
Abstract
Sweet potato [Ipomoea batatas (L.) Lam.], the world’s seventh most important food crop, is also a major industrial raw material for starch and ethanol production. In the plant starch biosynthesis pathway, ADP-glucose pyrophosphorylase (AGPase) catalyzes the first, rate-limiting step and plays a pivotal role in regulating this process. In spite of the importance of sweet potato as a starch source, only a few studies have focused on the molecular aspects of starch biosynthesis in sweet potato and almost no intensive research has been carried out on the AGPase gene family in this species. In this study, cDNAs encoding two small subunits (SSs) and four large subunits (LSs) of AGPase isoforms were cloned from sweet potato and the genomic organizations of the corresponding AGPase genes were elucidated. Expression pattern analysis revealed that the two SSs were constitutively expressed, whereas the four LSs displayed differential expression patterns in various tissues and at different developmental stages. Co-expression of SSs with different LSs in Escherichia coli yielded eight heterotetramers showing different catalytic activities. Interactions between different SSs and LSs were confirmed by a yeast two-hybrid experiment. Our findings provide comprehensive information about AGPase gene sequences, structures, expression profiles, and subunit interactions in sweet potato. The results can serve as a foundation for elucidation of molecular mechanisms of starch synthesis in tuberous roots, and should contribute to future regulation of starch biosynthesis to improve sweet potato starch yield.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sweet potato [Ipomoea batatas (L.) Lam.] is the world’s seventh most important food crop (Firon et al. 2013). In addition, the species is widely used as an industrial raw material for starch and ethanol production. Starch, the major storage polysaccharide in plants, is accumulated as granules in different organs, such as leaves, roots, shoots, fruits, and grains, for use as a carbon and energy source for plant growth. Starch biosynthesis in plants involves three main enzymes: ADP-glucose pyrophosphorylase (AGPase; EC 2.7.7.27), starch synthase, and starch branching enzyme. AGPase catalyzes the first, rate-limiting step in the starch biosynthesis pathway, namely, the conversion of glucose 1-phosphate and ATP to ADP-glucose and pyrophosphate (Ballicora et al. 2004). In view of this vital role in starch synthesis, AGPases of main crops such as rice (Dawar et al. 2013), wheat (Danishuddin et al. 2011), and potato (Barıs et al. 2009) and the model plant Arabidopsis (Bahaji et al. 2011) have been extensively studied. As a representative root crop, sweet potato is highly suitable for use in studies of starch biosynthesis in tuberous roots, but only a few investigations of sweet potato have focused on this aspect (Kwak et al. 2007; Kim et al. 2009; Ahn et al. 2010). A better understanding of AGPase and its expression patterns in sweet potato will provide guidance for regulating starch biosynthesis in tuberous roots.
In higher plants, AGPase is a heterotetramer (L2S2) consisting of two identical large subunits (LSs) and two identical small subunits (SSs) (Ballicora et al. 2004). Different approaches have been used in attempts to reveal the role of the two AGPase subunit types in higher plants. Numerous studies using various genetic, mutagenic, and biochemical strategies have strongly suggested that the SS has both catalytic and regulatory activities, and that the LS is mainly responsible for modulating the allosteric regulatory properties of the SS in the heterotetrameric enzyme (Ballicora et al. 1995; Salamone et al. 2000; Frueauf et al. 2003; Hwang et al. 2006). Although the LS is usually non-catalytic, recent studies have demonstrated that recombinant LSs (mutated or natural) show defective catalytic activity, both alone and in the presence of the SS in vitro (Ballicora et al. 2005; Ventriglia et al. 2008; Hwang et al. 2008; Petreikov et al. 2010). However, the binding of LS and SS to form a heterotetramer is essential for the enzyme to function efficiently in plants. Because the crystal structure of a nonnative, SS homotetramer derived from the potato tuber AGPase has been described (Jin et al. 2005), LS–SS interactions have been extensively studied by computational and experimental techniques, and specific regions from both the LS and the SS have been identified as important for subunit association and enzyme stability (Baris et al. 2009; Dawar et al. 2013).
The AGPase gene family has been characterized in several monocot and dicot plant species. One or two SS and several LS genes are typically present in plants, which allows for multiple forms of the active heterotetramer depending on the various combinations of LSs and SSs. An amino acid sequence comparison of AGPases from different plant species has revealed 85–95 % identity among all SSs and 50–60 % identity among LSs. Although LS and SS amino acid sequences share relatively fewer residues, significant similarity still exists between them; for example, potato tuber AGPase SS and LS sequences are 53 % identical (Ballicora et al. 2004). In light of the high degree of sequence similarity, higher plant AGPase SS- and LS-encoding genes are thought to have evolved from a common ancestor through gene duplication followed by divergence. It has been proposed that SSs in angiosperms have been subjected to a stricter evolutionary constraint than have LSs, as the former are less tissue-specific and must interact with different LSs (Georgelis et al. 2007).
Expression patterns of genes for AGPase subunits have been extensively studied. SSs are usually encoded by only one or two genes. SS gene expression is thus generally non-specific, although tissue-specific SS isoforms have been reported in bean and maize (Ballicora et al. 2004). On the contrary, the expressions of LS genes are usually tissue- and development- specific. The expression patterns and regulatory properties of different LS isoforms have been comprehensively studied in tomato (Park and Chung 1998), Arabidopsis (Crevillén et al. 2003, 2005), and rice (Lee et al. 2007), and the results of those studies strongly indicate that AGPase heterotetramers formed in different tissues or organs display distinct kinetic and regulatory properties to control starch biosynthesis. It is suggested that the LSs of agriculturally important crops such as potato and tomato, which have undergone significant selection under both domestication and natural selection, may have evolved novel modulatory characteristics (Petreikov et al. 2010). We speculate that sweet potato, which has undergone significant evolutionary development for starch accumulation in its tuberous root, may similarly possess a species-specific regulation mode rendered by different LSs to control source/sink tissue starch synthesis.
Korean researchers have previously cloned two cDNAs encoding sweet potato SSs and their corresponding genomic DNAs (Bae and Liu 1997; Noh et al. 2004) as well as the sweet potato LS cDNA sequence iAGPLI-1 (Harn et al. 2000). Analysis of the tissue-specific expression and subcellular location of the three subunits revealed that the two SS isoforms are non-organ-specific with high expression in storage roots (Harn et al. 2000; Kwak et al. 2007, 2008). The other three LSs have not yet been studied. No comprehensive investigation has been performed in sweet potato concerning the possible role played by the different AGPase isoforms.
In this study, we cloned six cDNAs and genomic DNAs encoding sweet potato AGPase isoforms and analyzed their gene expression patterns. Co-expression profile analysis and yeast two-hybrid experiments were conducted to elucidate the interactions between different SSs and LSs. We thereby generated comprehensive information on sequences, structures, expression profiles, and subunit interactions of AGPase genes in sweet potato. Our results lay a foundation for the elucidation of the key regulatory mechanism of starch synthesis in tuberous roots and provide guidance that can be used to regulate starch biosynthesis for increased starch content in sweet potato.
Materials and methods
Plant material
In this study, we used sweet potato cultivar Xushu 18, which is the leading cultivar in the Yangtze River Basin of China in terms of annual hectarage and total production.
Cloning of cDNA and genomic DNA sequences of AGPase subunits
A sweet potato transcriptome database was established from the cultivar Xushu 18 in our previous study (Tao et al. 2012). Of 51,763 annotated transcripts (≥100 bp), 6 were annotated as AGPase SSs and LSs with complete open-reading frames. The predicted sequences showed high identity to AGPase subunit sequences in GenBank. On the basis of the assembled transcript sequences, primers were designed from initiation codon to stop codon using Primer Premier 5.0 (Premier Biosoft International, Palo Alto, CA, USA) (Supplementary Table 1). After isolation with Trizol reagent (Invitrogen, Ambion, Carlsbad CA, USA), total RNA of sweet potato was treated with DNase I (Fermentas, Shenzhen, China). First-strand cDNA was synthesized from 2 μg of total RNA using Oligo (dT) primer. The cDNAs of AGPase SSs and LSs were PCR-amplified using KOD-Plus-Neo DNA polymerase (Toyobo, Tokyo, Japan). Genomic DNA was isolated from sweet potato leaf tissues using a modified CTAB method (He et al. 2011). PCR amplification of full-length genomic DNA of AGPase SSs and LSs was carried out using LA Taq DNA polymerase (Takara, Dalian, China). The resulting PCR products were cloned into a pEASY-T1 vector (Transgen, Beijing, China) and subjected to Sanger sequencing.
Sequence analysis
Homology analysis was performed using DNAMAN (Lynnon Biosoft) and a phylogenetic tree was constructed with MEGA 4.0 (www.megasoftware.net) (Tamura et al. 2007). Multiple sequence alignment of AGPase subunit sequences was carried out using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (Larkin et al. 2007). Gene structure organization was analyzed with the Gene Structure Display Server tool (http://gsds.cbi.pku.edu.cn/) (Guo et al. 2007). The subcellular location of isoforms was predicted by TargetP (http://www.cbs.dtu.dk/services/TargetP/) (Nielsen et al. 1997). Miniature inverted-repeat transposable elements (MITEs) were predicted and analyzed with the MITE Uncovering SysTem (MUST) tool (http://csbl1.bmb.uga.edu/ffzhou/MUST/) (Chen et al. 2009).
Digital gene expression (DGE) analysis of AGPase LSs and SSs from sweet potato
Illumina DGE tag profiling of different tissues from sweet potato, including young leaves, mature leaves, stems, fibrous roots, and tuberous roots at different developmental stages, was carried out in our previous studies (Tao et al. 2012). Since the number of DGE tags mapping to LSs and SSs may reflect LS and SS expression levels in different tissues and developmental stages, we mapped DGE tags to AGPase LSs and SSs using Bowtie 2.0. Using the Tag Per Million (TPM) algorithm (Robinson et al. 2010) to normalize the number of tags, we obtained expression levels of two SSs and four LSs of AGPase in six different tissues (Table 2).
Quantitative real-time PCR (qRT-PCR)
Using the cDNA and genomic DNA sequences, qRT-PCR primers were designed with Primer Premier 5.0 (Supplementary Table 2). PCR amplifications were performed using SYBR Premix Ex Taq II (Tli RNaseH Plus) (Takara).
Construction of expression plasmids in Escherichia coli
The E. coli-compatible expression vectors pRSFDuet-1 and pACYCDuet-1 (Novagen) were used to express mature forms of sweet potato AGPase LSs and SSs, respectively. cDNA fragments encoding SS and LS putative mature peptides were PCR-amplified from sequenced recombinant plasmids in the presence of the primers listed in Supplementary Table 1. The PCR amplifications were performed using KOD-Plus-Neo DNA polymerase (Toyobo). Amplified LSs IbAGPL1, IbAGPL2, and IbAGPL4 were digested with NcoI/EcoRI, while IbAGPL3 was digested with PscI/EcoRI; the digested fragments were cloned into pRSFDuet-1 expression vectors digested with NcoI/EcoRI to generate plasmids pRSF-IbAGPL1, pRSF-IbAGPL2, pRSF-IbAGPL3, and pRSF-IbAGPL4. cDNA fragments for SSs IbAGPS1 and IbAGPS2 were digested with NcoI/BamHI and cloned into pACYCDuet-1 expression vectors digested with NcoI/BamHI to generate plasmids pACYC-IbAGPS1 and pACYC-IbAGPS2.
Co-expression of AGPase LSs and SSs in E. coli
For co-expression studies, LS-harboring pRSFDuet-1 derivatives were co-transformed with SS-harboring pACYCDuet-1 derivatives into E. coli BL21(DE3) cells. Bacterial colonies harboring one or two expression plasmids were selected with corresponding antibiotics. Escherichia coli strains containing expression plasmids were grown overnight in Luria–Bertani medium at 37 °C and 250 rpm and then transferred to enriched Kornberg medium (1.1 % K2HPO4, 0.85 % KH2PO4, 0.6 % yeast extract, and 1 % glucose) (Baris et al. 2009) with suitable antibiotics until reaching an OD600 of 0.8–1.0. Inductions were carried out for 24 h with 1 mM isopropyl β-d-thiogalactoside at 18 °C. The induced cells were harvested for a glycogen accumulation assay and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The cells were washed and resuspended in distilled water, and the OD600 values were adjusted to be comparable among samples. After addition of 200 μL iodine solution (0.01 M I2 and 0.03 M KI) to 500 µL of cells, the solution was mixed and maintained at room temperature for 2 min. Staining differences were noted as follows: cells accumulating glycogen were brownish, whereas non-accumulating cells were faint yellow. Iodine staining for glycogen was repeated three times. To determine whether subunits were expressed, SDS-PAGE was performed on identical amounts of total protein from each induced cell sample.
Yeast two-hybrid analysis
The NcoI/EcoRI fragments containing cDNAs coding for the putative mature peptides of IbAGPL1 and IbAGPL2, the full length cDNA of IbAGPL4 and the PscI/EcoRI fragment coding for the putative mature peptide of IbAGPL3 were cloned into pGADT7-Rec yeast expression vectors (Clontech) digested with NcoI/EcoRI to give plasmids pGADT7-IbAGPL1, -IbAGPL2, -IbAGPL3, and -IbAGPL4. The NcoI/BamHI fragments containing cDNAs coding for the putative mature peptides of IbAGPS1 and IbAGPS2 were cloned into pGBKT7 plasmids (Clontech) digested with NcoI/BamHI to give plasmids pGBKT7-IbAGPS1 and -IbAGPS2. pGADT7-Rec- and pGBKT7-derived plasmids were co-transformed into Saccharomyces cerevisiae strain AH109 and subsequently used for a two-hybrid analysis according to the manufacturer’s instructions (Clontech). pGADT7-T and pGBKT7-53 (provided by Clontech) were co-transformed into S. cerevisiae strain AH109 as a positive control. The following vector pairs were co-transformed as negative controls: blank pGADT7-Rec and blank pGBKT7, blank pGADT7-Rec and pGBKT7-SS, and pGADT7-LS and blank pGBKT7. AH109 yeast cells expressing the designated plasmids were selected on a synthetic growth medium lacking Leu and Trp. The selection of subunit interactions was carried out on SD/-Leu -Trp -His -Ade medium and verified on SD/-Leu -Trp -His -Ade +X-α-gal medium.
Results
Cloning and analysis of sweet potato AGPase subunit cDNAs
Analysis of annotated transcripts in the sweet potato transcriptome generated and characterized in our previous studies (Tao et al. 2012) revealed the presence of AGPase gene family members, including two genes for SSs and four for LSs. Primers specific for the sequences encoding each large and small AGPase subunit were accordingly synthesized and used for full-length cDNA cloning and sequencing.
The cDNAs encoding the six AGPase SS and LS isoforms ranged in length from 1548 to 1578 bp, corresponding to 515–525 amino acids. The cDNAs for the four LSs were designated as IbAGPL1, IbAGPL2, IbAGPL3, and IbAGPL4, with the cDNAs for the two SSs named as IbAGPS1 and IbAGPS2. The general characteristics of these isoforms are shown in Table 1. The deduced amino acid sequences of the two SSs were highly similar, with the four LSs sharing comparatively lower identities (Fig. 1a). Residues important for enzyme activities, such as those at activator binding, substrate binding, and catalytic sites (Ballicora et al. 2004), were highly conserved in both SSs and LSs (Supplementary Fig. 1). A phylogenetic tree of deduced amino acid sequences of the four sweet potato AGPase LSs and LSs from some other plant species was constructed in MEGA 4.0. As shown in Fig. 1b, all the analyzed AGPase LSs could be classified into four groups, with the four LSs of sweet potato distributed into three of them (see Discussion for more details). The four LSs from sweet potato were most closely related to AGPase subunits from tomato and potato.

Homology analysis of sweet potato AGPase large and small subunits (a) and phylogenetic analysis of AGPase large subunits from sweet potato and other plants (b). Protein sequence alignment and calculation of phylogenetic distance were performed by ClustalW. The phylogenetic tree was generated by MEGA 4.0. Sequences data can be found in GenBank under accession numbers: IbAGPL1 (JQ797692.1), IbAGPL2 (JQ797693.1), IbAGPL3 (JQ797694.1), IbAGPL4 (JQ797695.1), IbAGPS1 (JQ797696.1), IbAGPS2 (JQ797697.1), A. thaliana ApL1 (NP_197423.1), A. thaliana ApL2 (AAM14190), A. thaliana ApL3 (CAA77173), A. thaliana ApL4 (AAM20291), S. lycopersicum AgpL1 (AAC49941), S. lycopersicum AgpL2 (AAC49942), S. lycopersicum AgpL3 (AAC49943), S. tuberosum agpS1 (CAA43490), S. tuberosum agpS2 (CAA52917), S. tuberosum agpS3 (CAA53741), O. sativa AGPL1 (NP_001056424.2), O. sativa AGPL2 (NP_001043654.1), O. sativa AGPL3 (AAT78793), and O. sativa AGPL4 (AK121036)
All dicotyledonous plant AGPases studied thus far are plastidic proteins, whereas cereal endosperm AGPase is both cytoplasmic and plastidic (Beckles et al. 2001; James et al. 2003). The subcellular location and transit peptide sequences of the six sweet potato AGPase isoforms were mainly predicted using TargetP (http://www.cbs.dtu.dk/services/TargetP/). This analysis demonstrated that a typical chloroplast transit peptide of 73 amino acids was presented in the IbAGPS2 isoform, with no typical subcellular locations identified in the other isoforms. Because N-terminal amino acid sequences of mature forms of AGPase subunits have been identified for spinach (Morell et al. 1987), potato (Ballicora et al. 1995), and tomato (Petreikov et al. 2010) by protein sequencing, we then compared amino acid sequences around the transit peptide cleavage site. By taking into consideration the high similarity of sweet potato AGPase subunits to subunits from potato (for SS) and tomato (for LS), we were able to deduce the transit peptide cleavage sites of IbAGPS1, IbAGPS2, IbAGPL1, IbAGPL2, and IbAGPL3 subunits (Fig. 2). In contrast, the cleavage site of IbAGPL4 could not be predicted because it had no significant homology with known sequences. The predicted lengths of the transit peptides are shown in Table 1.

The prediction of transit peptide cleavage sites of AGPase large and small subunits from sweet potato. The N-terminus of mature subunits from spinach, potato, and tomato identified by protein sequencing are indicated by asterisks (*). Underlined letters indicate the predicted N-terminus of the mature peptides of sweet potato AGPase large and small subunits. Letters in the shaded box indicate the N-terminus of the expressed protein in co-expression experiment and yeast two-hybrid experiment
Genomic organization of AGPase subunit genes from sweet potato
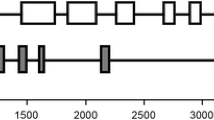
Genomic DNAs of the six AGPase subunits from sweet potato were amplified with the same primers designed for cDNA cloning (Supplementary Table 1). The resulting PCR products were cloned and sequenced by the Sanger method. The genomic DNAs of IbAGPS1 and IbAGPS2 had lengths of 3707 and 3747 bp, respectively. The two SS genes were 60.17 % identical and their cDNAs were 90.84 % identical, indicating that their intron sequences were more variable. The two genes shared a similar structure, with nine exons and eight introns located within the transcribed regions. Except for exon 1, the length of each corresponding exon was identical between IbAGPS1 and IbAGPS2. The locations of corresponding introns, but not their lengths, were identical between the two genes (Fig. 3).

Genomic organizations of large and small subunits of AGPase genes from sweet potato. Gene structure analysis was performed by GSDS (Gene Structure Display Server, http://gsds.cbi.pku.edu.cn/). The black boxes represent the exon region and the white boxes represent the intron region. The numbers on the top of the exons indicate the length of the exon. Sequences data can be found in GenBank under accession numbers: IbAGPL1 (JQ797698.1), IbAGPL2 (JQ797699.1), IbAGPL3 (JQ797700.1), IbAGPL4 (JQ797701.1), IbAGPS1 (KJ365312), and IbAGPS2 (KJ365313)
The lengths of the four LS genes varied from 3061 to 3595 bp. As shown in Fig. 3, IbAGPL1, IbAGPL2, and IbAGPL3 had similar gene structures consisting of 14 exons and 13 introns, while IbAGPL4 had 13 exons and 12 introns. The lengths of exons 4–14 were identical among IbAGPL1, IbAGPL2, and IbAGPL3, whereas a few differences were found with respect to exons 1, 2, and 3. We further compared the exon–intron constitutions of individual genes. As shown in Fig. 3, the combined length of exons 3 and 4 in IbAGPL3 was identical to that of exon 3 in IbAGPL4. Similarly, the combined length of exons 6 and 7 in IbAGPL3 was identical to that of exon 5 in IbAGPL4, and the length of exon 8 in IbAGPL3 was identical to the total length of exons 6 and 7 in IbAGPL4. These results suggest that intron loss and insertion have occurred during structural evolution of the IbAGPL4 gene.
In addition to intron numbers, marked changes in intron size were also noted between some corresponding introns. Previous studies have indicated that intron size expansion may primarily be due to the actions of MITEs, a class of non-autonomous DNA transposons (Hannah et al. 2001). The MUST system was applied to predict MITEs. The generated data suggested that MITEs have invaded four LS introns, namely, intron 6 of IbAGPL2, intron 2 of IbAGPL3, and introns 5 and 12 of IbAGPL4, and intron 1 of both SS genes (Supplementary Fig. 1). To identify these MITE sequences, we compared the six relevant intron sequences against TIGR plant repeat (www.tigr.org/tdb/e2k1/plant.repeats/index.shtml) and Repbase (http://www.girinst.org/censor/index.php) databases. Only one MITE sequence—in intron 5 of IbAGPL4—was confirmed; it was identified as an Ipomoea batatas microsatellite, gb|GU171584.1|.
Expression of AGPase subunits in sweet potato
In previous studies, we carried out Illumina DGE tag profiling of different sweet potato tissues, including young leaves, mature leaves, stems, fibrous roots, and tuberous roots at two developmental stages (initial and harvestable) (Tao et al. 2012). In the present study, we analyzed the expression patterns of six AGPase subunits that were previously uncovered by the DGE tag profiling (Table 2). These analyses showed that tuberous roots generally exhibited the highest AGPase expression levels of all studied tissues, with the harvestable root stage found to be the developmental phase with the highest levels among tuberous roots. AGPase subunit expression was low in young leaves and stems. SSs IbAGPS1 and IbAGPS2 were expressed in all tissues in this study, with IbAGPS1 always showing higher transcriptional levels than IbAGPS2. However, the ratio of IbAGPS1/IbAGPS2 varied among tissues: it was 1.5 in young leaves but approximately 8 in harvestable roots. IbAGPS1 had the highest expression in tuberous roots and the lowest in stems. The four LS isoforms exhibited very different transcriptional patterns. IbAGPL1 was the most abundant LS in roots and stems, with TPM levels more than 200, 49, and 35 times higher than those of IbAGPL2 in harvestable tuberous roots, fibrous roots, and stems, respectively. In mature leaves, the highest transcription was that of IbAGPL1, followed by IbAGPL4 and IbAGPL3. In young leaves, IbAGPL4 was the predominantly expressed LS, with transcription levels up to fivefold higher than the next highly expressed LS; however, expression levels of all AGPase genes were very low in this tissue compared with tuberous roots. The expression of IbAGPL4 was restricted in leaves. These data suggest a tissue specificity that may be related to the role of each LS isoform in the regulation of the activity of the heterotetrameric AGPase enzyme.
To validate the expression patterns obtained from the DGE analysis, we further analyzed the expression of these genes by qRT-PCR. Expression patterns uncovered by the qRT-PCR analysis were in accord with the DGE results (Supplementary Table 3), confirming that the differential expression analysis based on high-throughput RNA sequencing generated reliable expression data in this study.
Co-expression of sweet potato AGPase LS and SS cDNAs in E. coli
In a previous study, co-expression of cDNA sequences of potato tuber LSs and SSs in E. coli yielded a functional heterotetrameric enzyme that could be detected by iodine staining for glycogen, the product of AGPase. Such heterologous expression provides a powerful genetic approach to investigate the function of heterotetrameric enzymes formed by various SS and LS combinations.
Following the addition of an initiator methionine, the putative mature LS coding regions were cloned into the expression vector pRSFDuet-1, while those of SSs were inserted into pACYCDuet-1. Expression of these subunits in E. coli, individually and in various combinations, was analyzed by iodine staining. As shown in Fig. 4, the co-expression of mature SSs IbAGPS1 or IbAGPS2 with mature LSs IbAGPL1, IbAGPL2, or IbAGPL3 resulted in the formation of functional enzyme, as evidenced by glycogen accumulation, with cells displaying a brownish color when exposed to iodine staining. Engineered E. coli that expressed only individual subunits, whether small or large, showed no visible activity following iodide staining.

Iodine staining of different combinations of sweet potato AGPase large and small subunits expressed in E. coli. CK: control check, pRSF & pACYC; S1: IbAGPS1; S2: IbAGPS2; L1: IbAGPL1; L2: IbAGPL2; L3: IbAGPL3; L4: IbAGPL4; L1S1: IbAGPL1 & IbAGPS1; L2S1: IbAGPL2 & IbAGPS1; L3S1: IbAGPL3 & IbAGPS1; L4S1: IbAGPL4 & IbAGPS1; L1S2: IbAGPL1 & IbAGPS2; L2S2: IbAGPL2 & IbAGPS2; L3S2: IbAGPL3 & IbAGPS2; L4S2: IbAGPL4 & IbAGPS2. E. coli strains containing different expression plasmids were grown overnight in LB medium at 37 °C, 250 rpm, then transferred to Kornberg medium (1.1 % K2HPO4, 0.85 % KH2PO4, 0.6 % yeast extract and 1 % glucose) with suitable antibiotics until the OD600 reached 0.8–1.0. The induced expression was carried out with 1 mM isopropyl β-d-thiogalactoside (IPTG) at 18 °C for 24 h. The harvested cells were stained with iodine solution (0.01 M I2, 0.03 M KI) to analyze glycogen accumulation
Comparison of the color of stained cells harboring different heterotetramers revealed that co-expression of the same SS (IbAGPS1 or IbAGPS2) with different LSs (IbAGPL1, IbAGPL2, or IbAGPL3) resulted in different enzyme activities. This outcome suggests that SS catalytic activity is regulated by the LS, with the IbAGPL1 isoform possessing the largest regulative activity. When comparing the co-expression of the same LS with different SSs, such as L1S1 and L1S2, we additionally found that IbAGPS2 demonstrated higher catalytic activity than IbAGPS1. No enzyme activity could be detected with heterotetramers L4S1 and L4S2. The SDS-PAGE of the co-expression cells showed that all combinations (including L4S1 and L4S2) produced roughly the same amounts of target protein (Supplementary Fig. 3).
Analysis of interactions between sweet potato AGPase LSs and SSs
No enzyme activity was detected from heterotetramers containing the IbAGPL4 subunit in E. coli. This lack of activity was possibly due to the absence of essential amino acids involved in important regulatory functions and/or subunit association. To investigate the effect of IbAGPL4 on heterotetrameric assembly, we carried out a yeast two-hybrid assay.
Different pGADT7-LS plasmids were co-transformed along with various pGBKT7-SS plasmids into S. cerevisiae strain AH109. Interactions of SSs and LSs were detected on a synthetic growth medium lacking Leu, Trp, His, and Ade and were confirmed on an X-α-gal plate. As shown in Fig. 5, yeast cells carrying L4S1 or L4S2, unlike the other six SS–LS combinations, were unable to grow on the selective-interaction media in the yeast two-hybrid experiment. These results indicate that the lack of AGPase activity in heterotetramers containing the IbAGPL4 subunit may be due to the absence of interaction between IbAGPL4 and SSs.

Yeast two-hybrid analysis of sweet potato AGPase subunit interactions. CK1: positive control, pGADT7-T/pGBKT7-53; CK2: negative control, pGADT7/pGBKT7; CK3: negative control, pGADT7/pGBKT7-SS; CK4: negative control, pGADT7-LS/pGBKT7. L1S1: IbAGPL1 & IbAGPS1; L2S1: IbAGPL2 & IbAGPS1; L3S1: IbAGPL3 & IbAGPS1; L4S1: IbAGPL4 & IbAGPS1; L1S2: IbAGPL1 & IbAGPS2; L2S2: IbAGPL2 & IbAGPS2; L3S2: IbAGPL3 & IbAGPS2; L4S2: IbAGPL4 & IbAGPS2. The selection of subunit interactions was carried out on a SD/-Leu -Trp -His -Ade medium and confirmed on a SD/-Leu -Trp -His -Ade + X-α-gal medium
Discussion
Sweet potato is the world’s seventh most important food crop and the third most important root/tuber crop after potato and cassava (Firon et al. 2013). Sweet potato has recently been considered as a promising substrate for fuel ethanol production because it has a higher starch yield per unit of cultivated land than grains and displays strong adaptability (Zhang et al. 2013). Much research on sweet potato has thus focused on enhancing starch yield in the tuberous roots. Nevertheless, limited progress has been made with respect to the key enzymes involved in starch synthesis in sweet potato. AGPase, the first and rate-limiting enzyme in this pathway, is especially poorly characterized in sweet potato, with little known about AGPase gene properties such as sequence, structure, expression patterns, and subunit interactions.
As revealed by this study, the two genes encoding AGPase SSs in sweet potato are highly conserved, displaying an overall identity of 90.84 % and regional identities of 59.46 and 96 % for transit peptides and mature peptides, respectively. The expressions of the two SS genes were found to be non-specific, although expression levels varied among tissues depending on their differing starch synthesis requirements. Both IbAGPS1 and IbAGPS2 showed their highest expression in tuberous roots. IbAGPS1 was generally expressed at higher levels than IbAGPS2 in all tissues, especially in harvestable tuberous roots. In light of the higher catalytic activity of IbAGPS2 compared with IbAGPS1 as revealed by the co-expression experiment in E. coli, we speculate that both SSs are functional and important in all tissues. Not surprisingly, SSs showed the highest expression levels in tuberous roots—the main sink tissues, followed by mature leaves—the main source tissues.
The four genes encoding AGPase LSs that were characterized and cloned in sweet potato were found to share an amino acid sequence identity of 69.60 %. LS expressions differed significantly among sweet potato tissues. IbAGPL1 absolutely predominated in roots, especially in tuberous ones. However, the expression of IbAGPL2, which had the highest identity with IbAGPL1, was extremely low compared with that of IbAGPL1. Expression levels of the four AGPase LS genes were very low in leaves compared with those in roots. These observations confirm the idea that different AGPases in a given tissue have distinct regulatory properties depending on the specific LSs present (Ballicora et al. 2004). Plant AGPase LS genes could be categorized into five major groups according to their sequences and expression patterns (Georgelis et al. 2007). Group 1 comprised genes expressed in monocot and eudicot leaf tissue. Group 2 included genes expressed in eudicot source and sink tissues. Groups 3 and 4 contained genes expressed in monocot seed and eudicot sink tissues, respectively. Group 5 was composed of only two sequences whose tissue specificity has not been studied in detail. We compared the expression patterns of four sweet potato LSs with LSs of other plant species belonging to the same group and whose expression patterns have been well studied. The spatial expression patterns of sweet potato LSs were found to be similar, if not identical, to their homologs in potato and tomato. For instance, IbAGPL1, potato agpS1, and tomato AgpL1, all belonging to group 4, were abundantly expressed in sink tissues, such as tuberous roots in sweet potato, stems and roots in potato, and stems in tomato. IbAGPL4 and tomato AgpL3, both in group 1, were only expressed in leaf tissues. IbAGPL3, belonging to group 2, was expressed both in source and sink tissues, but with low mRNA levels. The ratio of LS to SS mRNA levels was 3–5 in tuberous roots and 0.3 in mature leaves. Further investigation will be required to determine how LSs expressed at low levels can coordinate with highly expressed SSs to assemble AGPase heterotetramers in mature leaves and to clarify the roles of the proteins.
Many AGPase genes have been characterized in monocot and dicot plants. In light of their high degree of sequence similarity, higher plant AGPase SS and LS genes are thought to have arisen by gene duplication followed by divergence. The transcribed regions of sweet potato IbAGPS1 and IbAGPS2 genes consist of nine exons and eight introns. Intron numbers and locations are identical between the two AGPase SS genes as well as their counterparts in potato and tomato; in Arabidopsis, however, the second sweet potato SS intron is absent (Supplementary Table 4). Intron size is quite similar between IbAGPS1 and IbAGPS2 genes; for example, intron 5 of both genes is 76 bp in length but with a shared identity of 51 %. We compared genomic structures of the two sweet potato AGPase SS genes inferred by this study with those previously reported from another cultivar by Noh et al. (2004) Gene sequences including the introns were quite conserved, with an overall identity of 98.28 % for IbAGPS1 and 95.76 % for IbAGPS2. In terms of intron sequence differences, IbAGPS1 appeared to be more conserved than IbAGPS2 between cultivars, possibly reflecting the more important role of IbAGPS1 in sweet potato.
Compared with SSs, higher plant AGPase LSs are less conserved (50–60 % identity). The higher heterogeneity seen among LS sequences probably reflects the differences in requirements for modulation of SS sensitivity to allosteric regulation that are posed by the various demands of tissues and species (Smith-White and Preiss 1992). The four AGPase LS genes identified in sweet potato may have originated from a common ancestor by duplication. IbAGPL1, IbAGPL2, and IbAGPL3 transcribed regions consist of 14 exons and 13 introns, whereas the transcribed region of IbAGPL4 comprises 13 exons and 12 introns. Given the correspondence between the length of exon 3 in IbAGPL4 and the combined lengths of exons 3 and 4 in IbAGPL3, but not those in IbAGPL1 or IbAGPL2 (Supplementary Table 2) as well as the higher similarity of amino acid sequences of IbAGPL3 and IbAGPL4, the IbAGPL4 gene can be assumed to be derived from an IbAGPL3 ancestor. The ancestral AGPase LS gene is thus suggested to have undergone a process of duplication, resulting in two copies. In one gene copy, gene duplication and divergence may have given rise to IbAGPL1 and IbAGPL2. In the other copy, gene duplication in one case was followed by drastic changes such as intron loss and gain to yield IbAGPL4; in the second case, the other product of duplication was still under strict selection pressure and developed into IbAGPL3.
Examination of tomato, potato, and Arabidopsis genomes allowed the genomic DNA sequences of the AGPase LS genes to be identified (Supplementary Table 4). Of the four LSs in Arabidopsis, ApL3 and ApL4 are both expressed in sink tissues and consist of 14 exons. ApL1, functioning in leaves and composed of 15 exons, is the product of an intron insertion in exon 8 of ApL3/4. ApL2, expressed in sink tissue, comprises 12 exons and originated from the combination of exons 11–13 of ApL3/4. With regard to the three LSs of tomato, AgpL1 and AgpL2 both contain 14 exons, whereas AgpL3, expressed in leaves, consists of 15 exons and may have resulted from an intron insertion in exon 8 of AGPL1/2. The same situation is true for the three LSs of potato. A similar intron insertion into exon 8 has occurred in IbAGPL4 of sweet potato, but without any other intron losses or gains. These results suggest that the LSs responsible for sink tissues are usually more conserved than those acting in other tissues and may have an earlier origin. Because the exon 8 split in group 1 (responsible for leaves in most cases) is identical in Arabidopsis, tomato, potato, and sweet potato, this event appears to have occurred before the divergence of these species. Interestingly, LSs expressed primarily in leaves are less conserved than LSs expressed in sink organs. These data are in accord with the idea that tissue identity has influenced the rate of evolution of both SSs and LSs of AGPase. Expression in multiple tissues would then compound the various selection pressures exerted by the different tissues (Georgelis et al. 2007).
According to the transcriptional level analysis, IbAGPL4 was the main isoform expressed in leaves, especially in young leaves, and was accordingly grouped with leaf-expressed LSs. As shown by our results, we detected no activity from the AGPase heterotetramer composed of IbAGPL4 and any SS. This lack of activity of IbAGPL4 was partially due to its loss of interaction with SSs.
The crystal structure of a nonnative, SS homotetramer derived from the potato tuber AGPase has been previously described (Jin et al. 2005). Drawing on the modeled heterotetrameric structure of potato AGPase, Baris et al. (2009) used computational and experimental techniques to identify important amino acid residues mediating LS–SS interactions. The LSs were found to interact with SSs in two ways: laterally and longitudinally. A total of 12 interface residues (8 and 4 involved in lateral and longitudinal interactions, respectively) were identified in LSs as hot spots for interaction with potato AGPase SSs. These residues were highly conserved among different species, including rice, barley, potato, and Arabidopsis (Baris et al. 2009). In the present study, a comparison of the amino acid sequences of the four sweet potato AGPase LS isoforms with those in potato and tomato revealed a close evolutionary relationship. As shown in Fig. 6, 11 of the 12 hot-spot residues involved in longitudinal and lateral LS–SS association were conserved across all isoforms or isoforms belonging to one group. The exception was the first hot-spot residue involved in lateral interactions, which was equivalent to Ser in sweet potato IbAGPL4 but Asn in all other isoforms. Previous research (Baris et al. 2009) has suggested that lateral interactions are mainly driven by hydrophobic amino acids, with the size and polarity change of the amino acids involved in subunit–subunit interactions possibly affecting heterotetrameric structure formation. In light of these findings, several questions are worthy of consideration. For instance, does the change from Asn to Ser directly influence the interaction of IbAGPL4 with SSs, or are other amino acids involved in the loss of the interactive function of IbAGPL4? Additionally, given the loss of interaction of IbAGPL4 with SSs and the lack of AGPase activity in the heterotetramers harboring the IbAGPL4 subunit, is IbAGPL4 a pseudogene in sweet potato, similar to the ApS2 gene in Arabidopsis? Finally, if this hypothesis is true, which LS plays a role in leaves? In view of the transcriptional levels of the four LSs, IbAGPL1 and IbAGPL3 likely complement the role of IbAGPL4 in mature leaves, while the low levels of the three LSs may fulfill the limited demand for AGPase for starch synthesis in young leaves. Nevertheless, further studies are required to confirm this hypothesis or to suggest other possible explanations.

Aligment of partial amino acid sequences of the sweet potato AGPase large subunits with potato and tomato large subunits. The sequences alignment was performed by ClustalW. The arrow indicated amino acid residues which were predicted to directly interact with small subunits according to Baris et al. (2009). Asterisks denote conserved amino acids, whilst ‘.’ or ‘:’ refer to conservative substitutions
This study has provided insights into DNA sequences and gene structures of six AGPase isoforms in sweet potato. We have also revealed the spatial and temporal expression patterns of these subunits in different tissues and at different developmental stages of sweet potato. The availability of these gene sequences and expression profiles will enable us to determine whether such tissue-specific enzymes have different properties. It is a general system by which starch synthesis is differentially controlled in sink and source tissues, and organs. In tuberous roots of sweet potato, the dominant AGPase heterotetramer participating in starch synthesis is L1S1, followed by L1S2. In addition, different AGPase isoforms can be expressed in a given tissue, with combinations of different LSs and SSs conferring even more plasticity on AGPase. Future studies of sweet potato AGPase subunit genes may eventually allow precise control of the starch synthesis pathway, with the potential to achieve significant increases in sweet potato starch yield.
References
Ahn YO, Kim SH, Kim CY, Lee JS, Kwak SS, Lee HS (2010) Exogenous sucrose utilization and starch biosynthesis among sweetpotato cultivars. Carbohydr Res 345:55–60
Bae JM, Liu JR (1997) Molecular cloning and characterization of two novel isoforms of the small subunit of ADP-glucose pyrophosphorylase from sweet potato. Mol Gen Genet 254:179–185
Bahaji A, Li J, Ovecka M, Ezquer I, Muñoz FJ, Baroja-Fernández E, Romero JM, Almagro G, Montero M, Hidalgo M, Sesma MT, Pozueta-Romero J (2011) Arabidopsis thaliana mutants lacking ADP-glucose pyrophosphorylase accumulate starch and wild-type ADP-glucose content: further evidence for the occurrence of important sources, other than ADP-glucose pyrophosphorylase, of ADP-glucose linked to leaf starch biosynthesis. Plant Cell Physiol 52(7):1162–1176
Ballicora MA, Laughlin MJ, Fu YB, Okita TW, Barry GF, Preiss J (1995) Adenosine 5′-diphosphate-glucose pyrophosphorylase from potato tuber. Significance of the N-terminus of the small subunit for catalytic properties and heat stability. Plant Physiol 109:245–251
Ballicora MA, Iglesias AA, Preiss J (2004) ADP-glucose pyrophosphorylase: a regulatory enzyme for plant starch synthesis. Photosynth Res 79:1–24
Ballicora MA, Dubay JR, Devillers CH, Preiss J (2005) Resurrecting the ancestral enzymatic role of a modulatory subunit. J Biol Chem 280:10189–10195
Baris I, Tuncel A, Ozber N, Keskin O, Kavakli IH (2009) Investigation of the interaction between the large and small subunits of potato ADP-glucose pyrophosphorylase. PLoS Comput Biol 5(10):e1000546
Beckles DM, Craig J, Smith AM (2001) ADP-glucose pyrophosphorylase is located in the plastid in developing tomato fruit. Plant Physiol 126(1):261–266
Chen Y, Zhou F, Li G, Xu Y (2009) MUST: a system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene 436:1–7
Crevillén P, Ballicora MA, Mérida A, Preiss J, Romero JM (2003) The different large subunit isoforms of Arabidopsis thaliana ADP-glucose pyrophosphorylase confer distinct kinetic and regulatory properties to the heterotetrameric enzyme. J Biol Chem 278:28508–28515
Crevillén P, Ventriglia T, Pinto F, Orea A, Mérida Á, Romero JM (2005) Differential pattern of expression and sugar regulation of Arabidopsis thaliana ADP-glucose pyrophosphorylase-encoding genes. J Biol Chem 280(9):8143–8149
Danishuddin M, Chatrath R, Singh R (2011) Insights of interaction between small and large subunits of ADP-glucose pyrophosphorylase from bread wheat (Triticum aestivum L.). Bioinformation 6(4):144–148
Dawar C, Jain S, Kumar S (2013) Insight into the 3D structure of ADP-glucose pyrophosphorylase from rice (Oryza sativa L.). J Mol Model 19:3351–3367
Firon N, LaBonte D, Villordon A, Kfir Y, Solis J, Lapis E, Perlman TS, Doron-Faigenboim A, Hetzroni A, Althan L, Nadir LA (2013) Transcriptional profiling of sweetpotato (Ipomoea batatas) roots indicates down-regulation of lignin biosynthesis and up-regulation of starch biosynthesis at an early stage of storage root formation. BMC Genom 14:460
Frueauf JB, Ballicora MA, Preiss J (2003) ADP-glucose pyrophosphorylase from potato tuber: site-directed mutagenesis of homologous aspartic acid residues in the small and large subunits. Plant J 33:503–511
Georgelis N, Braun EL, Shaw JR, Hannaha LC (2007) The two AGPase subunits evolve at different rates in Angiosperms, yet they are equally sensitive to activity-altering amino acid changes when expressed in bacteria. Plant Cell 19:1458–1472
Guo AY, Zhu QH, Chen X, Luo JC (2007) GSDS: a gene structure display server. Yi Chuan. 29(8):1023–1026
Hannah LC, Shaw JR, Giroux MJ, Reyss A, Prioul JL, Bae JM, Lee JY (2001) Maize genes encoding the small subunit of ADP-glucose pyrophosphorylase. Plant Physiol 127:173–183
Harn CH, Bae JM, Lee SS, Min SR, Liu JR (2000) Presence of multiple cDNAs encoding an isoform of ADP-glucose pyrophosphorylase large subunit from sweet potato and characterization of expression levels. Plant Cell Physiol 41(11):1235–1242
He X, Zheng T, Su J, Chen Z (2011) DNA extraction of 7 species plants of melastomacea using modified CTAB method. Chin J Trop Agric 31(10):73–77
Hwang SK, Hamada S, Okita TW (2006) ATP binding site in the plant ADP-glucose pyrophosphorylase large subunit. FEBS Lett 580:6741–6748
Hwang SK, Nagai Y, Kim D, Okita TW (2008) Direct appraisal of the potato tuber ADP-glucose pyrophosphorylase large subunit in enzyme function by study of a novel mutant form. J Biol Chem 283:6640–6647
James MG, Denyerz K, Myers AM (2003) Starch synthesis in the cereal endosperm. Plant Biol 6:215–222
Jin X, Ballicora MA, Preiss J, Geiger JH (2005) Crystal structure of potato tuber ADP-glucose pyrophosphorylase. EMBO J 24:694–704
Kim T-W, Goo Y-M, Lee C-H, Lee B-H, Bae J-M, Lee S-W (2009) The sweetpotato ADP-glucose pyrophosphorylase gene (ibAGP1) promoter confers high-level expression of the GUS reporter gene in the potato tuber. C R Biol 332:876–885
Kwak MS, Oh MJ, Lee SW, Shin JS, Paek KH, Bae JM (2007) A strong constitutive gene expression system derived from ibAGP1 promoter and its transit peptide. Plant Cell Rep 26:1253–1262
Kwak MS, Oh MJ, Paek KH, Shin JS, Bae JM (2008) Dissected effect of a transit peptide of the ADP-glucose pyrophosphorylase gene from sweet potato (ibAGP2) in increasing foreign protein accumulation. Plant Cell Rep 27:1359–1367
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948
Lee SK, Hwang SK, Han M, Eom JS, Kang HG, Han Y, Choi SB, Cho MH, Bhoo SH, An G, Hahn TR, Okita TW, Jeon JS (2007) Identification of the ADP-glucose pyrophosphorylase isoforms essential for starch synthesis in the leaf and seed endosperm of rice (Oryza sativa L.). Plant Mol Biol 65:531–546
Morell MK, Bloom M, Knowles V, Preiss J (1987) Subunit structure of spinach leaf ADP-glucose pyrophosphorylase. Plant Physiol 85:182–187
Nielsen H, Engelbrecht J, Brunak S, Heijne G (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10:1–6
Noh SA, Kwak MS, Lee HS, Huh GH, Liu JR, Shin JS, Bae JM (2004) Genomic organizations of two small subunit ADP-glucose pyrophosphorylase genes from sweetpotato. Gene 339:173–180
Park SW, Chung WI (1998) Molecular cloning and organ-specific expression of three isoforms of tomato ADP-glucose pyrophosphorylase gene. Gene 206:215–221
Petreikov M, Eisenstein M, Yeselson Y, Preiss J, Schaffer AA (2010) Characterization of the AGPase large subunit isoforms from tomato indicates that the recombinant L3 subunit is active as a monomer. Biochem J 428:201–212
Robinson MD, McCarthy DJ, Smyth GK (2010) edgerR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140
Salamone PR, Greene TW, Kavakli IH, Okita TW (2000) Isolation and characterization of a higher plant ADP-glucose pyrophosphorylase small subunit homotetramer. FEBS Lett 482:113–118
Smith-White BJ, Preiss J (1992) Comparison of proteins of ADP-glucose pyrophosphorylase from diverse sources. J Mol Evol 34:449–464
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Bio Evol 24:1596–1599
Tao X, Gu Y-H, Wang H-Y, Zheng W, Li X, Zhao C-W, Zhang Y-Z (2012) Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam.]. PLoS One 7(4):e36234
Ventriglia T, Kuhn ML, Ruiz MT, Ribeiro-Pedro M, Valverde F, Ballicora MA, Preiss J, Romero JM (2008) Two Arabidopsis ADP-glucose pyrophosphorylase large subunits (APL1 and APL2) are catalytic. Plant Physiol 148:65–76
Zhang P, Chen C, Shen Y, Ding T, Ma D, Hua Z, Sun D (2013) Starch saccharification and fermentation of uncooked sweet potato roots for fuel ethanol production. Bioresour Technol 128:835–838
Acknowledgments
This project was supported by grants from the National Science & Technology Pillar Program of China (No. 2007BAD78B03) and the National Natural Science Foundation of China (No. J1103518).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2015_1134_MOESM1_ESM.tif
Supplementary Fig. 1. Alignment of the amino acid residues sequences of the SSs and LSs of sweet potato AGPase with SSs and LSs of potato, tomato, A. thaliana AGPase. Sequences alignment was performed by DNAMAN. ‘↓’ denote amino acids are important for enzyme activity (TIFF 4625 kb)
438_2015_1134_MOESM2_ESM.tif
Supplementary Fig. 2. Prediction and structural analysis of miniature inverted-repeat transposable elements (MITE) in LS and SS genes of sweet potato. The MITE was predicted and analysed by MITE Uncovering SysTem (MUST) (http://csbl1.bmb.uga.edu/ffzhou/MUST/). The underlined sequence represents the whole MITE sequence, the bold Italic letters represent the target site duplication (TSD) and the black boxes represent the terminal inverted repeats (TIRs) (TIFF 1237 kb)
438_2015_1134_MOESM3_ESM.tif
Supplementary Fig. 3. SDS - PAGE analysis of the co-expressed products of different sweet potato AGPase LSs and SSs combinations in E.coli. M, Protein Marker; CK: control check, pRSF & pACYC; L1S1: IbAGPL1 & IbAGPS1; L2S1: IbAGPL2 & IbAGPS1; L3S1: IbAGPL3 & IbAGPS1; L4S1: IbAGPL4 & IbAGPS1; L1S2: IbAGPL1 & IbAGPS2; L2S2: IbAGPL2 & IbAGPS2; L3S2: IbAGPL3 & IbAGPS2; L4S2: IbAGPL4 & IbAGPS2 (TIFF 16156 kb)
Rights and permissions
About this article

Cite this article
Zhou, YX., Chen, YX., Tao, X. et al. Isolation and characterization of cDNAs and genomic DNAs encoding ADP-glucose pyrophosphorylase large and small subunits from sweet potato. Mol Genet Genomics 291, 609–620 (2016). https://doi.org/10.1007/s00438-015-1134-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1134-3