
Abstract
Skeletal muscle fibers are mainly categorized into red and white fiber types, and the ratio of red/white fibers within muscle mass plays a crucial role in meat quality such as tenderness and flavor. To better understand the molecular difference between the two muscle fibers, this study takes advantage of RNA-seq to compare differences in the transcriptome between extensor digitorum longus (EDL; white fiber) and soleus (Sol; red fiber) muscles of large white pigs. In total, 89,658,562 and 46,723,568 raw reads from EDL and Sol were generated, respectively. Comparison between the two transcriptomes revealed 561 differentially expressed genes, with 408 displaying higher and 153 lower levels of expression in Sol. Quantitative real-time polymerase chain reaction validated the differential expression of nine genes. Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis discovered several differentially enriched biological functions and processes of the two muscles. Moreover, transcriptome comparison between EDL and Sol identified many muscle-related genes (CSRP3, ACTN2, MYL1, and MYH6) and pathways related to myofiber formation, such as focal adhesion, tight junction formation, extracellular matrix (ECM)–receptor pathway, calcium signaling, and Wnt signaling. In addition, 58,362 and 58,359 single nucleotide polymorphisms were identified in EDL and Sol, respectively, and the sequence of 9069 genes was refined at the 5′, 3′ or both ends. Numerous novel transcripts and alternatively spliced RNAs were also identified. Our transcriptome analysis constitutes valuable sequence resource for uncovering important genes and pathways involved in muscle fiber type determination, and might help further our understanding of the molecular mechanisms in different types of muscle.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vertebrate skeletal muscles are mainly comprised of two myofiber types: red (type I and IIa) and white (type IIb) (Lefaucheur et al. 2004). In many pig breeds, muscles with different fiber type composition vary in postmortem properties, thus affecting meat quality (Choi et al. 2013). Specifically, favorable meat quality determinants, such as tenderness and color, show tight and positive correlation with the abundance of red (or oxidative) muscle fibers (Maltin et al. 2003). Profiling differentially expressed genes (DEGs) among different muscles with varying fiber type compositions enables molecular differences between the two fiber types and factors dictating fiber type determination to be discovered. It will also contribute to improvement of meat quality in the animal husbandry industry.
Transcriptome studies have been performed in the hope of revealing the complete network that regulates the phenotypic traits of different types of muscles. cDNA microarray analysis of porcine skeletal muscle by Bai et al. (2003) identified a large number of candidate genes for muscle phenotype determination, which included several members of the casein kinase 2 signaling pathway. In 2012, de la Serrana et al. (2012) performed a de novo assembly of 454 reads of the transcriptome of the fast skeletal muscle of gilthead sea bream and identified numerous gene paralogs, microsatellite repeats and splice variants. Recently, a genome-wide investigation between the slow (soleus) and fast (longissimus dorsi) skeletal muscle of Chinese Meishan pigs identified 28 key signaling pathways, such as insulin and mitogen-activated protein kinase (MAPK) pathways that contribute to metabolic discrepancy (Li et al. 2010). However, the aforementioned methods also come with some limitations, such as an over-reliance on the present understanding of relative genome sequences, and irregular high background levels caused by cross-hybridization. Furthermore, the dynamic range detection is also impeded by both background and saturation of signals (Wang et al. 2009).
Next generation sequencing technologies (e.g., RNA-seq) are powerful tools for unraveling transcriptome complexity, identification of genes, and for alternative splice and transcriptome profiling. RNA-seq displays several advantages over other existing methods, as it is not constrained to identifying transcripts that relate to existing genome sequence and also has very low background signals, enabling the whole transcriptome to be studied in a high-throughput and quantitative manner (Wang et al. 2009). Previously, Palstra et al. (2013) utilized an RNA-seq approach to classify slow and fast muscle transcriptomes of rainbow trout and found 1085 and 1228 novel sequences in the muscle types, respectively, covering most of the key genes for skeletal muscle function.
The large white, an intensively selected porcine breed, shows better growth performance than many other pig breeds (Ruusunen et al. 2012). Breeding and selection can increase skeletal muscle mass by significantly changing both the number and diameter of myofibers. However, a byproduct of breeding and selection aiming to increase growth rate and lean meat production is variation in skeletal muscle traits, such as myofiber composition. This is important because muscle fiber composition is a major property of meat quality (Rehfeldt et al. 2008). Therefore, a better understanding of the molecular mechanisms defining this trait will provide insight into improving meat production and quality.
In this study, RNA-seq was used to investigate differences in skeletal muscle transcriptomes between red (soleus, Sol) and white (extensor digitorum longus, EDL) fiber muscles of the large white pig. We also explored the effects of DEGs in transcriptome networks between different types of myofiber. Our study provides an analysis of the mechanisms underlying muscle fiber type determination, and expands our existing knowledge on the molecular networks regulating meat quality and production.
Materials and methods
Animals and tissue sampling
Three 180-day-old large white pigs from the same litter were slaughtered by electrical stunning and exsanguination, in accordance with the national regulations for commercial slaughtering. EDL and Sol muscle samples were isolated and then snap-frozen in liquid nitrogen for further analysis.
RNA isolation, library construction and RNA-seq analysis
EDL and Sol muscle samples from the three 180-day-old pigs were, respectively, pooled, and total RNA was collected from each pooled sample using Trizol reagent (Invitrogen, USA) in accordance with the manufacturer’s protocol. The quality and concentration of RNAs were determined by denaturing gel electrophoresis and spectrophotometry (Thermo, USA). The pooled RNA samples from three pigs are tested by RNA-seq and we carry out qPCR validation with these pigs for three times separately.
Total RNA was treated with DNase I and mRNA isolated with Oligo (dT) magnetic beads. The mRNA was fragmented into short fragments using fragmentation buffer and cDNA was synthesized using the mRNA fragments as templates. The purified short fragments were incubated with EB buffer for end reparation and single nucleotide A (adenine) addition, and then the short fragments were ligated to adaptors. After agarose gel electrophoresis, the proper fragments were isolated as polymerase chain reaction (PCR) templates. Quantification and quality control of the sample libraries were performed using an Agilent 2100 Bioanalyzer and the ABI StepOnePlus Real-time PCR System.
The library was sequenced using an Illumina HiSeq™ 2000, and primary sequencing data (raw reads) were subjected to quality control to filter clean reads from raw reads. Clean reads were aligned to reference sequences using SOAPaligner/SOAP2 (Li et al. 2009b). The alignment data were utilized to calculate read distribution and to perform coverage analysis.
Sequence annotation and analysis of differential gene expression
After quality control and filtering of raw data, clean reads were annotated to reference sequences using SOAPaligner/SOAP2 with up to five mismatches allowed. Mapping was performed against the entire genome sequence archived in the Ensemble database (Sus scrofa, http://ensembl.org/pub/release-71/fasta/sus_scrofa/), and the statistics of alignment results are presented for each reference. We used a rigorous algorithm by BGI-Shenzhen to detect DEGs between EDL and Sol muscles based on the reads per kilobase transcriptome per million mapped reads (RPKM) method (Mortazavi et al. 2008), and the formula is:
Given to be the expression level of reference gene X, N represents the whole number of uniquely aligned reads for all genes, C to be number of uniquely mappable reads that aligned to gene X, and L shows the number of bases on gene X. The RPKM algorithm is utilized to minimize the impact of sequencing discrepancy as well as different gene length for calculating the expression level. As a result, the relative gene expression could be directly applied for comparing the genes with differentially expressed levels among detected samples.
DEG analysis tests thousands of hypotheses simultaneously; therefore, correlation for false positive and false negative errors was performed using the False Discovery Rate (FDR) method. We defined genes as DEGs that had absolute fold-change values between EDL and Sol of greater than 2.0 and an FDR of less than 0.001 (Wang et al. 2010).
GO and pathway enrichment analysis of DEGs
To thoroughly assess the properties of genes and their products in the two different types of muscle, Gene Ontology (GO) and pathway enrichment analysis were performed to categorize the considerably enriched functional classification or metabolic pathways in which DEGs operate. First all DEGs were aligned to GO terms in the database (http://www.geneontology.org/), and gene numbers calculated for every term. Hypergeometric tests then filtered considerably enriched GO terms from the input list of DEGs, based on GO::TermFinder. The calculated p value was assessed by Bonferroni correction, and GO terms were considered as significantly enriched when corrected p values ≤ 0.05. Pathway analysis was used to identify the significant pathways in which DEGs are involved according to the KEGG.
Refinement of gene structures
Transcripts were assembled from reads using Cufflink software (Roberts et al. 2011). Gene structure was determined on the basis of the distribution of reads, reference gene annotations and paired-end sequences. Having performed transcript reconstruction and aligned reads to the reference genome, we obtained the genomic regions that have continuous and uniquely mapped reads, called transcription-active regions (TARs). With the paired-end data, different TARs are utilized to construct a potential gene model, and optimization of 5′ and 3′ boundaries was decided by comparing the potential gene model with existing gene annotations.
Single nucleotide polymorphism analysis
We utilized SOAPsnp software (Li et al. 2009a) to detect putative single nucleotide polymorphisms (SNPs) between EDL and Sol samples. According to alignment of the sequencing reads on existing reference sequences, this program assembles the consensus sequence for the transcriptome of a newly sequenced individual. SNPs were then detected on this consensus sequence after being compared with the reference. The program calculates the probability of each genotype at each site according to the alignment results of short reads to a reference sequence accompanied by the corresponding sequencing quality scores. Finally, it deduces the genotype with the highest posterior probability at each site on the basis of Bayes’ theorem.
Identification of novel transcript units and alternative splicing events
To identify novel transcribed regions, assembled transcripts and annotated genomic transcripts were compared with reference sequences in the Ensemble database. Transcript units identified in intergenic regions more than 200 bp away from annotated genes and with a continuous mapping length ≥180 bp and the sequence depth ≥2 were defined as putative novel transcript units. Coding Potential Calculator software (CPC; http://cpc.cbi.pku.edu.cn/) was employed to assess protein-coding ability.
According to the structure of exons, we classified alternative splicing (AS) events into seven different types, namely exon skipping, intron retention, alternative 5′ splicing, alternative 3′ splicing, alternative first exon, alternative last exon, and mutually exclusive exon, using SOAPsplice software (Huang et al. 2011). SOAPsplice implements an alignment step by mapping all of the complete reads to the reference genome, then the initially unmapped reads are mapped using a spliced alignment algorithm.
Protein–protein interaction network construction
To improve understanding of the complex relationships between genes in the EDL and Sol libraries, a network analysis of protein–protein interactions was performed. We analyzed 18 muscle-related DEGs and converted the gene IDs to the symbols of corresponding encoded proteins using bioDBnet software (http://biodbnet.abcc.ncifcrf.gov/db/db2db.php). Then we aligned gene symbols to the human functional interaction network in the BioGRID database (http://thebiogrid.org/) and generated a network using Cytoscape (http://www.cytoscape.org/). In this interaction network, proteins are represented as nodes and intermolecular interactions are indicated as edges between nodes.
Real-time quantitative PCR
Total tissue RNAs were isolated using Trizol reagent (Takara, Japan) and the quality of RNA was checked by agarose gel electrophoresis. RNA concentration was calculated using a spectrophotometer (Thermo, USA). cDNA was synthesized using a reverse transcription kit with random six-mers and oligo-dT primer (Takara, Japan) and approximately 500 ng of total RNA per reaction. Real-time quantitative PCR analysis was performed in triplicate using a SYBR green kit on a Bio-Rad iQ™5 system with GAPDH as the internal reference. The 2−ΔΔCt algorithm was used to calculate the expression level of target genes. Supplementary Table S12 lists the primer sequences, melting temperature and predicted product sizes for each gene studied. The expression difference of each gene between the two types of muscles was analyzed using the t test. A value of p < 0.05 was defined to be significant and p < 0.01 highly significant.
Results
Overview of RNA-Seq data
High-throughput mRNA sequencing was performed to obtain expressed short reads for EDL and Sol muscles of large white pigs. Two libraries were constructed by pooling RNA collected from three EDL muscles and from three Sol muscles. As shown in Table 1, RNA-seq analysis yielded 89,658,562 total reads for EDL muscle and 46,723,568 total reads for Sol muscle. After filtering to remove low-quality reads and adaptor sequences, we obtained 60,582,089 and 30,748,017 unique match reads for EDL and Sol samples, respectively.
Expressed short reads were mapped to 32,432 reference genes (16,415/16,017, EDL/Sol) of the Sus scrofa genome assembly in the Ensemble database (Supplementary Table S1). As shown in Fig. 1a, b, relatively large numbers of genes were highly expressed in both EDL and Sol muscle with RPKM values fluctuating from 1.0 to 50. Figure 1c, d shows the percentage of genes in the EDL and Sol libraries with different RPKM values. Only a small proportion of genes have very high expression levels (>1000 RPKM), while many genes are not expressed or only transcribed at a low level in both EDL and Sol. The gene ACTA1 (ENSSSCG00000010190) was highly expressed in both types of muscle with RPKM values of 49,175.14 and 60,481.23, respectively. In summary, in both muscle samples only a few genes were expressed at high levels, whereas most genes displayed medium or relatively low expression levels.

Distribution of genes based on RPKM. a–b The RPKM range detected in EDL and Sol libraries. c–d The percentage of RPKM range occupied in the whole reference genes between EDL and Sol samples
Identification of differentially expressed transcripts between EDL and Sol skeletal muscle
In this study, a rigorous formula was applied to categorize different expression levels of genes in the two different types of muscle based on “The significance of digital gene expression profiles”. Transcripts with a twofold or greater difference (FDR ≤ 0.001 and absolute values of \({\text{log}}_{2} \left( {\text{ratio}} \right) \ge 1\)) between the two muscle libraries were identified. The global expression pattern of Sol was compared with that of EDL and revealed that up to 2152 genes were differentially expressed between the two types of muscle. Overall, we identified 1534 up-regulated and 618 down-regulated genes (FDR ≤ 0.001 and |log2 Ratio| ≥ 1). Moreover, 561 transcripts with at least twofold difference (408 up-regulated and 153 down-regulated) were identified with the defined significance level (Supplementary Table S2).
Expression levels of muscle-related genes
To validate the two libraries, we investigated the transcription levels of 57 genes related to muscle characteristics (Table 2). In summary, there was no significant discrepancy for 22 muscle-related genes between EDL and Sol libraries (at most 1.5-fold up- or down-regulation). There were five muscle-related transcripts expressed at relatively high levels in both EDL and Sol (RPKM value > 1000), and the gene CXADR (ENSSSCG00000012012) was expressed at considerably lower levels in the Sol library than in the EDL library (Supplementary Table S3). The differentially expressed transcripts participate in contractile fiber and sarcomere formation, myofibril and myosin complex constitution, and muscle cell development. These RNA-seq results reaffirmed the differential expression of several genes between fast and slow skeletal muscles, such as GATA-6, HSP20, PGM, fibronectin and MyHC II b. In addition, several important transcription factors, including CSRP3, ACTG2, HOXD8 and HBM, which play crucial roles in muscle development, were observed to be differentially expressed. The newly identified DEGs might function in transcriptional regulation of metabolic or contractile characteristics in the different muscle fiber types.
qPCR validation of RPKM fold changes in EDL and Sol muscle
To validate the expression levels of DEGs observed in our RNA-seq assay, nine randomly selected transcripts shown to be differentially expressed on the basis of RPKM values were validated by qPCR. These genes included four with higher expression (MYOM3, CSRP3, MYH7B and COMP) and five with lower expression (ACTG2, HOXD8, HBM, IGFN1 and IFIT1) in EDL muscle compared with Sol muscle. Fold changes from qPCR analyses were compared with those from the RPKM values of the RNA-seq results. As shown in Fig. 2, the qPCR results confirmed our RNA-seq analysis, with all nine genes displaying similar expression patterns in both methods. Taken together, these results indicate the high reliability and accuracy of the RNA-seq data.

qRT-PCR validation of nine differentially expressed genes between EDL and Sol. Black indicates the tissue of EDL and white indicates the Sol. The data presented in Y axis represented the relative mRNA expression of both RPKM and qRT-PCR and expressed as means of three pigs ± SD. *p < 0.05, **p < 0.01. N = 3
Gene ontology analysis of DEGs
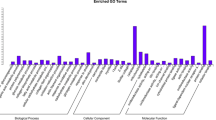
To illustrate the relationship between differential expression patterns of genes and their phenotypic variance, we performed GO analysis on genes that are differentially expressed between the two muscle types. In summary, 1422, 1481, and 1526 genes were mapped to one or multiple GO terms for biological process, molecular function and cellular component, respectively (Supplementary Table S4). According to biological process, the most abundant GO terms in both fast and slow muscles consisted of cellular process, single-organism cellular process, metabolic process, single-organism process, and biological regulation (Fig. 3). As for molecular function, GO terms for fast and slow muscle were predominantly composed of binding, catalytic activity, protein binding, heterocyclic compound binding, organic cyclic compound binding and ion binding. In terms of cellular component, GO terms in both fast and slow muscle mainly converged on cell and cell part, intracellular and intracellular part, organelle and intracellular organelle. Comparing the fast muscle transcriptome against that of the slow muscle identified a variance of GO term distribution with the threshold of corrected p ≤ 0.05. This level of significance indicates significantly enriched GO terms in DEGs. Significant expression discrepancy was identified for biological processes such as those correlated with muscle cell development and muscle structure development. With regard to molecular function, cytoskeletal protein binding and oxidoreductase activity were identified on the basis of essential differential expression. Genes involved in cellular components that were differentially expressed between fast and slow muscle were relevant to myofibril, contractile fiber, actin cytoskeleton and sarcomere. Our results provide further evidence for a relationship between muscle fiber type transformation and DEGs.

List of top ten enriched Gene Ontology terms based on GO classifications. GO annotations was based on Sus scrofa RefSeq and GO-terms were processed by GO::TermFinder and categorized into three functional groups such as biological process, molecular function and cellular component
Pathway analysis
KEGG pathway analysis was performed for annotated genes. Specific enrichment of DEGs was observed in 241 signaling pathways using the KEGG pathway database and 1673 genes were assigned with one or more KEGG annotation. As shown in Supplementary Table S5, 230 and 216 pathways were influenced by up- and down-regulated DEGs, respectively. The ten most enriched pathways between EDL and Sol are listed in Table 3. Notably, metabolic pathways occupied the top pathways influenced by both up- and down-regulated DEGs. The other enriched pathways mainly involved up-regulated DEGs, and included focal adhesion, dilated cardiomyopathy and regulation of actin cytoskeleton.
Pathway analysis of DEGs indicated that transcripts up-regulated in Sol muscle were mostly concerned with hypertrophic cardiomyopathy, vascular smooth muscle contraction, regulation of actin cytoskeleton and p53 signaling. Meanwhile, genes up-regulated in EDL muscle were involved with cardiac muscle contraction, tight junctions, vascular smooth muscle contraction and MAPK signaling. These results showed that different sub-branches of related signaling pathways might lead to the distinct gene expression observed in these two muscle types.
Construction of protein–protein interaction network
We next studied potential protein–protein interactions of 18 muscle-related DEGs. A comprehensive functional interaction network is not available for pigs; therefore, Homo sapiens interaction datasets were utilized to construct protein–protein interacting networks according to the BioGRID database. Cytoscape software produced a protein–protein network of the candidate proteins and their candidate targets; we obtained 404 target proteins (including proteins that self-interact) involved in this network (Supplementary Table S6). In summary, 412 nodes (18 candidate proteins and 394 candidate targets) and 464 edges made up the network. Many candidate targets are connected by only one candidate protein, but some can be regulated by multiple target proteins to affect their expression levels. As shown in Fig. 4, VCL, CRYAB and CCNB1 have more than 70 interactions with other proteins. However, MYL1, MYH7 and MYPN, which are closely associated with muscle growth and development, showed fewer interactions compared with other candidate proteins.

Protein–protein interaction network of 18 muscle-related DEGs. Network of 18 candidate proteins (Triangle) predicted to have 405 protein targets (Circle). The red and green dots delineate the up-regulated and down-regulated genes, respectively. Blue dots represent non-different expressed genes and the lines indicate the interactions between the proteins and their targets
Novel transcript prediction
To discover novel transcribed regions, we compared our assembled transcripts with annotated genomic transcripts from reference sequences. As shown in Supplementary Table S7, 10,962 and 9686 novel transcripts were identified in EDL and Sol muscles, respectively. The average length of a novel transcript unit was 1835 bp, with the size ranging from 180 to 22,355 bp. In EDL and Sol libraries, 81.91 and 82.60 % of novel transcript units were composed of multiple exons, and the longest novel transcript unit for EDL and Sol contained 98 and 85 exons. To study the function of novel transcripts, we further distinguished protein-coding RNAs from non-coding RNAs. Using CPC software, we found 9537 and 8636 coding transcripts in EDL and Sol muscle, respectively. Generally, protein-coding transcripts are associated with a higher CPC score, and thus better overall quality. In EDL and Sol muscle, 24.06 and 24.01 % of these coding transcripts, respectively, possess relatively high potential for protein coding ability (CPC scores ≥ 10).
Optimization of annotated gene structures
To annotate the boundaries of genes more accurately, we refined the 5′ and 3′ ends of genes, which is important for determining genomic loci for further investigation. After genes were compared with existing gene annotations, we analyzed the up- and down-stream regions of relevant transcripts acquired by RNA-seq analysis. We assigned 4870 and 4199 reference transcripts to EDL and Sol, respectively, and the extended results are shown in Supplementary Table S8. In the EDL sample, 3271 genes were refined in the 5′-end region, and 1599 genes had an extension at the 3′-end. In the Sol sample, 2814 genes were detected to be extended at the 5′-end, and 1385 genes had at least an extension at the 3′-end. In the optimized results, 1548 (17.1 %) genes were extended in at least two regions in both muscle samples.
Identification of alternative splicing events
AS is considered to be a significant factor in regulating gene expression and increasing functional complexity. To elucidate a complete picture of AS events for EDL and Sol libraries, SOAPsplice software was used to identify all theoretical splicing junctions. In summary, 9334 and 8585 AS events were observed in EDL and Sol libraries, respectively. As shown in Fig. 5, different types of AS events and the associated gene numbers were determined. In this study, we found that 61.8 and 61.4 % of the alternatively spliced genes underwent multiple AS events in EDL and Sol libraries, respectively, indicating the complexity of the Sus scrofa transcriptome. Of seven types of AS event, exon skipping was the most common, accounting for 37.3 % (3478) and 26.0 % (2230) of all AS events identified in the EDL and Sol libraries, respectively. In contrast, mutually exclusive exons were not detected in either library. Supplementary Table S9 shows all AS events in detail for each sample.

Statistics of alternative splicing and genes in EDL and Sol samples. The green bars illustrate the distribution of genes for each type of alternative splicing model. The red bars indicate the number of alternative splicing events identified in each gene
SNPs exploitation
Based on the alignment of the short reads to the Sus scrofa reference genome and the corresponding scores of sequence quality, we obtained the likelihood of each genotype at each site using SOAPsnp software and then identified candidate SNPs. We identified 58,362 and 58,359 potential SNPs in the EDL and Sol libraries (Supplementary Table S10). The most common change was C/Y, followed by G/R and A/R (Table 4). We mapped most of the SNPs on the 19 pig chromosomes (18 autosomes and 1 allosome). The two muscle samples displayed similar mapping results, including most of the SNPs distributed on chromosomes 2, 6 and 14. In addition, the number of SNPs was moderately higher in EDL than in Sol among four chromosomes (Fig. 6).

The distribution of SNPs in Sus scrofa. 116,721 putative SNPs distributed on the 18 pig autosomes and X allosome from two libraries are shown. All of the SNPs distributed predominantly on chromosomes 2, 6 and 14
Discussion
In this study, we utilized RNA-seq to provide a detailed view of the transcriptome of red- and white-fiber skeletal muscle of the large white pig. We generated >100 million sequence reads, corresponding to 13.6 Gb of raw sequence data, by performing Illumina sequencing of mRNA from EDL and Sol skeletal muscle. We identified 2152 DEGs and classified them into GO and KEGG categories; 230 and 216 pathways in EDL and Sol skeletal muscle, respectively, were influenced by up- and down-regulated DEGs. Among these DEGs, we identified a large body of genes and related pathways implicated in muscle fiber type determination, which may offer insight into further improvement of meat quality. By comparing the two types of muscle sample, we found numerous novel transcripts, AS events, SNPs and refinements of genes structure, which may play crucial roles in the different types of muscle.
The large white, as the lean type pig, shows better growth potential and stable meat quality in commercial use. The growth, carcass, fiber type and meat quality characteristics in lager white pigs remained stable and indicates relatively low standard error in the certain group. Selecting the live weight at the same age is one of the significant indicators to improve and control meat quality without reducing the growth and carcass performance (Choi et al. 2013). Thus, the pooled EDL and Sol samples from three pigs in the certain age group would possibly represent the characteristics of large white pigs in some extent.
Muscle-related genes
In both EDL and Sol muscle, 110 annotated genes were categorized as muscle-related genes, most of which are implicated in contractile fiber, myofibril, sarcomere, contractile fiber part, myosin complex and myosin II complex (Supplementary Table S11). Myofibril was the most significant GO term in muscle fiber type determination process.
CSRP3 is a component of the Z-disk of sarcomeres. CSRP family members are LIM proteins and participate in several regulatory functions indispensable for skeletal muscle development and myogenic differentiation (Weiskirchen and Gunther 2003). GO terms related to CSRP3 included contractile fiber, myofibril and cation binding. CSRP3 is a muscle-specific isoform and belongs to the cysteine and glycine-rich protein family, which plays an important role in muscle fiber differentiation (Xu et al. 2010). The RPKM value of CSRP3 in Sol muscle (5268.9) was nearly 3.5-fold higher than that in EDL muscle (1174.2), which is consistent with CSRP3 being constitutively expressed in slow muscle of rat but having a low expression level in fast muscle (Schneider et al. 1999). CSRP3 was previously shown to be expressed only in striated muscle and its expression pattern coincides with myogenic differentiation (Arber et al. 1994). In the indigenous Chinese Tongcheng pig breed, the relative mRNA expression of CSRP3 was up-regulated during the development of embryonic skeletal muscle, indicating a potential role in muscle growth.
Alpha actinins are actin-binding proteins found in both muscle and non-muscle cells, and interact with dystrophin and spectrins (Tiso et al. 1999). In skeletal muscle, ACTN2 and ACTN3 act as the key structural components of sarcomeric Z-lines and function to anchor actin. ACTN2 has varying expression levels in all types of myofibers, whereas ACTN3 is localized to a fast-twitch-specific type, found only in type II muscle fibers (Mills et al. 2001). The GO annotations related to these genes included contractile fiber, myofibril, sarcomere and actin filament. Previous research has indicated partial redundancy between ACTN3 and ACTN2 as in the ACTN3−/− mouse, the loss of ACTN3 in fast fibers is compensated for by an up-regulation of ACTN2, whose expression pattern changed from predominantly oxidative fibers to uniform expression in all fibers (MacArthur et al. 2007).
Pathways in different types of muscle
We have utilized RNA-seq analysis to thoroughly examine the skeletal muscle transcriptome of the large white pig, which will help elucidate the molecular mechanisms determining muscle fibers. From the results of pathway analysis, we have identified several significant pathways related to myofiber type and muscle contraction, including focal adhesion, ECM–receptor pathway, calcium signaling pathway, and Wnt signaling pathway.
Focal adhesion and ECM–receptor interaction pathways encompass a large proportion of the DEGs between the muscle types. The pathway analysis revealed 75 focal adhesion genes and 41 ECM–receptor interaction genes related to the muscle types. Collagens, as the major constituent of the ECM-receptor interaction, account for the strength and form of tissues (Leitinger 2011). Moreover, collagen is found to be expressed abundantly in connective tissue, which is a pivotal molecule for regulation of meat tenderness and texture (Lepetit 2007). Six genes from the collagen family were found according to the pathway analysis, including COL12A1, COL11A2, COL11A1, COL13A1, COL28A1 and COL6A3. GO annotations related to these genes were fibrillar collagen, extracellular region and extracellular matrix. Focal adhesions, composed of large, dynamic protein complexes, can connect the cytoskeleton of neighboring cells or join the cytoskeleton with the ECM. Their diverse components consist of scaffolding molecules, GTPases, and various enzymes, such as phosphatases, kinases, proteases and lipases (Geiger and Bershadsky 2002). Focal adhesions not only function as anchorage for the cell, but also act as signal carriers to transmit information about the condition of the ECM and thus influence the behavior of adjacent cells (Riveline et al. 2001). In this study, higher expression of several collagen encoding genes (COL12A1, COL11A2, COL11A1 and COL13A1) was detected in Sol muscle compared with EDL muscle, suggesting a distinction in collagen constituents between different types of myofibers.
Calcineurin is composed of calcineurin A catalytic subunit, calcineurin B regulatory subunit as well as the calcium-binding protein, calmodulin. It serves as a calcium mediated serine–threonine phosphatase and is expressed in many types of tissues. Calcineurin participates in many biological functions, such as T-lymphocyte activation, vascular and cardiac development, and skeletal muscle development (Chin et al. 1998). The expression of myofiber-type-specific genes in skeletal muscle was previously reported to be controlled by signaling pathways depending on calcineurin, which acted as a cyclosporin-sensitive, calcium-regulated serine/threonine phosphatase. Stimulation of calcineurin in skeletal muscle cells specifically activates the promoters of slow-fiber-specific genes. Transcriptional activation of slow-fiber-specific factor seems to be mediated by a combinatorial mechanism including NFAT and MEF2 families (Naya et al. 2000; Wu et al. 2000). RNA-seq analysis showed that 36 genes were implicated in this pathway, and some of them have relatively high expression levels in EDL and Sol muscle. This finding indicates a complete signaling pathway linking DEGs to selective changes in muscle fiber type.
Wnt signaling pathways represent a group of signal transduction pathways that respond to proteins outside the cell that mediate their signal to the inside of the cell via cell surface receptors (Sethi and Vidal-Puig 2010). Canonical Wnt signaling has been documented to modulate the differentiation of muscle stem cells in adult skeletal muscle, while non-canonical signals regulate the self-renewal of satellite cells and the development of myofibers (von Maltzahn et al. 2012). In this study, 29 enriched genes were identified in the Wnt signaling pathway. Multiple studies have investigated the role of canonical Wnt signaling in myofiber type determination. In the development of avian wing muscles, Wnt5a induces slow MHC-positive muscle fibers, while Wnt11 enhances the numbers of fast MHC-positive muscle fibers. The distinct influences of Wnt5a and Wnt11 are in accordance with their different expression patterns, which contribute to the ultimate distribution of different types of myofibers in the wing (Anakwe et al. 2003). Moreover, it has been confirmed that Wnt4 over-expression in chicken embryos results in both enhanced muscle mass, and a significant shift to fast type myofibers. Wnt4 acts as a stimulator during myoblast proliferation and differentiation, as shown in the C2C12 model, especially for fast-type fiber differentiation (Takata et al. 2007).
The RNA-seq analysis presented here increases the existing gene annotation for large white pigs, mainly for novel transcripts, AS events and SNP identification. It has been shown that AS plays a significant role in muscle fiber type determination according to many previous studies (Wei and Jin 2011; Sebastian et al. 2013). In this research, 9334 and 8585 AS events were detected in EDL and Sol libraries, respectively. We also identified 58,362 and 58,359 potential SNPs in the EDL and Sol libraries and the most common change was C/Y, followed by G/R and A/R. It is reported that SNPs are widely utilized linkage mapping, and they can potentially be tightly related to functional genes which may control muscle fiber type (Mortazavi et al. 2008; Lee et al. 2012; Lim et al. 2015). In addition, 10,962 and 9686 novel transcripts were found between EDL and Sol muscles from our analysis. The identification of novel transcripts, AS events and SNP will contribute to a better understanding of the mechanisms of transcription regulation in large white pigs.
Muscles with a higher percentage of type IIB fibers were reported to produce a higher amount of lactate and more rapid pH decline at the early postmortem period, contributing to a generally poorer meat quality than a higher percentage of type I fibers (Choi et al. 2010). Meat animals selected for their muscle growth capacity produce muscles with a higher percentage of larger type IIB fibers in pigs (Ruusunen and Puolanne 2004). Our RNA-seq analysis identified 2,152 DEGs and classified them into GO and KEGG categories; 230 and 216 pathways in EDL and Sol skeletal muscle were influenced by up- and down-regulated DEGs. Among these DEGs, CSPR3, ACTN2 and ACTN3 were selected to be implicated in muscle fiber type determination, and we construct the interaction network to see how these muscle-related DEGs affect the potential target genes. In addition, some important pathways such as wnt signaling, calcineurinm, focal adhesion and ECM–receptor interaction pathways, were identified to study the function in different muscle fiber types in further research. In conclusion, this study provides a comprehensive analysis of the EDL and Sol transcriptome in large white pigs, and further functional analysis of these potential may help us elucidate the mechanism of muscle fiber type determination.
Abbreviations
- EDL:
-
Extensor digitorum longus
- Sol:
-
Soleus
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- SNPs:
-
Single nucleotide polymorphisms
- CPC:
-
Coding potential calculator
- FDR:
-
False discovery rate
- AS:
-
Alternative splicing
References
Anakwe K, Robson L, Hadley J, Buxton P, Church V, Allen S, Hartmann C, Harfe B, Nohno T, Brown AMC, Evans DJR, Francis-West P (2003) Wnt signalling regulates myogenic differentiation in the developing avian wing. Development 130:3503–3514
Arber S, Halder G, Caroni P (1994) Muscle lim protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell 79:221–231
Bai Q, McGillivray C, da Costa N, Dornan S, Evans G, Stear MJ, Chang KC (2003) Development of a porcine skeletal muscle cDNA microarray: analysis of differential transcript expression in phenotypically distinct muscles. BMC Genom 4:8
Chin ER, Olson EN, Richardson JA, Yano Q, Humphries C, Shelton JM, Wu H, Zhu WG, Bassel-Duby R, Williams RS (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Gene Dev 12:2499–2509
Choi YM, Lee SH, Choe JH, Rhee MS, Lee SK, Joo ST, Kim BC (2010) Protein solubility is related to myosin isoforms, muscle fiber types, meat quality traits, and postmortem protein changes in porcine longissimus dorsi muscle. Livest Sci 127:183–191
Choi YM, Nam KW, Choe JH, Ryu YC, Wick MP, Lee K, Kim BC (2013) Growth, carcass, fiber type, and meat quality characteristics in large white pigs with different live weights. Livest Sci 155:123–129
Garcia de la Serrana D, Estevez A, Andree K, Johnston IA (2012) Fast skeletal muscle transcriptome of the gilthead sea bream (Sparus aurata) determined by next generation sequencing. BMC Genom 13:181
Geiger B, Bershadsky A (2002) Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell 110:139–142
Huang S, Zhang J, Li R, Zhang W, He Z, Lam TW, Peng Z, Yiu SM (2011) SOAPsplice: genome-wide ab initio detection of splice junctions from RNA-seq data. Front Genet 2:46
Lee EA, Kim JM, Lim KS, Ryu YC, Jeon WM, Hong KC (2012) Effects of variation in porcine MYOD1 gene on muscle fiber characteristics, lean meat production, and meat quality traits. Meat Sci 92:36–43
Lefaucheur L, Milan D, Ecolan P, Le Callennec C (2004) Myosin heavy chain composition of different skeletal muscles in large white and Meishan pigs. J Anim Sci 82:1931–1941
Leitinger B (2011) Transmembrane collagen receptors. Annu Rev Cell Dev Biol 27:265–290
Lepetit J (2007) A theoretical approach of the relationships between collagen content, collagen cross-links and meat tenderness. Meat Sci 76:147–159
Li RQ, Li YR, Fang XD, Yang HM, Wang J, Kristiansen K, Wang J (2009a) SNP detection for massively parallel whole-genome resequencing. Genome Res 19:1124–1132
Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, Wang J (2009b) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li Y, Xu ZY, Li HY, Xiong YZ, Zuo B (2010) Differential transcriptional analysis between red and white skeletal muscle of Chinese Meishan pigs. Int J Biol Sci 6:350–360
Lim KS, Lee SH, Lee EA, Kim JM, Hong KC (2015) Effects of intergenic single nucleotide polymorphisms in the fast myosin heavy chain cluster on muscle fiber characteristics and meat quality in Berkshire pigs. Meat Sci 110:224–229
MacArthur DG, Seto JT, Raftery JM, Quinlan KG, Huttley GA, Hook JW, Lemckert FA, Kee AJ, Edwards MR, Berman Y, Hardeman EC, Gunning PW, Easteal S, Yang N, North KN (2007) Loss of ACTN3 gene function alters mouse muscle metabolism and shows evidence of positive selection in humans. Nat Genet 39:1261–1265
Maltin C, Balcerzak D, Tilley R, Delday M (2003) Determinants of meat quality: tenderness. P Nutr Soc 62:337–347
Mills MA, Yang N, Weinberger RP, Vander Woude DL, Beggs AH, Easteal S, North KN (2001) Differential expression of the actin-binding proteins, alpha-actinin-2 and-3, in different species: implications for the evolution of functional redundancy. Hum Mol Genet 10:1335–1346
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN (2000) Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem 275:4545–4548
Palstra AP, Beltran S, Burgerhout E, Brittijn SA, Magnoni LJ, Henkel CV, Jansen HJ, van den Thillart GE, Spaink HP, Planas JV (2013) Deep RNA sequencing of the skeletal muscle transcriptome in swimming fish. PLoS One 8:e53171
Rehfeldt C, Henning M, Fiedler I (2008) Consequences of pig domestication for skeletal muscle growth and cellularity. Livest Sci 116:30–41
Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD (2001) Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol 153:1175–1185
Roberts A, Pimentel H, Trapnell C, Pachter L (2011) Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 27:2325–2329
Ruusunen M, Puolanne E (2004) Histochemical properties of fibre types in muscles of wild and domestic pigs and the effect of growth rate on muscle fibre properties. Meat Sci 67:533–539
Ruusunen M, Puolanne E, Sevon-Aimonen ML, Partanen K, Voutila L, Niemi J (2012) Carcass and meat quality traits of four different pig crosses. Meat Sci 90:543–547
Schneider AG, Sultan KR, Pette D (1999) Muscle LIM protein: expressed in slow muscle and induced in fast muscle by enhanced contractile activity. Am J Physiol 276:C900–C906
Sebastian S, Faralli H, Yao ZZ, Rakopoulos P, Palii C, Cao Y, Singh K, Liu QC, Chu A, Aziz A, Brand M, Tapscott SJ, Dilworth FJ (2013) Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Gene Dev 27:1247–1259
Sethi JK, Vidal-Puig A (2010) Wnt signalling and the control of cellular metabolism. Biochem J 427:1–17
Takata H, Terada K, Oka H, Sunada Y, Moriguchi T, Nohno T (2007) Involvement of Wnt4 signaling during myogenic proliferation and differentiation of skeletal muscle. Dev Dyn 236:2800–2807
Tiso N, Majetti N, Stanchi F, Rampazzo A, Zimbello R, Nava A, Danieli GA (1999) Fine mapping and genomic structure of ACTN2, the human gene coding for the sarcomeric isoform of alpha-actinin-2, expressed in skeletal and cardiac muscle. Biochem Bioph Res Commun 265:256–259
von Maltzahn J, Chang NC, Bentzinger CF, Rudnicki MA (2012) Wnt signaling in myogenesis. Trends Cell Biol 22:602–609
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang LK, Feng ZX, Wang X, Wang XW, Zhang XG (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wei B, Jin JP (2011) Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch Biochem Biophys 505:144–154
Weiskirchen R, Gunther K (2003) The CRP/MLP/TLP family of LIM domain proteins: acting by connecting. BioEssays 25:152–162
Wu H, Naya FJ, McKinsey TA, Mercer B, Shelton JM, Chin ER, Simard AR, Michel RN, Bassel-Duby R, Olson EN, Williams RS (2000) MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J 19:1963–1973
Xu XW, Qiu HF, Du ZQ, Fan B, Rothschild MF, Yuan F, Liu B (2010) Porcine CSRP3: polymorphism and association analyses with meat quality traits and comparative analyses with CSRP1 and CSRP2. Mol Biol Rep 37:451–459
Acknowledgments
We thank the Beijing Genomics Institute (BGI)-Shenzhen for helping us with technical assistance in DGE and Edanz for providing language editing service.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors declare any conflict of interest.
Ethical approval
The large white pigs were supplied by the Northwest A&F University Experiment Station (Yangling, China). Experiments were handled in accordance with the guidelines of the Administration of Affairs Concerning Experimental Animals and authorized by the Institutional Animal Care and Use Committee of the College of Animal Science and Technology, Northwest A&F University, Shaanxi, China. All applicable international, national, and institutional guidelines for the care and use of animals were followed.
Funding
This study was supported by the National Basic Research Program of China (Grant No. 2015CB943102), the National Natural Science Foundation of China (Grant No. 31272410), and the Scientific and Technological Innovation Project for Youths of Northwest A&F University (Grant No. ZD2012007). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional information
Communicated by S. Hohmann.
Jiayu Zhu and Xin’e Shi contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhu, J., Shi, X., Lu, H. et al. RNA-seq transcriptome analysis of extensor digitorum longus and soleus muscles in large white pigs. Mol Genet Genomics 291, 687–701 (2016). https://doi.org/10.1007/s00438-015-1138-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1138-z