
Abstract
Dwarfing is the development trend of pepper breeding. It is of great practical and scientific value to generate new dwarf germplasms, and identify new genes or alleles conferring dwarf traits in pepper. In our previous study, a weakly BR-insensitive dwarf mutant, E29, was obtained by EMS mutagenesis of the pepper inbred line 6421. It can be used as a good parent material for breeding new dwarf varieties. Here, we found that this dwarf phenotype was controlled by a single recessive gene. Whole-genome resequencing, dCAPs analysis, and VIGs validation revealed that this mutation was caused by a nonsynonymous single-nucleotide mutation (C to T) in CaBRI1. An enzyme activity assay, transcriptome sequencing, and BL content determination further revealed that an amino-acid change (Pro1157Ser) in the serine/threonine protein kinase and catalytic (S_TKc) domain of CaBRI1 impaired its kinase activity and caused the transcript levels of two important genes (CaDWF4 and CaROT3) participating in BR biosynthesis to increase dramatically in the E29 mutant, accompanied by significantly increased accumulation of brassinolide (BL). Therefore, we concluded that the novel single-base mutation in CaBRI1 conferred the dwarf phenotype and resulted in brassinosteroid (BR) accumulation in pepper. This study provides a new allelic variant of the height-regulating gene CaBRI1 that has theoretical and practical values for the breeding of the plants suitable for the facility cultivation and mechanized harvesting of pepper varieties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dwarfing is an important trait in crop breeding that has attracted a great deal of attention because of its advantages of allowing an increased planting density, improving the photosynthetic efficiency per unit area, lowering water consumption, conferring lodging resistance, and facilitating close planting and once-over mechanical harvesting. Excellent varieties selected from dwarf germplasm resources significantly increase the production of major crops. In particular, the breeding of varieties of Oryza sativa (Suh 1978) and Triticum aestivum (Evans et al. 1998) in the twentieth century led to a “green revolution” clearly demonstrating the potential of dwarf crops for production. The advantages of dwarf varieties that are suitable for facility cultivation and once-over mechanical harvesting among vegetable crops such as cucumber, pumpkin, and watermelon have been demonstrated (Tang et al. 2017).
Identifying new genes or alleles conferring dwarf traits is an important prerequisite for efficient breeding of new dwarf varieties and clarified of dwarfing mechanisms. Since the 1990s, a number of important dwarf genes related to the synthesis, metabolism, and signal transduction of phytohormones gibberellin (GA) (Silverstone and Sun 2000; Wang et al. 2012; Perez et al. 2014), indoleacetic acid (IAA) (Rouse et al. 1998), and strigolactone (SL) (Tsuchiya and McCourt 2009) have been cloned. In wheat, semi-dwarfism can be controlled by the Rht (for ‘reduced height’) gene, which encodes a DELLA protein involved in the GA signalling pathway (Peng et al. 1999). In rice, the reduced plant height associated with the sd1 alleles is caused by a mutation of the GA20 oxidase (GA20ox-2) involving in GA biosynthesis (Sasaki et al. 2002). BRZ and DW3 genes widely used in dwarf breeding of maize and sorghum are associated with IAA signal transduction (Xu and Li 2006). In rice and barley, the regulation of BRI1 gene expression can make the plant type more compact, improve the light utilization rate of crops, and confer a greater production potential in high-density crop-planting conditions (Yamamuro et al. 2000; Chono et al. 2003; Morinaka et al. 2006; Nakamura et al. 2006).
The varieties with lodging resistance, high yield, and good quality selected by dwarf genes play an important role in the production of major crops.
Pepper is an important vegetable crop and condiment that is widely cultivated around the world. In China, pepper is the most commonly cultivated vegetable, with an area of 1.5–2 million hectares. The applied cultivation methods include facility and field cultivation in the main producing areas in China. At present, most of the pepper varieties used for facility cultivation are easy to grow and do not resist lodging. Field cultivars exhibit a long-harvesting period and need to be harvested several times in batches. Facility suitability for cultivation and once-over mechanical harvesting has become an important factor in the selection of new varieties of pepper. Special pepper varieties suitable for facility cultivation and mechanized harvesting are urgently needed for breeding. However, pepper breeders mainly pay attention to the yield and disease resistance, there are a few reports on the cloning of pepper dwarf genes, and let alone apply them to genetic improvement. It is of great significance to identify new dwarf genes or alleles and generate new dwarf germplasms in pepper.
In a previous study, we generated a weakly BR-insensitive dwarf mutant, E29, through EMS mutagenesis of the pepper inbred line 6421. E29 showed a shortened stem and a compact plant type and could produce seeds (Yang et al. 2017). This mutant can be used as parent material for facility cultivation and mechanized harvesting and provides a good study material for the investigation of BR signal transduction. Here, whole-genome resequencing, dCAPS analysis, and virus-induced gene silencing (VIGS) validation were used to map the dwarf gene. An enzyme activity assay, transcriptome sequencing, and BL content determination further revealed the molecular mechanism of the dwarfism and BR accumulation in E29. This study provides a good basis for understanding the molecular mechanism of the mutation in pepper and for breeding varieties suitable for mechanized harvesting.
Materials and methods
Plant materials
The dwarf mutant E29 was identified through screening of an EMS mutagenized population derived from the pepper inbred line 6421. An F1 generation was generated from a cross between E29 and 6421, and an F2 population was generated by selfing the F1 plants. All plants were grown in a greenhouse at the Vegetable Institute of Hunan Academy of Agricultural Science, Changsha, China.
Histological observations
At the 12-leaf stage, the fourth stem internodes of 6421 and E29 were collected and cut into 2 mm segments. Each sample was rinsed in 0.01 M phosphate-buffered saline three times and then fixed in 2.5% formaldehyde solution for 2 h at 4 °C. The material was then rinsed as before and dehydrated through increasing concentrations of ethanol (10 min each in 50, 75, 90, and 100% ethanol). The samples were dried in an HCP-2 (Hitachi) critical point-dryer, attached to stubs with colloidal carbon and coated with gold–palladium in a sputtering device. Specimens were examined and photographed with a scanning transmission electron microscope (SU8010, Hitachi Ltd, Tokyo, Japan) operated at 15 kV.
Determination of the chlorophyll (Chl) content
Fully developed leaves were selected from the second to the fourth leaves of the plants. A total of 2.0 g fresh leaves were homogenized using quartz sand, CaCO3, and 3 mL of 100% acetone, and then extracted with 80% acetone. After centrifugation at 2500 rpm for 2 min, the supernatant was measured using a spectrophotometer (Ruili UV-2100, Beijing, China) at 665 and 649 nm. The contents of Chl a, Chl b, and total Chl (Chl (a + b)) were calculated using the equations below: Chl a = 13.95 OD665—6.88 OD649; Chl b = 24.96 OD649—7.32 OD665; Chl (a + b) = Chl a + Chl b.
Determination of endogenous BL levels
Leaves of 6421, E29, CaBRI1-silenced 6421 plant, and TRV-infiltrated 6421 plant (1.0 g) were ground into a powder with liquid nitrogen, and then, the powder was extracted with 10 mL of 80% aqueous methanol in an ultrasonic bath at 4 °C for 2 h. The homogenate was centrifuged at 10,000 rpm for 5 min at 4 °C, and the supernatant was loaded onto the Bond Elut C18 column (100 mg packing material, Agilent). Washing was performed with 3 mL of aqueous methanol, and then, the samples were loaded onto the solid-phase extraction (SPE) columns (Strata-X8B-S100-UAK). Finally, the samples were redissolved with 3 mL of aqueous methanol. After being dried with an N2 stream, the samples were redissolved with aqueous methanol and filtered (0.22 µm, regenerate cellulose) before injection. The ESI–HPLC–MS/MS system used consisted of a Qtrap6500 mass spectrometer coupled to an Agilent 1290 HPLC (Agilent Technologies Italian S.P.A., Cernusco Sul Naviglio, Milano, Italy). The separation was performed at a flow rate of 0.35 mL/min with a ZORBAX SB-C18 column (2.1 mm × 150 mm, 3.5 µm, Agilent) with an injection volume of 5 μL and a column temperature of 35 °C. The mobile phase consisted of a combination of solvent A (0.1% formic acid in acetonitrile) and solvent B (0.1% formic acid in distilled water). The gradient was set as follows: from 5% A at 0 min to 80% A at 2 min, then to 95% A at 2 min to 3.5 min, to 95% A at 3.5 min to 6 min, followed by a re-equilibration step (80% A) at 6 min to 10 min. For full mass spectrometer (MS) analysis, default settings for the electrospray ionization (ESI) source were used: curtain gas press of 15 psi, nebulizer gas pressure of 65 psi, the auxiliary gas pressure of 70 psi, drying gas flow rate of 7 L/min, and drying gas temperature of 350 °C. The capillary voltage between the MS and nebulizer was 5500 V. All remaining ion-transport parameters were determined by the target mass (TM) parameter set by the operator. The TM was optimized for each component, and the optimal value was used during measurements of ionization efficiency. A standard curve was prepared with BL standard (Olchemim, purity > 95%) solution with concentration ranging from 0.5 to 50 ng/mL. BL content was calculated from the standard curve and expressed as ng/g.
Bulked-segregant analysis (BSA) sequencing and SNP detection
Genomic DNA was extracted as previously described (Murray and Thompson 1980) from fresh leaves of the parent 6421 and the F2 population. Three DNA pools, a parent 6421 pool and two offspring pools (the wild-type pool and the mutant pool), were constructed by mixing equal amounts of DNA from 10 parent plants, 20 mutant plants, and 20 wild-type plants from the F2 population, respectively. Paired-end DNA libraries with insert sizes of approximately 350 bp were constructed using the Illumina Genomic DNA Sample Preparation Kit and sequenced on an Illumina HiSeq 2500 platform following the manufacturer’s instructions (Illumina, CA, USA).
Raw reads of fastq format were first processed through a series of quality control (QC) procedures using in-house Perl scripts. Briefly, reads with ≥ 10% unidentified nucleotides (N) or > 50% bases having Phred quality < 5 or > 10 nt aligned to the adaptor sequencing allowing ≤ 10% mismatches were first removed. PCR duplicates generated by PCR amplification in the library construction process, which were defined as those with identical paired-end reads, were then removed. BWA (Burrows-Wheeler Aligner) (Li and Durbin 2009) was used to align the final cleaned paired-end reads of the three DNA pools to the pepper genome (Zunla-1, version 2.0) (Qin et al. 2014) with default parameters. The alignment files were converted to BAM format using SAMtools (Li et al. 2009).
Variant calling was performed for the three DNA pools using the HaplotypeCaller function in GATK3.8 (McKenna et al. 2010) and the parameter “-stand_call_conf” was set as 30. SNPs were filtered using VariantFiltration in GATK3.8 with the following parameters: QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < − 12.5, ReadPosRankSum < − 8.0. Only the SNPs that exhibit G → A or C → T transitions were retained, which were the most frequent changes caused by EMS mutagenesis. ANNOVAR (Wang et al. 2010) was used to annotate SNPs based on the GFF3 file of the reference genome.
The read depth information for SNPs in the offspring pools was used to calculate the SNP index according to the method described in Takagi et al. (2013). At each SNP locus, the genotype of the parent 6421 was used as the reference to derive the number of reads corresponding to the genotype of 6421 and the genotypes of each of the two offspring pools. Then, the ratio of the number of reads corresponding to the 6421 genotype to the total number of reads covering the SNP locus was calculated and considered the SNP index of that SNP site. Sites with an SNP index in the offspring pools of less than 0.3 were filtered out. An average SNP index of the two offspring pools was calculated using a 5 Mb sliding window with a step size of 50 kb. We also calculated the statistical 99% confidence intervals of the ΔSNP index under the null hypothesis following the method described in Takagi et al. (2013).
Verification of the causative SNP using dCAPS markers
The causative SNP was confirmed by Sanger sequencing, and a dCAPS marker was designed to assess its cosegregation with the mutant phenotype. A dCAPS PCR primer containing one mismatched base in the sequence was designed using the online software dCAPS FINDER 2.0 (Neff et al. 1998). The other primer was designed using Primer3. The products were digested with the appropriate restriction enzymes at 37 °C for 4 h, and then analyzed by electrophoresis on 2.5% agarose gels. The PCR system and conditions were as follows: 10 × PCR buffer 3 μL, dNTPs 3 μL, forward primers 0.9 μL (10 μM), reverse primers 0.9 μL (10 μM), Taq enzyme 0.3 μL (5 U/μL), DNA 1.5 μL (50 ng/μL), ddH2O 20.4 μL; 94 °C for 5 min followed by 35 cycles of at 94 °C for 35 s, 58 °C for 35 s, and 72 °C for 30 s, with a final extension at 72 °C for 10 min. The digestion system was as follows: 10 × H buffer 2.0 μL, restriction enzyme 0.3 μL (10 U/μL), PCR products 15.0 μL, ddH2O 2.7 μL. The parents, and F1 and F2 populations were tested with the dCAPS marker. The primers and restriction enzymes used here are listed in the Supplemental Table S1.
VIGS of CaBRI1 in 6421 and E29 and RT-qPCR detection
pTRV2:CaBRI1 was constructed from a 346 bp fragment containing the mutation site of CaBRI1, which was PCR-amplified from a pepper 6421 cDNA template. The resulting product was cloned into the SacI-BamHI-cut pTRV2 vector to form pTRV2:CaBRI1. The sequence of pTRV2:CaBRI1 was confirmed by sequencing. For the VIGS assay, pTRV1, pTRV2, and pTRV2:CaBRI1 were introduced separately into Agrobacterium tumefaciens GV3101. The bacterial culture solution (5 mL) was grown in LB medium-containing the appropriate antibiotics (50 mg/L kanamycin, 50 mg/L rifampicin, and 50 mg/L gentamycin) and shaken at 28 °C overnight. The Agrobacterium cells were collected by centrifugation (3000 rpm, 10 min) and then suspended in buffer containing 10 mM MgCl2, 10 mM MES, and 200 µM acetosyringone. After adjusting the concentration of the bacteria to an optical density of 0.5 at 600 nm, Agrobacteria carrying the pTRV1 and pTRV2 or pTRV2:CaBRI1 were subsequently mixed at a 1:1 ratio and incubated for 5 h at 25 °C, and then infiltrated into the abaxial sides of the cotyledons of 6421 and E29 using a 1 mL sterilized syringe without a needle. The Agrobacterium-infiltrated pepper plants were transferred to 18 °C in the dark for 2 days and grown in a growth chamber at 25 °C under a 16-h light/8-h dark photoperiod cycle with 60% relative humidity.
RT-qPCR was used to quantify the abundance of pTRV2:CaBRI1 transcripts in the leaves of inoculated pepper plants. The total RNA was extracted from the leaves of silenced and nonsilenced (infiltrated with empty pTRV1 and pTRV2 vectors) at 30 days after infiltration using Easy Pure Plant RNA Kit (Tiangen, China), following the manufacturer’s protocol. Reverse transcription was performed with 1 μg of total RNA using the PrimeScript RT Kit (TAKARA Biotech). The RT-qPCR assays using the TransStart® Green qPCR SuperMix kit (Transgene Biotech) were conducted on a Fluorescence quantitative PCR instrument (Light Cycler® 480, Roche, Switzerland). Primers were designed according to the CaBRI1 sequences, and the housekeeping gene β-actin was used as an internal control (Supplemental Table S2). Three technical replicates were run for each biological replicate.
In vitro kinase assay
To enhance the expression level of CaBRI1-S_TKc (wild type) and CaBRI1-S_TKc (dw) (mutant) in the E. coli host cells in a synchronous manner, we optimized the DNA sequences based on E. coli codon usage. In addition, 21 bases (CATCATCATCATCACCATTAA) were added at the end of the sequence, so that the translated protein sequence C-terminus contained a 6 × His-Tag, which facilitates the detection and affinity purification of recombinant proteins. The codon-optimized target gene was cloned into the multiple cloning sites Nde I and BamH I of the prokaryotic expression vector pMAL-c5x (New England Biolabs). The recombinant plasmid was then used to transform E. coli Arctic Express into host cells and expression of the fusion protein was induced by supplementing the culture with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 37 °C for 4 h. The fusion protein was further purified using a Ni-IDA-Sepharose CI-6B affinity column (Qiagen, Germany) according to the manufacturer’s protocols. The purified samples were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and then, the proteins were transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk at 37 °C for 1 h and then incubated with a His antibody (1:1000 diluted in 5% nonfat dry milk) at 4 °C overnight. The membrane was removed the next day and washed four times with PBST (1:25 diluted in PBS-0.05% Tween-20) for 5 min each time. Then, the membrane was incubated with a secondary antibody (goat anti-rabbit IgG, HRP-conjugate, freshly diluted 1:5000 in 5% nonfat dry milk) at 37 °C for 1 h. The blotted membrane was washed four times for 5 min each in TBST prior to electro-chemiluminescence (ECL) detection in the dark. Kinase activity was assayed according to the instructions of the Kinase-Lumi Plus Luminescent Kinase Assay Kit (Beyotime, China). Myelin basic protein (MBP), which is an efficient artificial substrate for numerous protein kinases including BRI1 (Zhao et al. 2013), was used as a substrate. The relative luminescence unit (RLU) value of each well was measured using a SpectraMax M5 microplate reader (Molecular Devices, USA). The kinase activity was calculated per mg (protein) in U according to the kit instructions.
RNA-seq analysis and RT-qPCR detection
Total RNA from the fresh leaves of 6421 and E29 (three seedlings each) was extracted with the Easy Pure Plant RNA Kit (Tiangen, China). The mRNA was purified and fragmented. Using the mRNA fragments as templates, double-stranded cDNA was synthesized, end repaired, and adaptor ligated. Finally, a cDNA library was created after PCR amplification and purification, and sequenced on an Illumina HiSeq 2500 platform.
The raw RNA-Seq reads were processed with in-house Perl scripts to remove the adaptor and low-quality sequences. Reads with more than 15% low-quality bases (Phred quality value less than 19) or with more than 5% N bases were treated as low-quality reads and therefore discarded. For the paired-end sequencing data, both reads were filtered out if one of the paired-end reads was removed. The obtained high-quality cleaned RNA-Seq data were used for downstream analyses. The cleaned RNA-Seq reads were aligned to the pepper reference genome (Zunla-1, Version 2.0) using TopHat v2.0.12 (Trapnell et al. 2012). The Fragment count for each gene in each sample was derived by HTSeq v0.6.0 (Anders et al. 2015) and then normalized to FPKM (fragments per kilobase per million mapped fragments) (Mortazavi et al. 2008) to estimate the expression levels of the genes in each sample. DESeq2 (Love et al. 2014) was used for differential gene expression analysis between the mutant E29 and the wild-type 6421. Genes with adjust p values ≤ 0.05 and |log2 fold-change| ≥ 1 were identified as differentially expressed genes (DEGs).
The GO (Gene Ontology; http://geneontology.org/) terms and KEGG (Kyoto Encyclopedia of Genes and Genomes; http://www.kegg.jp/kegg) terms enriched in DEGs were identified using the hypergeometric test, in which p value was calculated and adjusted as q value, and the genes of the entire genome were used as the background. GO terms or KEGG terms with q < 0.05 were considered to be significantly enriched.
The expression of the genes identified using RNA-Seq data was validated in the leaves of 6421 and E29 using RT-qPCR. Three biological replicates were performed. The reaction system and program were the same as those used from CaBRI1 VIGS. Primers are listed in Supplemental Table S2.
Results
Phenotypic characterization of the E29 mutant
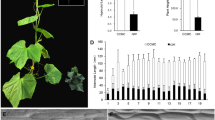
The E29 mutant had a short stature, compact plant type, and wide, thick, dark-green-bladed leaves (Fig. 1a–c). We histologically examined the stems at the seedling stage to determine the cause of the shortened stem in E29. Longitudinal sections of the stems revealed that the cell length in E29 was reduced, while the cell width was increased (Fig. 1d, e). The epidermal stem cells were shorter and thicker in E29 than in 6421 (Fig. 1f, g). These results indicated that the shorter and thicker stem in E29 was due to a reduction in cell length and an increase in cell width.

Plant phenotype and scanning electron microscopic observation of 6421 and E29 stems sectioned. a, b Seedlings of 6421 (left) and E29 (right), Scale Bar = 1 cm. (c) Plants of 6421 (left) and E29 (right), Scale Bar = 10 cm; (d, e) longitudinal sections on the fourth stem internodes of 6421 (d) and E29 (e), Scale Bar = 100 µm. f, g The epidermal cells on the fourth stem internodes of 6421 (f) and E29 (g), Scale Bar = 100 µm. h–m Phenotypic characterization of 6421 and E29 at the adult stage. n–p The chlorophyll levels of 6421 and E29. Data in H to P are given as means ± standard deviation (SD) (n = 6) (*p < 0.05, **p < 0.01 according to unpaired t test)
We then investigated the main phenotypes of 6421 and E29 at the adult stage. The plant height, plant width, and main stem length of E29 were significantly lower than those of 6421 by approximately 36.7%, 63.4%, and 50.6%, respectively (Fig. 1h–j). The main stem diameter and leaf area of E29 were significantly higher than those of 6421, approximately 1.15- and 1.41-fold, respectively (Fig. 1k, l). The internode elongation of E29 was severely inhibited, which caused a decrease in the number of flowers. As a result, the fruit yield per plant was decreased by 74.7% in E29 compared with 6421 (Fig. 1m). In addition, compared with those of 6421, the Chl a, Chl b, and Chl (a + b) levels of E29 were significantly increased by 33.2%, 321.1,% and 55.8%, respectively, indicating that higher chlorophyll levels caused the dark green leaves of E29 (Fig. 1n–p).
The signal-base mutation of CaBRI1 led to stunted growth in E29 mutant
The phenotypes of F1 and F2 progeny were analyzed. All of the F1 progeny showed a wild-type phenotype, indicating that the mutation was recessive. Among the 800 F2 progeny, the mutant phenotype segregated at a 1:3 ratio (wild-type phenotype:dwarf phenotype = 593:207, χ2 = 0.281, p < 0.05). We thereby concluded that the mutant phenotype was caused by a single recessive gene.
Whole-genome sequencing resulted in 245184846, 495574798, and 521749880 short reads from the parent 6421 pool (11.42 × average depth coverage), wild-type pool (22.76 × average depth coverage), and mutant pool (23.73 × average depth coverage), respectively. These short reads were aligned to the Zunla-1 reference genome and 33,859 single-nucleotide polymorphisms (SNPs) were identified. SNP index graphs were generated for the wild-type pool and mutant pool by plotting the average SNP index against the position of each sliding window in genome assembly (Fig. 2a). By combining the SNP index information from the wild-type and mutant pools, Δ(SNP_index) was calculated and plotted against the genome position (Fig. 2b). A 99% confidence level was chosen as the threshold for screening, and SNP sites with significant SNP index differences between the two progeny groups were selected from the genome. A total of 312 candidate SNPs were found and annotated between the two bulks via analysis of the sequencing data (Supplemental Table S3). Finally, we focused on the SNPs that caused stop-loss, stop-gain, or nonsynonymous mutations or splice-site variants as the candidate SNPs and found one SNP, SNP12G174801930, which was located in the exon of Capana12G001867 (Fig. 2c).

Identification of the candidate gene conferring the dwarf phenotype. a, b SNP_index graphs of wild-type pool, dwarf mutant pool (a) and Δ(SNP_index) graph (b) from whole-genome sequencing analysis. The x-axis represents the position of twelve chromosomes and the y-axis represents the SNP_index. c Chromosome 12. Red arrow indicates the candidate gene Capan12G001867. d Exon of Capana12G001867 (CaBRI1). Red arrow indicates the mutation site. The dot lines indicate the sequences encoding S_TKc domain. e Sequence alignment of the S_TKc domain with CaBRI1 homologs from diverse species. The 272-amino-acid (residues 894-1165) of CaBRI1 is the S_TKc domain. Amino-acid residues displaying similarity among the homologs are shaded black. The yellow mark indicates C-to-T mutation causes the Pro1157Ser mutation in S_TKc domain (colour figure online)
To confirm the causal SNP, the coding regions of Capana12G001867 (3642 bp) in 6421 and E29 were cloned and confirmed by Sanger sequencing. The sequencing result was consistent with that of the genome-wide resequencing analysis (data not shown). 129 plants were examined in the E29 × 6421 F2 population for the presence of this SNP. All mutant individual plants showed homozygous T at this site, while wild-type individual plants exhibited homozygous C or heterozygous C/T (Supplemental Figure S1), suggesting that SNP12G174801930 (C-to-T) cosegregated with the dwarf phenotype. Deep resequencing of 6421 revealed that SNP12G174801930 was C, and the same position in Zunla-1 was also C. We then examined another 200 dwarf plants in the F2 population for the presence of SNP12G174801930, and found that all mutant individual plants showed homozygous T at this site (data not shown). Therefore, we inferred that SNP12G174801930 was the causative mutation underlying the dwarf phenotype. SNP12G174801930 was located in the exon of the gene Capana12G001867, which encoded a homolog of the BR receptor BRI1, designated CaBRI1 here.
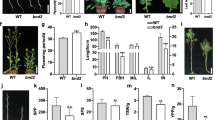
To further investigate the effect of C-to-T mutation of CaBRI1 on plant height, VIGS was used to silence the CaBRI1 gene in 6421 and E29. At 30 days after infiltration, the phenotype of plants treated with Agrobacterium carrying VIGS constructs targeting CaBRI1 was investigated. In 6421, compared with the TRV-infiltrated plants, plants infiltrated with the TRV:CaBRI1 generally became stunted and exhibited dwarfed morphology, a typical BR mutant effect, with reduced CaBRI1 transcript level (Fig. 3a, c, d, f). However, in E29, the plants infiltrated with the TRV:CaBRI1 had the same phenotype as the TRV-infiltrated plants, with reduced CaBRI1 transcript level too (Fig. 3b, c, e, f), suggesting that this point mutation in CaBRI1 was highly likely to confer a dwarf phenotype.

VIGS of CaBRI1 in 6421and E29. a The TRV-infiltrated 6421 plant and CaBRI1-silence 6421 plant. b The TRV-infiltrated E29 plant and CaBRI1-silence E29 plant. c Plant height of TRV-infiltrated plants and CaBRI1-silence plants. d The internodes length of TRV-infiltrated 6421 plant and CaBRI1-silence 6421 plant. e The internode length of TRV-infiltrated E29 plant and CaBRI1-silence E29 plant. f Relative mRNA levels of CaBRI1 in RTV-infiltrated plants and CaBRI1-silence plants. Data in c, f are given as means ± SD (n = 3) (**p < 0.01 according to unpaired t test)
The signal-base mutation impaired the kinase activity of CaBRI1
The C-to-T mutation at SNP12G174801930 was predicted to result in an amino-acid change from proline to serine at residue 1157 (Pro1157Ser) of CaBRI1 (Fig. 2d, e). Alignment of the CaBRI1 amino-acid sequences with those of homologous proteins from Arabidopsis, tomato, tobacco, potato, and Brassica napus (Mayer et al. 1999; Montoya et al. 2002; Scheer and Ryan 2002; Malinowski et al. 2009) showed that Pro1157Ser was located in the S_TKc domain and was highly conserved among these homologous proteins (Fig. 2e), indicating that E29 contained a novel allelic mutation in the S_TKc domain of CaBRI1. To investigate the possible effect of the Pro1157Ser substitution in the S_TKc domain of CaBRI1, purified proteins of CaBRI1-S_TKc and CaBRI1-S_TKc (dw) were used for the in vitro kinase activity assay. The recombinant plasmid (Fig. 4a) showed a 6658 bp band before enzyme digestion and exhibited two bands, 5677 bp and 981 bp, after digestion (Figure S2). SDS-PAGE and western blot analysis showed that CaBRI1-S_TKc and CaBRI1-S_TKc (dw) could be highly expressed in E. coli Arctic Express. The purified protein showed a molecular weight of 79 kD, which was consistent with the expected molecular weight (Fig. 4b, c, Figure S2). The CaBRI1-S_TKc showed strong activity, while this activity was dramatically decreased in CaBRI1-S_TKc (dw) harboring the Pro1157Ser substitution (Fig. 4d). Therefore, the mutation in the S_TKc domain impaired the kinase activity of CaBRI1.

In vitro kinase activity of CaBRI1 assay. a pMAL-c5x-CaBRI1-S_TKc/pMAL-c5x-CaBRI1-S_TKc (dw) recombinant plamid structure. b The SDS-PAGE results of CaBRI1-S_TKc and CaBRI1-S_TKc (dw) purified proteins. M: protein marker; 1 and 2: The eluted proteins of CaBRI1-S_TKc (1) and CaBRI1-S_TKc (dw) (2). c The west blot results of CaBRI1-S_TKc/and CaBRI1-S_TKc (dw) purified proteins. M: protein marker; 1 and 2: The purified proteins of CaBRI1-S_TKc (1) and CaBRI1-S_TKc (dw) (2). d The in vitro kinase activity of CaBRI1-S_TKc and CaBRI1-S_TKc (dw); Data in d are given as means ± SD (n = 3) (**p < 0.01 according to unpaired t test). The full-length blots and gels are presented in Supplementary Figure S2
The kinase inactive of CaBRI1 led to increased expression of BR biosynthetic genes and BR accumulation
In BR-mediated signaling pathway, BRI1 can activate BSKs and BSU1 to inactive BIN2, resulting in the activation of downstream transcription factors (TF) (Kim et al. 2009). The TF BES1/BZR1 can negatively regulate the expression of genes involved in BR biosynthesis, such as CPD, DWF4, ROT3, and BR6ox (He et al. 2005; Yin et al. 2005; Vert and Chory 2006). Here, we used RNA-seq to identify the BR biosynthetic DEGs between 6421 and E29 and to further clarify the molecular mechanism of CaBRI1 in pepper. A total of 238 DEGs, with 159 upregulated genes and 79 downregulated genes, respectively, were identified between 6421 and E29 (Supplemental Table S4; Figure S3). RT-qPCR was performed on 12 randomly selected DEGs and these 12 genes showed similar expression patterns between RNA-seq and RT-qPCR (Supplemental Figure S4), confirming that the RNA-Seq data were reliable. GO term enrichment analysis was performed to study the specific function of these DEGs. The most enriched GO terms were the “cellular process” of the biological process, the “cell part” of the cellular component, and the “catalytic” of the molecular function (Fig. 5). KEGG enrichment analysis discovered 269 KEGG pathways, including the BR biosynthetic pathway (Supplemental Figure S5).

GO analysis of DEGs obtained through RNA-Seq of 6421 and E29. The abscissa of the bar plot represents the gene count within each GO category. All listed processes have enrichment p < 0.05
Two key BR biosynthetic DEGs, CaDWF4 (Capana02g002685) and CaROT3 (Capana02g002751), significantly upregulated in E29 mutant. RT-qPCR analysis showed that the expression levels of CaDWF4 and CaROT3 in E29 were increased approximately 2.3- and 3.1-fold of those in 6421 (Fig. 6a, b). It indicated that CaBES1/CaBZR1 cannot negatively regulate the expression of these two key BR biosynthetic genes in E29 mutant because of its kinase inactive of CaBRI1.

The expression levels of two BR biosynthetic genes, BL levels and the simplified models of BR signal transduction and downstream events. a, b The expression levels of CaDWF4 (a) and CaROT3 (b) in 6421 and E29. C The BL levels of 6421, E29 (left), TRV-infiltrated 6421 plants, and CaBRI1-silence 6421 plants (right). d The kinase active of CaBRI1 caused the CaBES1/CaBZR1 to negatively regulate the expression of CaDWF4 and CaROT3, while the kinase inactive of CaBRI1 cannot provide negative feedback regulation. The red letters indicate the upregulated DEGs; Data in a–c are given as means ± SD (n = 3) (**p < 0.01 according to unpaired t test) (colour figure online)
In the BR biosynthetic pathway, the 22α-hydroxylase encoded by DWF4 appears to catalyze rate-limiting steps (Choe et al. 1998). ROT3 gene is required for the late steps (Kim et al. 2005) and can catalyze C-23 hydroxylation of various 22-hydroxylated BRs with markedly different catalytic efficiencies (Ohnishi et al. 2006). The increased levels of these two genes may promote the synthesis of BR, resulting in the accumulation of a high level of BR. Here, we found that the level of BL in E29 mutant was significantly higher than that of 6421 (3.0-fold). Moreover, the level of BL in CaBRI1-silence 6421 plant was also significantly higher than that of TRV-infiltrated 6421 plant (3.7-fold) (Fig. 6c).
Discussion
The whole-genome sequencing method used in this study takes advantage of high-throughput whole-genome resequencing and BSA. This technology is advanced and rapid, and it serves as a tool for quickly identifying mutations caused by EMS (Abe et al. 2012; Chen et al. 2014; Takagi et al. 2015; Thole and Strader 2015; Xu et al. 2015; Tribhuvan et al. 2018). In this study, the wild-type 6421 and the EMS mutant line E29 derived from it can be considered to be near-isogenic lines. The resequencing of bulk segregant pools from an F2 population derived from two near-isogenic lines identified only one candidate causative SNP (SNP12G174801930). dCAPS marker analysis revealed that SNP12G174801930 cosegregated with the dwarf phenotype. SNP12G174801930 was located in the exon of the CaBRI1. The VIGS system, which involves TRV1 and TRV2, is a powerful tool for the functional characterization of genes in vivo during seed germination or other stages of early plant growth, and has been applied for dwarf phenotype identification (Van Schie et al. 2007; Liu et al. 2016; Thyssen et al. 2017). We applied the VIGS technique to identify whether a single-base mutation in CaBRI1 caused the dwarfing of pepper plants. The CaBRI1-silenced 6421 seedlings became stunted and exhibited a dwarf morphology, while CaBRI1-silenced E29 seedlings exhibited the same phenotype as TRV-infiltrated plants, suggesting that this point mutation in CaBRI1 was highly likely to confer a dwarf phenotype.
BRs regulate a wide range of plant developmental and physiological processes, such as cell elongation and division, photomorphogenesis, seed germination, flowering, male fertility, senescence, and tolerance to environmental stress (Wang et al. 2012; Perez et al. 2014). BR signaling is recognized by BRI1 to induce a phosphorylation-mediated cascade that regulates downstream genes expression. BRI1 is a receptor kinase belonging to a large family of plant leucine-rich repeat (LRR) receptor-like kinases (RLKs) (Li and Chory 1997; Vert et al. 2005). In Arabidopsis, more than 20 bri1 mutants with different mutation sites have been identified. bri1-6 (Gly644Asp), bri1-7 (Gly613Ser), bri1-9 (Ser662Phe), bri1-9 (Thr750Ile), and bri1-116 (Gln583Stop) were the bri1 mutants harboring missense or nonsense mutations in the 70-amino-acid ID (Noguchi et al. 1999; Friedrichsen et al. 2000; Hong et al. 2008). bri1-1 (Ala909Thr), bri1-8/108/112 (Arg983Gln), bri1-101 (Glu1078Lys), bri1-7 (Gly613Ser), bri1-103/104 (Ala1031Thr), bri1-115 (Gly1048Asp), bri1-117/118 (Asp1139Asn), bri1-301 (Gly989Ile), and bri1-105/106/107 (Gln1059Stop) were the bri1 mutants harboring missense or nonsense mutations in the kinase domain (Clouse et al. 1996; Li and Chory 1997; Noguchi et al. 1999; Friedrichsen et al. 2000; Hong et al. 2008; Xu et al. 2008). These mutants showed severe serious defects in growth and development processes including extremely dwarfed stature, wide and dark green leaf, and cannot be rescued by BR supplementation, which were similar to the E29 mutant trait.
In this study, the allelic mutation in CaBRI1 was also located in the kinase domain. The C-to-T mutation resulted in an amino-acid change from proline to serine at residue 1157 (Pro1157Ser) of CaBRI1. This site was a new mutation site, which was different from what was reported by previous researchers. The result can provide new insights into the details of early events of BR signaling transduction.
The amino-acid change might affect the kinase activity of BRI1. In Arabidopsis, the BRI1 kinase with a Glu1078Lys mutation exhibited greatly reduced kinase activity in bri1-101 (Friedrichsen et al. 2000). BRI1 kinase with a Ser1049Ala, Ser1044Ala, or Thr1045Ala mutation completely lost its activity in vitro, and the presence of these BRI1 mutations in transgenic plants failed to rescue the dwarf phenotype in bri1-5 (Wang et al. 2005; Yang et al. 2011; Hao et al. 2013). In rice, the Fn189 mutant exhibiting a change in a conserved residue (Ile1843Phe) in the kinase domain also showed dramatically decreased kinase domain activity of OsBRI1 (Zhao et al. 2013). In our study, we assayed the in vitro kinase activity of CaBRI1 and found that the mutation of Pro1157Ser also reduced the kinase activity of CaBRI1. The kinase-dead BRI1 mutants are unable to activate the downstream transcription factor BES1/BZR1. The plants cannot normally grow and develop, because the inactivated BES1/BZR1 was unable to provide negative feedback regulation of biosynthetic gene expression (Kim et al. 2009; He et al. 2005; Yin et al. 2005; Vert and Chory 2006). In this study, we found that two key BR biosynthetic genes, CaDWF4 and CaROT3, were upregulated in E29 by transcriptome sequencing. In Arabidopsis, BRI1 and BAK1 can interact with each other through their kinase domains, depending on the kinase activity of BRI1. Kinase-dead BRI1 cannot interact with BAK1, resulting in the inability of BR signal to be transmitted. The mutant exhibited BR-insensitive characteristics (Wang et al. 2005; 2008). In Arabidopsis mutants bri1-4, bri1-5, bri1-6 (Noguchi et al. 1999) and pea mutant lka (Nomura et al. 1997, 1999), a high level of BL accumulated in BR-insensitive mutants. In this study, a large amount of BL was also accumulated in E29 and CaBRI1-silenced 6421 plants. We inferred that the kinase-inactive CaBRI1 prevented CaBES1/CaBZR1 from negatively regulating the expression of CaDWF4 and CaROT3, causing BR accumulation in pepper.
Therefore, we concluded that the new allelic variation of CaBRI1 conferred a dwarf phenotype and BR accumulation in pepper. This study has theoretical and practical value for obtaining pepper varieties suitable for facility cultivation and mechanized harvesting. In the next step, we will establish a genetic transformation system for pepper to confirm whether CaBRI1 (C-to-T) can complement the phenotype of E29 mutant and use the developed SNP marker (SNP12G174801930) to rapidly select pepper dwarf varieties.
Data availability
The transcriptome sequencing data have been deposited into sequence read archive (SRA) database under Accession Number PRJNA540896.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178. https://doi.org/10.1038/nbt.2095
Anders S, Pyl PT, Huber W (2015) HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. https://doi.org/10.1093/bioinformatics/btu638
Chen Z, Yan W, Wang N (2014) Cloning of a rice male sterility gene by a modified MutMap method. Hereditas 36:85–93. https://doi.org/10.3724/SP.J.1005.2014.00085
Choe S, Dilkes BP, Fujioka S, Takatsuto S, Sakurai A, Feldmann KA (1998) The DWF4 gene of Arabidopsis encodes a cytochrome P450 that mediates multiple 22ɑ-hydroxylation steps in brassinosteroid biosynthesis. Plant Cell 10:231–243. https://doi.org/10.1105/tpc.10.2.231
Chono M, Honda I, Zeniya H, Yoneyama K, Saisho D, Takeda K, Takatsuto S, Hoshino T, Watanabe Y (2003) A semidwarf phenotype of barley uzu results from a nucleotide substitution in the gene encoding a putative brassinosteroid receptor. Plant Physiol 133:1209–1219. https://doi.org/10.1104/pp.103.026195
Clouse SD, Langford M, McMorris TC (1996) A brassinosteroid-insensitive mutant in Arabidopsis thaliana exhibits multiple defects in growth and development. Plant Physiol 111:671–678. https://doi.org/10.1104/pp.111.3.671
Evans LT (1998) Feeding the ten billion: plants and population growth. Cambridge University Press, Cambridge
Friedrichsen DM, Joazeiro CAP, Li J, Hunter T, Chory J (2000) Brassinosteroid-insensitive-1 is a ubiquitously expressed leucine-rich repeat receptor serine/threonine kinase. Plant Physiol 123:1247–1256. https://doi.org/10.1104/pp.123.4.1247
Hao J, Yin Y, Fei SZ (2013) Brassinosteroid signaling network: implications on yield and stress tolerance. Plant Cell Rep 32:1017–1030. https://doi.org/10.1007/s00299-013-1438-x
He JX, Gendron JM, Sun Y, Gampala SSL, Gendron N, Sun CQ, Wang ZY (2005) BZR1 is a transcriptional repressor with dual roles in brassinosteroid homeostasis and growth responses. Science 307:1634–1638. https://doi.org/10.1126/science.1107580
Hong Z, Jin H, Tzfira T, Li J (2008) Multiple mechanism-mediated retention of a defective brassinosteroid receptor in the endoplasmic reticulum of Arabidopsis. Plant Cell 20:3418–3429. https://doi.org/10.1105/tpc.108.061879
Kim GT, Fujioka S, Kozuka T, Tax FE, Takatsuto S, Yoshida S, Tsukaya H (2005) CYP90C1 and CYP90D1 are involved in different steps in the brassinosteroid biosynthesis pathway in Arabidopsis thaliana. Plant J 41:710–721. https://doi.org/10.1105/tpc.108.061879
Kim TW, Guan S, Sun Y, Deng Z, Tang W, Shang JX, Sun Y, Burlingame AL, Wang ZY (2009) Brassinosteroid signal transduction from cell-surface receptor kinases to nuclear transcription factors. Nat Cell Biol 11:1254–1260. https://doi.org/10.1038/ncb1970
Li J, Chory J (1997) A putative leucine-rich repeat receptor kinase involved in brassinosteroid signal transduction. Cell 90:929–938. https://doi.org/10.1016/S0092-8674(00)80357-8
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Liu Y, Huang X, Li M, He P, Zhang Y (2016) Loss-of-function of Arabidopsis receptor-like kinase BIR1 activates cell death and defense responses mediated by BAK1 and SOBIR1. New Phytol 212:637–645. https://doi.org/10.1111/nph.14072
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Malinowski R, Higgins R, Luo Y, Piper L, Nazir A, Bajwa VS, Clouse SD, Thompson PR, Stratmann JW (2009) The tomato brassinosteroid receptor BRI1 increases binding of systemin to tobacco plasma membranes, but is not involved in systemin signaling. Plant Mol Biol 70:603–616. https://doi.org/10.1007/s11103-009-9494-x
Mayer K, Schüller C, Wambutt R, Murphy G, Volckaert G, Pohl T, Düsterhöft A, Stiekema W, Entian KD, Terryn N, Harris B, Ansorge W, Brandt P, Grivell L, Rieger M, Weichselgartner M, de Simone V, Obermaier B, Mache R, Müller M, Kreis M, Delseny M, Puigdomenech P, Watson M, Schmidtheini T, Reichert B, Portatelle D, Perez-Alonso M, Boutry M, Bancroft I, Vos P, Hoheisel J, Zimmermann W, Wedler H, Ridley P, Langham SA, McCullagh B, Bilham L, Robben J, Van der Schueren J, Grymonprez B, Chuang YJ, Vandenbussche F, Braeken M, Weltjens I, Voet M, Bastiaens I, Aert R, Defoor E, Weitzenegger T, Bothe G, Ramsperger U, Hilbert H, Braun M, Holzer E, Brandt A, Peters S, van Staveren M, Dirkse W, Mooijman P, Lankhorst RK, Rose M, Hauf J, Kötter P, Berneiser S, Hempel S, Feldpausch M, Lamberth S, Van den Daele H, De Keyser A, Buysshaert C, Gielen J, Villarroel R, De Clercq R, Van Montagu M, Rogers J, Cronin A, Quail M, Bray-Allen S, Clark L, Doggett J, Hall S, Kay M, Lennard N, McLay K, Mayes R, Pettett A, Rajandream MA, Lyne M, Benes V, Rechmann S, Borkova D, Blöcker H, Scharfe M, Grimm M, Löhnert TH, Dose S, de Haan M, Maarse A, Schäfer M, Müller-Auer S, Gabel C, Fuchs M, Fartmann B, Granderath K, Dauner D, Herzl A, Neumann S, Argiriou A, Vitale D, Liguori R, Piravandi E, Massenet O, Quigley F, Clabauld G, Mündlein A, Felber R, Schnabl S, Hiller R, Schmidt W, Lecharny A, Aubourg S, Chefdor F, Cooke R, Berger C, Montfort A, Casacuberta E, Gibbons T, Weber N, Vandenbol M, Bargues M, Terol J, Torres A, Perez-Perez A, Purnelle B, Bent E, Johnson S, Tacon D, Jesse T, Heijnen L, Schwarz S, Scholler P, Heber S, Francs P, Bielke C, Frishman D, Haase D, Lemcke K, Mewes HW, Stocker S, Zaccaria P, Bevan M, Wilson RK, de la Bastide M, Habermann K, Parnell L, Dedhia N, Gnoj L, Schutz K, Huang E, Spiegel L, Sehkon M, Murray J, Sheet P, Cordes M, Abu-Threideh J, Stoneking T, Kalicki J, Graves T, Harmon G, Edwards J, Latreille P, Courtney L, Cloud J, Abbott A, Scott K, Johnson D, Minx P, Bentley D, Fulton B, Miller N, Greco T, Kemp K, Kramer J, Fulton L, Mardis E, Dante M, Pepin K, Hillier L, Nelson J, Spieth J, Ryan E, Andrews S, Geisel C, Layman D, Du H, Ali J, Berghoff A, Jones K, Drone K, Cotton M, Joshu C, Antonoiu B, Zidanic M, Strong C, Sun H, Lamar B, Yordan C, Ma P, Zhong J, Preston R, Vil D, Shekher M, Matero A, Shah R, Swaby I’K, Shaughnessy AO’, Rodriguez M, Hoffman J, Till S, Granat S, Shohdy N, Hasegawa A, Hameed A, Lodhi M, Johnson A, Chen E, Marra M, Martienssen R, McCombie WR (1999) Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature 402:769–777. https://doi.org/10.1038/47134
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Montoya T, Nomura T, Farrar K, Kaneta T, Yokota T, Bishop GJ (2002) Cloning the tomato curl3 gene highlights the putative dual role of the leucine-rich repeat receptor kinase tBRI1/SR160 in plant steroid hormone and peptide hormone signaling. Plant Cell 14:3163–3176. https://doi.org/10.1105/tpc.006379
Morinaka Y, Sakamoto T, Inukai Y, Agetsuma M, Kitano H, Ashikari M, Matsuoka M (2006) Morphological alteration caused by brassinosteroid insensitivity increases the biomass and grain production of rice. Plant Physiol 141:924–931. https://doi.org/10.1104/pp.106.077081
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. https://doi.org/10.1038/nmeth.1226
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4326. https://doi.org/10.1093/nar/8.19.4321
Nakamura A, Fujioka S, Sunohara H, Kamiya N, Hong Z, Inukai Y, Miura K, Takatsuto S, Yoshida S, Ueguchi-Tanaka M, Hasegawa Y, Kitano H, Matsuoka M (2006) The role of OsBRI1 and its homologous genes, OsBRL1 and OsBRL3, in rice. Plant Physiol 140:580–590. https://doi.org/10.1104/pp.105.072330
Neff MM, Neff JD, Chory J, Pepper AE (1998) dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J 14:387–392. https://doi.org/10.1046/j.1365-313X.1998.00124.x
Noguchi T, Fujioka S, Choe S, Takatsuto S, Yoshida S, Yuan H, Feldmann KA, Tax FE (1999) Brassinosteroid-insensitive dwarf mutants of Arabidopsis accumulate brassinosteroids. Plant Physiol 121:743–752. https://doi.org/10.1104/pp.121.3.743
Nomura T, Nakayam M, Reid JB, Takeuchi Y, Yokota T (1997) Blockage of brassinosteroid biosynthesis and sensitivity causes dwarfism in garden pea. Plant Physiol 113:31–37. https://doi.org/10.1104/pp.113.1.31
Nomura T, Kitasaka Y, Takatsuto S, Reid JB, Fukami M, Yokota T (1999) Brassinosteroid/Sterol synthesis and plant growth as affected by lka and lkb mutations of pea. Plant Physiol 119:1517–1526. https://doi.org/10.1104/pp.119.4.1517
Ohnishi T, Szatmari AM, Watanabe B, Fujita S, Bancos S, Koncz C, Lafos M, Shibata K, Yokota T, Sakata K, Szekeres M, Mizutani M (2006) C-23 hydroxylation by Arabidopsis CYP90C1 and CYP90D1 reveals a novel shortcut in brassinosteroid biosynthesis. Plant Cell 18:3275–3288. https://doi.org/10.1105/tpc.106.045443
Peng JR, Richards DE, Hartley NM, Murphy GP, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AJ, Pelica F, Sudhakar D, Christou P, Snape JW, Gale MD, Harberd NP (1999) ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 400:256–261. https://doi.org/10.1038/22307
Perez MBM, Zhao J, Yin Y, Hu J, Fernandez MGS (2014) Association mapping of brassinosteroid candidate genes and plant architecture in a diverse panel of Sorghum bicolor. Theor Appl Genet 127:2645–2662. https://doi.org/10.1007/s00122-014-2405-9
Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y, Yang Y, Wu Z, Mao L, Wu H, Hu-Ling C, Zhou H, Lin H, González-Morales S, Trejo-Saavedra DL, Tian H, Tang X, Zhao M, Huang Z, Zhou A, Yao X, Cui J, Li W, Chen Z, Feng Y, Niu Z, Bi S, Yang X, Li W, Cai H, Luo X, Montes-Hernández S, Leyva-González MA, Xiong ZQ, He X, Bai L, Tan S, Tang X, Liu D, Liu J, Zhang S, Chen M, Zhang L, Zhang L, Zhang Y, Liao W, Zhang Y, Wang M, Lv X, Wen B, Liu H, Luan H, Zhang Y, Yang S, Wang X, Xu J, Li X, Li S, Wang J, Palloix A, Bosland PW, Li Y, Krogh A, Rivera-Bustamante RF, Herrera-Estrella L, Yin Y, Yu J, Hu K, Zhang Z (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci 111:5135–5140. https://doi.org/10.1073/pnas.1400975111
Rouse D, Mackay P, Stirnberg P, Estelle M, Leyser O (1998) Changes in auxin response from mutations in an AUX/IAA gene. Science 279:1371–1373. https://doi.org/10.1126/science.279.5355.1371
Sasaki A, Ashikari M, Ueguchi-Tanaka M, Itoh H, Nishimura A, Swapan D, Ishiyama K, Saito T, Kobayashi M, Khush GS, Kitano H, Matsuoka M (2002) Green revolution: a mutant gibberellin-synthesis gene in rice. Nature 416:701. https://doi.org/10.1038/416701a
Scheer JM, Ryan CA Jr (2002) The systemin receptor SR160 from Lycopersicon peruvianum is a member of the LRR receptor kinase family. P Natl Acad Sci 99:9585–9590. https://doi.org/10.1126/science.279.5355.1371
Silverstone AL, Sun TP (2000) Gibberellins and the green revolution. Trends Plant Sci 5:1–2. https://doi.org/10.1016/s1360-1385(99)01516-2
Suh HS (1978) The segregation mode of plant height in the cross of rice varieties. II. Linkage analysis of the semi-dwarfness of rice variety “Tongil”. Korean J Breeding 10:1–6
Sun C, Li J (2017) Biosynthesis, catabolism, and signal transduction of brassinosteroids. Plant Physiol J 53:291–307. https://doi.org/10.13592/cnki/ppj.2017.1002
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183. https://doi.org/10.1111/tpj.12105
Takagi H, Tamiru M, Abe A, Yoshida K, Uemura A, Yaegashi H, Obara T, Oikawa K, Utsushi H, Kanzaki E, Mitsuoka C, Natsume S, Kosugi S, Kanzaki H, Matsumura H, Urasaki N, Kamoun S, Terauchi R (2015) MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol 33:445–449. https://doi.org/10.1038/nbt.3188
Tang M, Zeng H, Ren J, Zhang N, Li Y (2017) Research progress on dwarf character of cucurbit plants. J Changjiang Veg 2:41–44. https://doi.org/10.3865/j.issn.1001-3547.2017.02.016
Thole JM, Strader LC (2015) Next-generation sequencing as a tool to quickly identify causative EMS-generated mutations. Plant Signal Behav 10:1–4. https://doi.org/10.1080/15592324.2014.1000167
Thyssen GN, Fang DD, Turley RB, Florane CB, Li P, Mattison CP, Naoumkina M (2017) A Gly65Val substitution in an actin, GhACT_LI1, disrupts cell polarity and F-actin organization resulting in dwarf, lintless cotton plants. Plant J 90:111–121. https://doi.org/10.1111/tpj.13477
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflink. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Tribhuvan KU, Kumar K, Sevanthi AM, Gaikwad K (2018) MutMap: a versatile tool for identification of mutant loci and mapping of genes. Indian J Plant Physiol 23:612–621. https://doi.org/10.1007/s40502-018-0417-1
Tsuchiya Y, McCourt P (2009) Strigolactones: a new hormone with a past. Curr Opin Plant Biol 12:556–561. https://doi.org/10.1016/j.pbi.2009.07.018
Van Schie CCN, Ament K, Schmidt A, Lange T, Haring MA, Schuurink RC (2007) Geranyl diphosphate synthase is required for biosynthesis of gibberellins. Plant J 52:752–762. https://doi.org/10.1111/j.1365-313X.2007.03273.x
Vert G, Chory J (2006) Downstream nuclear events in brassinosteroid signalling. Nature 441:96–100. https://doi.org/10.1038/nature04681
Vert G, Nemhauser JL, Geldner N, Hong F, Chory J (2005) Molecular mechanisms of steroid hormone signaling in plants. Annu Rev Cell Dev Biol 21:177–201. https://doi.org/10.1146/annurev.cellbio.21.090704.151241
Wang X, Li X, Meisenhelder J, Hunter T, Yoshida S, Asami T, Chory J (2005) Autoregulation and homodimerization are involved in the activation of the plant steroid receptor BRI1. Dev Cell 8:855–865. https://doi.org/10.1016/j.devcel.2005.05.001
Wang X, Kota U, He K, Blackbum K, Li J, Goshe MB, Huber SC, Clouse SD (2008) Sequential transphosphorylation of the BRI1/BAK1 receptor kinase complex impacts early events in brassinosteroid signaling. Dev Cell 15:220–235. https://doi.org/10.1016/j.devcel.2008.06.011
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. https://doi.org/10.1093/nar/gkq603
Wang ZY, Bai MY, Oh E, Zhu JY (2012) Brassinosteroid signaling network and regulation of photomorphogenesis. Annu Rev Genet 46:701–724. https://doi.org/10.1146/annurev-genet-102209-163450
Xu Z, Li J (2006) Plant hormones research in China: past, present and future. Chinese Bull Bot 23:433–442
Xu W, Huang J, Li B, Li J, Wang Y (2008) Is kinase activity essential for biological functions of BRI1? Cell Res 18:472–478. https://doi.org/10.1038/cr.2008.36
Xu M, Wang S, Zhang S, Cui Q, Gao D, Chen H, Huang S (2015) A new gene conferring the glabrous trait in cucumber identified using MutMap. Hortic Plant J 1:29–34. https://doi.org/10.16420/j.issn.2095-9885.2015-0003
Yamamuro C, Ihara Y, Wu X, Noguchi T, Fujioka S, Takatsuto S, Ashikari M, Kitano H, Matsuoka M (2000) Loss of function of a rice brassinosteroid insensitive1 homolog prevents internode elongation and bending of the lamina joint. Plant Cell 12:1591–1605. https://doi.org/10.1105/tpc.12.9.1591
Yang CJ, Zhang C, Lu YN, Jin JQ, Wang XL (2011) The mechanisms of brassinosteroids’ action: from signal transduction to plant development. Mol Plant 4:588–600. https://doi.org/10.1093/mp/ssr020
Yang BZ, Zhou SD, Yang LL, Ma YQ, Zou XX (2017) Phenotypic characteristic of a dwarf mutant in pepper and its response to exogenous hormones. J Hunan Agric Univ 43:518–523. https://doi.org/10.13331/j.cnki.jhau.2017.05.009
Yin Y, Vafeados D, Tao Y, Yoshida S, Asami T, Chory J (2005) A new class of transcription factors mediates brassinosteroid-regulated gene expression in Arabidopsis. Cell 120:249–259. https://doi.org/10.1016/j.cell.2004.11.044
Zhao J, Wu C, Yuan S, Yin L, Sun W, Zhao Q, Zhao B, Li X (2013) Kinase activity of OsBRI1 is essential for brassinosteroids to regulate rice growth and development. Plant Sci 199:113–120. https://doi.org/10.1016/j.plantsci.2012.10.011
Funding
This study was funded by the National Natural Science Foundation of China (Grant No. 31601757).
Author information
Authors and Affiliations
Contributions
BZY and SDZ performed the experiments and wrote this manuscript. LJO and FL helped data analysis. LYY, JYZ, WCC, ZQZ, and SY assisted in the experiments. YQM and XXZ supervised the study. All authors read, commented, and approved the submitted and final versions.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Stefan Hohmann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, B., Zhou, S., Ou, L. et al. A novel single-base mutation in CaBRI1 confers dwarf phenotype and brassinosteroid accumulation in pepper. Mol Genet Genomics 295, 343–356 (2020). https://doi.org/10.1007/s00438-019-01626-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01626-z