S, Helmut Sigel RKOS (eds) Interrelations between essential metal ions and human diseases. Springer, Dordrecht, pp 81–137
Cao JY, Dixon SJ (2016) Mechanisms of ferroptosis. Cell Mol Life Sci 73:2195–2209. https://doi.org/10.1007/s00018-016-2194-1
Casas E, Duan Q, Schneider MJ, Shackelford SD, Wheeler TL, Cundiff LV, Reecy JM (2014) Polymorphisms in calpastatin and mu-calpain genes are associated with beef iron content. Anim Genet 45:283–284. https://doi.org/10.1111/age.12108
Chen J, Long F (2018) mTOR signaling in skeletal development and disease. Bone Res 6:1. https://doi.org/10.1038/s41413-017-0004-5
Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, Hanspers K, Isserlin R, Kelley R, Killcoyne S, Lotia S, Maere S, Morris J, Ono K, Pavlovic V, Pico AR, Vailaya A, Wang P-L, Adler A, Conklin BR, Hood L, Kuiper M, Sander C, Schmulevich I, Schwikowski B, Warner GJ, Ideker T, Bader GD (2007) Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2:2366–2382. https://doi.org/10.1038/nprot.2007.324
Davis M, Clarke S (2013) Influence of microRNA on the maintenance of human iron metabolism. Nutrients 5:2611–2628. https://doi.org/10.3390/nu5072611
Davis MR, Hester KK, Shawron KM, Lucas Smith EBJ, Clarke SL (2012a) Comparisons of the iron deficient metabolic response in rats fed either an AIN-76 or AIN-93 based diet. Nutr Metab (Lond) 9:95. https://doi.org/10.1186/1743-7075-9-95
Davis MR, Rendina E, Peterson SK, Lucas EA, Smith BJ, Clarke SL (2012b) Enhanced expression of lipogenic genes may contribute to hyperglycemia and alterations in plasma lipids in response to dietary iron deficiency. Genes Nutr 7:415–425. https://doi.org/10.1007/s12263-011-0278-y
Diniz WJS, Coutinho LL, Tizioto PC, Cesar ASM, Gromboni CF, Nogueira ARA, de Oliveira PSN, de Souza MM, de Regitano LCA (2016) Iron content affects lipogenic gene expression in the muscle of Nelore Beef Cattle. PLoS ONE 11:e0161160. https://doi.org/10.1371/journal.pone.0161160
Diniz WJS, Mazzoni G, Coutinho LL, Banerjee P, Geistlinger L, Cesar ASM, Bertolini F, Afonso J, de Oliveira PSN, Tizioto PC, Kadarmideen HN, de Regitano LCAA (2019) Detection of co-expressed pathway modules associated with mineral concentration and meat quality in Nelore Cattle. Front Genet 10:210. https://doi.org/10.3389/FGENE.2019.00210
de Oliveira PSN, Coutinho LL, Tizioto PC, Cesar ASM, de Oliveira GB, da Diniz WJ, S, De Lima AO, Reecy JM, Mourão GB, Zerlotini A, Regitano LCA, (2018) An integrative transcriptome analysis indicates regulatory mRNA–miRNA networks for residual feed intake in Nelore cattle. Sci Rep 8:17072. https://doi.org/10.1038/s41598-018-35315-5
de Souza MM, Zerlotini A, Geistlinger L, Tizioto PC, Taylor JF, Rocha MIP, Diniz WJS, Coutinho LL, Regitano LCA (2018) A comprehensive manually-curated compendium of bovine transcription factors. Sci Rep 8:13747. https://doi.org/10.1038/s41598-018-32146-2
Dengler VL, Galbraith M, Espinosa JM (2014) Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 49:1–15. https://doi.org/10.3109/10409238.2013.838205
El AH, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, McClellan EA, Poels E, Sluimer JC, van den Hoogenhof MMG, Armand A-S, Yin X, Langley S, Bourajjaj M, Olieslagers S, Krishnan J, Vooijs M, Kurihara H, Stubbs A, Pinto YM, Krek W, Mayr M, da Martins PA, C, Schrauwen P, De Windt LJ, (2013) The hypoxia-inducible microRNA cluster miR-199a∼214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab 18:341–354. https://doi.org/10.1016/j.cmet.2013.08.009
FASTX-Toolkit (2009) FASTX-Toolkit. https://hannonlab.cshl.edu/fastx_toolkit/. Accessed 2 Apr 2018
Feng Y, Xing Y, Liu Z, Yang G, Niu X, Gao D (2018) Integrated analysis of microRNA and mRNA expression profiles in rats with selenium deficiency and identification of associated miRNA–mRNA network. Sci Rep 8:1–9. https://doi.org/10.1038/s41598-018-24826-w
Fischer D, Sironen A (2016) An Introduction to hoardeR. https://cran.r-project.org/web/packages/hoardeR/vignettes/hoardeR-vignette.pdf. Accessed 2 Apr 2018
Fleet JC, Replogle R, Salt DE (2011) Systems genetics of mineral metabolism. J Nutr 141:520–525. https://doi.org/10.3945/jn.110.128736
Friedländer MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N (2008) Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26:407–415. https://doi.org/10.1038/nbt1394
Friedman RC, Farh KK-H, Burge CB, Bartel DP (2008) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105. https://doi.org/10.1101/gr.082701.108
Frost RJA, Olson EN (2011) Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci 108:21075–21080. https://doi.org/10.1073/pnas.1118922109
Gambacciani C, Kusmic C, Chiavacci E, Meghini F, Rizzo M, Mariani L, Pitto L (2014) miR-29a and miR-30c negatively regulate DNMT 3a in cardiac ischemic tissues: implications for cardiac remodelling. microRNA Diagn Ther. https://doi.org/10.2478/micrnat-2013-0004
Garmyn AJ, Hilton GG, Mateescu RG, Morgan JB, Reecy JM, Tait RG, Beitz C, Duan Q, Schoonmaker JP, Mayes MS, Drewnoski ME, Liu Q (2011) Estimation of relationships between mineral concentration and fatty acid composition of longissimus muscle and beef palatability traits. J Anim Sci 89:2849–2858. https://doi.org/10.2527/jas.2010-3497
Hao R, Hu X, Wu C, Li N (2014) Hypoxia-induced miR-15a promotes mesenchymal ablation and adaptation to hypoxia during lung development in chicken. PLoS ONE 9:e98868. https://doi.org/10.1371/journal.pone.0098868
He J, Jiang B-H (2016) Interplay between reactive oxygen species and microRNAs in cancer. Curr Pharmacol Rep 2:82–90. https://doi.org/10.1007/s40495-016-0051-4
Kaczmarek M, Cachau RE, Topol IA, Kasprzak KS, Ghio A, Salnikow K (2009) Metal ions-stimulated iron oxidation in hydroxylases facilitates stabilization of HIF-1 alpha protein. Toxicol Sci 107:394–403. https://doi.org/10.1093/toxsci/kfn251
Kappeler BIG, Regitano LCA, Poleti MD, Cesar ASM, Moreira GCM, Gasparin G, Coutinho LL (2019) MiRNAs differentially expressed in skeletal muscle of animals with divergent estimated breeding values for beef tenderness. BMC Mol Biol 20:1. https://doi.org/10.1186/s12867-018-0118-3
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73. https://doi.org/10.1093/nar/gkt1181
Langfelder P, Horvath S (2007) Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol 1:54. https://doi.org/10.1186/1752-0509-1-54
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. https://doi.org/10.1186/1471-2105-9-559
Langfelder P, Zhang B, Horvath S (2008) Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24:719–720. https://doi.org/10.1093/bioinformatics/btm563
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. https://doi.org/10.1186/gb-2009-10-3-r25
Li Q, Chen H, Huang X, Costa M (2006) Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1α) and HIF-regulated genes. Toxicol Appl Pharmacol 213:245–255. https://doi.org/10.1016/j.taap.2005.11.006
Li Y, Lin L, Li Z, Ye X, Xiong K, Aryal B, Xu Z, Paroo Z, Liu Q, He C, Jin P (2012) Iron Homeostasis regulates the activity of the microRNA pathway through Poly(C)-binding protein 2. Cell Metab 15:895–904. https://doi.org/10.1016/j.cmet.2012.04.021
Magenta A, Dellambra E, Ciarapica R, Capogrossi MC (2016) Oxidative stress, microRNAs and cytosolic calcium homeostasis. Cell Calcium 60:207–217. https://doi.org/10.1016/J.CECA.2016.04.002
Mamdani M, Williamson V, McMichael GO, Blevins T, Aliev F, Adkins A, Hack L, Bigdeli T, van der Vaart DA, Web BT, Bacanu S-A, Kalsi G, Kendler KS, Miles MF, Dick D, Riley BP, Dumur C, Vladimirov VI (2015) Integrating mRNA and miRNA weighted gene co-expression networks with eQTLs in the nucleus accumbens of subjects with alcohol dependence. PLoS ONE 10:e0137671. https://doi.org/10.1371/journal.pone.0137671
Massart J, Sjögren RJO, Lundell LS, Mudry JM, Franck N, O’Gorman DJ, Egan B, Zierath JR, Krook A (2017) Altered miR-29 expression in type 2 diabetes influences glucose and lipid metabolism in skeletal muscle. Diabetes 66:1807–1818. https://doi.org/10.2337/db17-0141
Mateescu RG, Garrick DJ, Reecy JM (2017) Network analysis reveals putative genes affecting meat quality in Angus cattle. Front Genet. https://doi.org/10.3389/fgene.2017.00171
Oh K-J, Han H-S, Kim M-J, Koo S-H (2013) CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep 46:567–574. https://doi.org/10.5483/BMBRep.2013.46.12.248
Oliveira GB, Regitano LCA, Cesar ASM, Reecy JM, Degaki KY, Poleti MD, Felício AM, Koltes JE, Coutinho LL (2018) Integrative analysis of microRNAs and mRNAs revealed regulation of composition and metabolism in Nelore cattle. BMC Genom 19:126. https://doi.org/10.1186/s12864-018-4514-3
Otto T, Fandrey J (2008) Thyroid hormone induces hypoxia-inducible factor 1α gene expression through thyroid hormone receptor β/retinoid X receptor α-dependent activation of hepatic leukemia factor. Endocrinology 149:2241–2250. https://doi.org/10.1210/en.2007-1238
Peña KA, Kiselyov K (2015) Transition metals activate TFEB in overexpressing cells. Biochem J 470:65–76. https://doi.org/10.1042/BJ20140645
Qiu R, Li W, Liu Y (2018) MicroRNA-204 protects H9C2 cells against hypoxia/reoxygenation-induced injury through regulating SIRT1-mediated autophagy. Biomed Pharmacother 100:15–19. https://doi.org/10.1016/J.BIOPHA.2018.01.165
Ripa R, Dolfi L, Terrigno M, Pandolfini L, Savino A, Arcucci V, Groth M, Terzibasi Tozzini E, Baumgart M, Cellerino A (2017) MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol 15:9. https://doi.org/10.1186/s12915-017-0354-x
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47–e47. https://doi.org/10.1093/nar/gkv007
Ritchie W, Rajasekhar M, Flamant S, Rasko JEJ (2009) Conserved expression patterns predict microRNA targets. PLoS Comput Biol 5:1000513. https://doi.org/10.1371/journal.pcbi.1000513
Ritchie H, Roser M (2018) Micronutrient deficiency. https://ourworldindata.org/micronutrient-deficiency. Accessed 22 Aug 2018
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5:ra42. https://doi.org/10.1126/scisignal.2002790
Sengar GS, Deb R, Singh U, Junghare V, Hazra S, Raja TV, Alex R, Kumar A, Alyethodi RR, Kant R, Jakshara S, Joshi CG (2018) Identification of differentially expressed microRNAs in Sahiwal (Bos indicus) breed of cattle during thermal stress. Cell Stress Chaperones 23:1019–1032. https://doi.org/10.1007/s12192-018-0911-4
Shalgi R, Lieber D, Oren M, Pilpel Y (2007) Global and local architecture of the mammalian microRNA–transcription factor regulatory network. PLoS Comput Biol 3:e131. https://doi.org/10.1371/journal.pcbi.0030131
Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, Yu L, Li L, Ruan K, Hu H, Huang Y, Hui L, Xie D, Wang F, Hu R (2014) Iron metabolism regulates p53 signaling through direct Heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep 7:180–193. https://doi.org/10.1016/j.celrep.2014.02.042
Speer RE, Karuppagounder SS, Basso M, Sleiman SF, Kumar A, Brand D, Smirnova N, Gazaryan I, Khim SJ, Ratan RR (2013) Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: From ferroptosis to stroke. Free Radic Biol Med 62:26–36. https://doi.org/10.1016/J.FREERADBIOMED.2013.01.026
Su W-L, Kleinhanz RR, Schadt EE (2014) Characterizing the role of miRNAs within gene regulatory networks using integrative genomics techniques. Mol Syst Biol 7:490–490. https://doi.org/10.1038/msb.2011.23
Suttle N (2010) Mineral nutrition of livestock, 4a. CABI, Wallingford
Tarazona S, Furió-Tarí P, Turrà D, Di PA, Nueda MJ, Ferrer A, Conesa A (2015) Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. https://doi.org/10.1093/nar/gkv711
Tizioto PC, Gromboni CF, Nogueira ARDA, de Souza MM, Mudadu MDA, Tholon P, Rosa ADN, Tullio RR, Medeiros SR, Nassu RT, Regitano LCDA (2014) Calcium and potassium content in beef: influences on tenderness and associations with molecular markers in Nellore cattle. Meat Sci 96:436–440. https://doi.org/10.1016/j.meatsci.2013.08.001
Tizioto PC, Taylor JF, Decker JE, Gromboni CF, Mudadu MA, Schnabel RD, Coutinho LL, Mourão GB, Oliveira P, Souza MM, Reecy JM, Nassu RT, Bressani FA, Tholon P, Sonstegard TS, Alencar MM, Tullio RR, Nogueira A, Regitano L (2015) Detection of quantitative trait loci for mineral content of Nelore longissimus dorsi muscle. Genet Sel Evol 47:15. https://doi.org/10.1186/s12711-014-0083-3
Watson A, Lipina C, McArdle HJ, Taylor PM, Hundal HS (2016) Iron depletion suppresses mTORC1-directed signalling in intestinal Caco-2 cells via induction of REDD1. Cell Signal 28:412–424. https://doi.org/10.1016/J.CELLSIG.2016.01.014
Xu J, Ji J, Yan X (2012) Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr 52:373–381. https://doi.org/10.1080/10408398.2010.500245
Xu Z, Shi Z, Li Y (2013) The crosstalk between micro RNA and iron homeostasis. Int J Genom Med 01:1–8. https://doi.org/10.4172/2332-0672.1000112
Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. https://doi.org/10.2202/1544-6115.1128
Acknowledgements
We are thankful to the EMBRAPA Multiuser Bioinformatics Laboratory (Laboratório Multiusuário de Bioinformática da Embrapa—LMB) for providing high-performance computational infrastructure; Dr. Bruno G. N. Andrade for the server management and support in the EMBRAPA Pecuária Sudeste; and the Technical University of Denmark (DTU) for accepting the first author as a visiting scholar.
Funding
This study was conducted with funding from EMBRAPA (Macroprograma 1, 01/2005), São Paulo Research Foundation (FAPESP) (Grant #2012/23638-8) and by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. ARAN, LCAR, and LLC were granted CNPq fellowships. JA was granted CAPES fellowship. WJSD was granted FAPESP (Grant #2015/09158-1 and #2017/20761-7) scholarship.
Author information
Authors and Affiliations
Contributions
WJSD, LCAR, LLC, and HNK conceived the idea of this research. ARAN and CFG carried out the mineral measurement; WJSD, PB, and GM carried out the bioinformatics and data analysis. ASMC carried out miRNA data analysis (quality control, mapping, and counting). WJSD, PB, GM, ASMC, HNK, JA, and LCAR collaborated with the interpretation of results, discussion and review of the manuscript. WJSD and PB drafted the manuscript. All authors have reviewed, discussed, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
Experimental procedures involving the animals used in this study were carried out in accordance with the Institutional Animal Care and Use Committee Guidelines of the Empresa Brasileira de Pesquisa Agropecuária (EMBRAPA—Pecuária Sudeste) (approval code CEUA 01/2013).
Additional information
Communicated by S. Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
Module-trait association analysis. Modules are labeled by color on the y-axis and traits on the x-axis. For significantly associated modules, the coefficient from the linear model is given within the cell. Only associations with p < 0.05 are shown (PDF 6 kb)
Supplementary Fig. S2
Regulatory network of negative miRNA-mRNA pairs in Nelore muscle. The edges are colored according to miRNA module (miR.MEbrown, miR.MEcyan, miR.MEgreen, miR.MElightyellow, miR.MEmagenta, miR.MEmidnightblue, miR.MEred, and miR.MEtan) Transcription factors are represented by lightpurple diamond shape (TIFF 10471 kb)
Rights and permissions
About this article

Cite this article
da Silva Diniz, W.J., Banerjee, P., Mazzoni, G. et al. Interplay among miR-29 family, mineral metabolism, and gene regulation in Bos indicus muscle. Mol Genet Genomics 295, 1113–1127 (2020). https://doi.org/10.1007/s00438-020-01683-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-020-01683-9