
Abstract
Despite advances by genome-wide association studies (GWAS), much of heritability of common human diseases remains missing, a phenomenon referred to as ‘missing heritability’. One potential cause for ‘missing heritability’ is the rare susceptibility variants overlooked by GWAS. Atrial fibrillation (AF) is the most common arrhythmia seen at hospitals and increases risk of stroke by fivefold and doubles risk of heart failure and sudden death. Here, we studied one large Chinese family with AF and hypertrophic cardiomyopathy (HCM). Whole-exome sequencing analysis identified a mutation in TNNI3, R186Q, that co-segregated with the disease in the family, but did not exist in >1583 controls, suggesting that R186Q causes AF and HCM. High-resolution melting curve analysis and direct DNA sequence analysis were then used to screen mutations in all exons and exon–intron boundaries of TNNI3 in a panel of 1127 unrelated AF patients and 1583 non-AF subjects. Four novel missense variants were identified in TNNI3, including E64G, M154L, E187G and D196G in four independent AF patients, but no variant was found in 1583 non-AF subjects. All variants were not found in public databases, including the ExAC Browser database with 60,706 exomes. These data suggest that rare TNNI3 variants are associated with AF (P = 0.03). TNNI3 encodes troponin I, a key regulator of the contraction–relaxation function of cardiac muscle and was not previously implicated in AF. Thus, this study may identify a new biological pathway for the pathogenesis of AF and provides evidence to support the rare variant hypothesis for missing heritability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia in the clinical set. AF is electrocardiographically characterized by the absence of P waves, irregular RR intervals, and a fast atrial rate of up to 300 beats/min (Fye 2006). AF affects 2.6 million Americans, 6 million Europeans and 8 million Chinese people (Zhang 2009; Chugh et al. 2014). AF has a population prevalence rate of 1 % (Camm et al. 2012). AF is becoming a costly health problem in the near future with aging of the population because its rate increases up to 8 % in the population with the age of 80 years or above (Hu and Sun 2008). AF increases cardiovascular morbidity (Krahn et al. 1995), doubles the mortality (Kannel et al. 1998), and increases the risk of stroke fivefold (Wolf et al. 1991). Epidemiological studies revealed a strong genetic component significantly contributed to the incidence of AF, especially to lone AF not associated with any structural heart disease (Wang 2008). A study in Danish twins suggested that the heritability of AF was as high as 62 % (Christophersen et al. 2009).
Mutations associated with familial AF were initially identified in genes encoding ion channels, including potassium channels and sodium channels. Subsequent studies demonstrated that mutations in non-ion channel genes such as NPPA and NUP155 can also causer AF (Hodgson-Zingman et al. 2008; Zhang et al. 2008; Ren et al. 2010). Several genome-wide association studies (GWAS) on AF showed that a group of loci containing single-nucleotide polymorphisms (SNPs) confer risk to AF (Gudbjartsson et al. 2007; Benjamin et al. 2009; Gudbjartsson et al. 2009; Ellinor et al. 2010, 2012; Sinner et al. 2014). A candidate gene approach is another major strategy in identifying rare mutations in AF patients. For example, using the candidate gene approach our group successfully identified a functional dominant-negative AF mutation in sodium channel subunit gene SCN3B and suggested that SCN3B is a new pathogenic gene of AF (Wang et al. 2010b). Variants in non-ion channel genes GATA4, GATA6, LMNA, GREM2, and NKX2-5 were identified as AF-associated variants using the candidate gene approach (Olesen et al. 2014). Somatic mutations in GJA1 and GJA5 were first identified in atrial tissue of lone AF patients, while germline mutations in GJA1 and GJA5 were also identified by resequencing in lone AF cohorts (Olesen et al. 2014). Long QT syndrome 3-associated SCN5A variants were identified at a high frequency in patients with early-onset lone AF (Olesen et al. 2012). However, known gene mutations and risk loci can explain only a limited proportion of AF heritability because of the complex genetic architecture of AF, a phenomenon referred to as ‘missing heritability’. It is postulated that the remaining heritability for complex diseases such as AF may include rare variants, structural variants such as copy number variants (CNVs), epigenetics, and gene–environment interactions (Manolio et al. 2009; Eichler et al. 2010). In this study, we provide evidence to support the rare variant hypothesis to explain ‘missing heritability’ for AF.
The troponin complex of the thin filament of striated muscle consists of three subunits, including troponin I (TnI), troponin T (TnT) and troponin C (TnC) (Takeda et al. 2003). Troponin I, the inhibitory subunit of the troponin complex, serves as a calcium-sensitive molecular switch in the thin filament regulatory system (Farah and Reinach 1995). The troponin I subfamily includes three proteins: TnI-skeletal-fast-twitch, TnI-skeletal-slow-twitch, and TnI-cardiac (cTnI). Cardiac TnI is encoded by the TNNI3 gene located on human chromosome 19q13.4 and is expressed strongly and specifically in cardiac muscle tissues (Bhavsar et al. 1996). Mutations in TNNI3 were reported in patients with hypertrophic cardiomyopathy (HCM, CMH7), restrictive cardiomyopathy (RCM) and dilated cardiomyopathy (DCM) (Lu et al. 2013). In this study, whole-exome sequencing analysis identified a TNNI3 mutation that caused both HCM and AF in a large Chinese family. We further showed that rare TNNI3 variants, in aggregate, were associated with AF by sequencing analysis of a large case–control cohort of AF.
Methods
Study subjects and isolation of human genomic DNA samples
All study subjects were from the large GeneID database with collection of DNA samples and clinical data from over 80,000 study subjects with cardiovascular diseases in the Chinese Han population (Wang et al. 2011). We ascertained a large Chinese family with combined AF and HCM (Figs. 1, 2). In addition, a total of 1127 AF patients and 1583 controls without AF were enrolled into the present study. This study was approved by the Ethics Committee of College of Life Science and Technology, Huazhong University of Science and Technology. This study conformed to the principles set forth by the Declaration of Helsinki. Written informed consent was obtained from the participants.

An electrocardiogram from patient II:2 in the Chinese family with both HCM and AF. Typical electrocardiographic features with a diagnosis of AF were observed in patient II:2

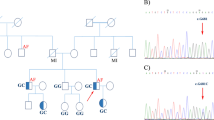
The pedigree structure of the Chinese with both HCM and AF. Males and females are shown with squares and circles, respectively. Filled symbols indicate ‘affected’, and open symbols are ‘unaffected’. A symbol with a question mark shows a family member without a definitive diagnosis of AF. The genotype for TNNI3 mutation c.G557A (p.R186Q) is indicated below each symbol with DNA available for genotyping
The diagnosis of AF was based on the standards as reported in the ACC/AHA/ESC AF guidelines (Fuster et al. 2006). AF was diagnosed using electrocardiograms (ECG) and/or Holter recordings as described by us previously (Shi et al. 2009; Wang et al. 2010b). AF occurring in subjects without evidence of structural heart disease and thyroid dysfunction was classified as lone AF. The control group consisted of healthy individuals without AF and other cardiovascular disease. HCM was diagnosed based on data from echocardiography as described (Gersh et al. 2011).
Genomic DNA samples were isolated from peripheral blood samples using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacture’s standard protocol.
Whole exome sequencing analysis
To identify the responsible mutation in the Chinese family with AF and HCM, we performed whole exome sequencing analysis for three affected individuals (II:2, III:7 and IV:2 in Fig. 2) and one unaffected individual (II:5 in Fig. 2) using our ABI SOLiD™ 5500xl Genetic Analysis System. The preparation of a SOLiD® barcoded fragment library was performed with the 5500 SOLiD™ Fragment Library Core Kit (Part no. 4464412) according to manufacturer’s protocol (Life Technologies). Three μg of genomic DNA from each individual as the starting material was sheared into small fragments with a mean fragment size of 165 bp by using the Covaris® System. Sheared DNA was end-repaired (5500 SOLiD™ Fragment Library Enzyme Module) and then selected by the Agencourt AMPure® XP Reagent (Beckman Coulter) to retain DNA fragments between 100 and 250 bp. After addition of a dA-tail to the size-selected DNA fragments, adaptors with specific sequences were linked to the DNA fragments. Adaptor-ligated, purified DNA fragments were PCR-amplified for six cycles before exome-enrichment (5500 SOLiD™ Fragment Library Amplification Module). The expected size distribution of the DNA fragment library was verified to be around 260 bp with Agilent Technologies 2100 Bioanalyzer™. The verified library fragments were then captured in a solution using the TargetSeq™ Exome Enrichment System. The TargetSeq™ system contained ~2 million TargetSeq™ capture probes and blocker DNA sequences to ensure hybridization specificity.
Following the exome capture step, the probe-hybridized DNA fragments were pulled down using Dynabeads M-270 streptavidin attached to the probes, washed and PCR-amplified with the 5500 SOLiD™ Fragment Library Amplification Module.
Quantitative real-time PCR analysis with six control primer pairs was used to measure exome enrichment. The successfully enriched exome library was subjected to the SOLiD® EZ Bead™ System to prepare templated beads. Emulsion PCR was then performed on an E80 scale with a titration point of 0.6 pM Targetseq library. After emulsion cleanup and bead enrichment, 3′-ends of the templated beads were modified (SOLiD Pre Deposition Kit).
Templated beads were quantified using a NanoDrop® 2000 Spectrophotometer to determine the appropriate volume of sample beads to be deposited. Before depositing the beads on the FlowChip, the templated beads were washed three times with SOLiD® FlowChip Deposition Buffer 1. Massively parallel sequencing of the templated beads on the FlowChip was accomplished on the SOLiD® 5500xl Genetic Analysis System.
The raw data in the format of *.xsq was imported to LifeScope™ Genomic Analysis Software to perform secondary (SAET, mapping) and tertiary (small indel, SNP finding, annotations) analyses. Parameters for target resequencing analysis were set to default values as recommended. Variation annotation of genes and exons was performed against refGene.hg19 (20101221). SNP annotation was performed with reference to dbSNP 132.
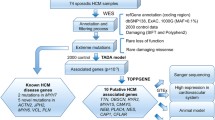
Variants identified in the study were filtered or selected according to the following criteria: (1) exclusion of variants present in dbSNP build 132 as polymorphisms; (2) selection of heterozygous variants and exclusion of homozygous variants because the disease is inherited in a dominant mode in the family; (3) selection of variants shared by three affected individuals and not present in the unaffected individual II:5; (4) exclusion of variants present in 40 individuals without HCM or AF and with whole exome sequencing data completed in our laboratory as polymorphisms. The derived list of variants was then analyzed using the wANNOVAR program (http://wannovar.usc.edu/) to annotate functional consequences of these variants (Wang et al. 2010a; Chang and Wang 2012). The wANNOVAR is a web interface to the ANNOVAR software and one of the most widely used functional annotation tools for high-throughput sequencing data. Following the annotation by wANNOVAR, polymorphisms were further filtered out using the data from the 1000 genome project (www.1000genomes.org/) and the NHLBI Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/). All variants that changed protein coding (non-synonymous variants, splicing site mutations, indels) were selected for follow-up studies.
For co-segregation analysis, direct Sanger sequencing analysis was performed for all family members with DNA samples available using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies) as previously described by us (Tian et al. 2004; Du et al. 2005; Zhang et al. 2008).
Mutation screening of TNNI3 mutations
In the AF patient group, high-resolution melting (HRM) curve analysis and direct DNA sequence analysis were used to screen for genomic variants in exons 1–6 and exons 7–8 of TNNI3 (NM_000363.4), respectively. The eight exons and exon–intron boundaries of TNNI3 were amplified by polymerase chain reactions (PCR) with primers listed in Table 1.
The primers were designed using Primer3web (version 4.0.0) (Koressaar and Remm 2007; Untergasser et al. 2012). PCR analysis was performed in a 25 μl volume containing 30 ng of genomic DNA, 10 pmol of each primer, 10 mM of deoxynucleotide triphosphates, 2.5 μl of 10 × PCR buffer with 1.5 mM MgCl2, and 1 unit of Taq DNA polymerase (TaKaRa Biotechnology Co. Dalian, China). For HRM analysis, 1.5 M SYTO® 9 green fluorescent nucleic acid stain was added to the PCR mixture (Life Technologies). The PCR profile was 94 °C for 5 min, 35–40 cycles of 94 °C for 20 s, annealing temperature for a specific pair of primers for 20 s, 72 °C for 20–30 s, and final extension at 72 °C for 10 m. For DNA sequencing analysis, PCR products after 35 cycles were purified by 2 % agarose gel electrophoresis, and sequenced using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies).
For HRM analysis, the PCR products after 40 cycles were loaded onto the RotorGene Q real-time PCR cycler (Qiagen), and their melting characteristics at different temperatures were generated as curves. Samples showing abnormal HRM curves were identified. The potential mutation represented by the abnormal HRM pattern was PCR-amplified from the original DNA samples again and identified by direct DNA sequence analysis as described above.
Mutational analysis of TNNI3 in the control group was performed using HRM curve analysis for all eight exons and exon–intron boundaries as described by us previously (Shi et al. 2009; Ren et al. 2010).
The detected variants were analyzed against the 6500 individuals whose exome variants data were reported in the Exome Variant Server, NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/; August 2012), the 1000 genome project data, and other existing SNP databases.
Each identified variant was then further analyzed using bioinformatic software, including SIFT and PROVEAN, to predict whether the amino acid substitution affects the function of cardiac troponin I. The SIFT (Sorting Intolerant From Tolerant) program predicts the impact of an amino acid substitution based on the degree of conservation of amino acid residues (Kumar et al. 2009). The PROVEAN (Protein Variation Effect Analyzer) program provides a generalized approach to predict the functional impact of protein variations including single or multiple non-synonymous and indel variants (Choi et al. 2012).
Results
Clinical features of a Chinese family with HCM and AF
We identified a large Chinese HCM family with twenty family members (9 males and 11 females; ages from 6 to 60 years) in three living generations (Table 2; Fig. 2). Ten family members were affected with HCM, while three of them (II:1, II:2 and III:1) were affected with both HCM and AF (Table 2). Patient II:1 sample was not available for this study. Patient II:2 was a 60-year-old female diagnosed with both HCM and AF. Echocardiography of patient II:2 showed segmental wall motion abnormalities, left and right atrial enlargement, mitral regurgitation and tricuspid regurgitation. The ECG of patient II:2 showed the typical features of AF (Fig. 1). Individual III:1 was diagnosed with both HCM and AF at the age of 32, and died suddenly at the age of 43. The youngest affected member was a 19-year-old female (Table 2).
Whole exome sequencing analysis identified a mutation in TNNI3 in the family with HCM and AF
Whole exome sequencing analysis of three affected individuals (II:2, III:7 and IV:2 in Fig. 2) and one unaffected individual (II:5, father of III:7) was performed using ABI SOLiD™ 5500xl Genetic Analysis System. We employed genomic resequencing analysis modules in LifeScope™ Genomic Analysis Software to analyze raw data (*.xsq files). The numbers of different variants identified by whole exome sequencing analysis were shown in Table 3. Variants that were shared by three patients but not by the unaffected individual were selected for further analysis. After all polymorphisms were filtered out, six heterozygous variants that changed protein coding were selected as candidate variants (listed in Table 4). Direct Sanger sequencing analysis was used to analyze all six candidate variants for co-segregation with the disease in the large Chinese family with HCM and AF. Variant p.D74N in the PSG1 gene was excluded as an error during whole exome sequencing (Table 4). Variants p.A78V, p.S101P, and p.L338F were excluded because obligate recombinant(s) were identified (i.e. patients without the variant).
The two remaining variants, p.R186Q in TNNI3 and p.E52D in MEIG1, were found to co-segregate with all patients in the family although non-obligate recombinants (unaffected individuals carrying the variant) were identified, which may be due to incomplete penetrance or young age (not reaching age of onset). The substitution of the E residue at position 52 by a D residue, i.e., p.E52D variant, in MEIG1 is considered to be a mild substitution and unlikely to be a pathogenic mutation. Moreover, the R186Q mutation was not present in 1583 controls. All together, we concluded that R186Q is the pathogenic mutation that causes AF and HCM in the Chinese family.
Identification of novel genomic variants of TNNI3 in patients with AF
Because above studies demonstrated that TNNI3 mutation R186Q causes both AF and HCM, we hypothesized that TNNI3 mutations may be found in patients with AF alone. To test this hypothesis, we screened TNNI3 mutations with a panel of 1127 patients with a definitive diagnosis of AF selected from the GeneID database (Table 5). The average onset age of AF was 66.57 (±14.57) years. 33.7 % of the AF patients can be classified as lone AF cases without concomitants structural heart disease. The mean age of 380 lone AF patients was 56.78 ± 10.46 years.
Mutation screening of all eight exons and exon–intron boundaries of TNNI3 in 1127 AF patients revealed four non-synonymous genomic variants (E64G, M154L, E187G, D196G) (Figs. 3, 4). All four variants were not detected in 1583 controls without AF. These four variants were not found in existing public exome databases, including the ExAC Browser database with 60,706 exomes. The four variants were identified in four independent patients with persistent AF. The clinical and demographic characteristics of the four AF patients are shown in Table 7, and all the four AF patients did not have HCM. All four variants led to amino acid substitutions at residues which were highly conserved across species (Fig. 5), suggesting that these amino acid residues are of functional importance. Bioinformatic analysis using SIFT and PROVEAN predicted that the variants had a damaging or deleterious effect on the function of cardiac troponin I (Table 6). No other TNNI3 variants were found in 1583 non-AF controls by mutation screening of all exons and exon–intron boundaries of TNNI3. Statistical analysis revealed a significant association between TNNI3 variants in aggregate and risk of AF in the cohort (P = 0.03 by Fisher’s exact test).

Sequencing data for mutations of TNNI3 identified in four different AF patients

Schematic structure of cardiac troponin I. a Structure of cardiac troponin I with the location of the mutations identified in this study indicated. b Schematic structure showing interactions among troponin I, troponin T, and troponin C in the calcium saturated thin filament. Cardiac troponin I is marked in red. α-Helices are shown with cylinders (Wang et al. 2012) (color figure online)

All five AF mutations in cardiac troponin I occur on amino acid residues that are highly conserved among different species during evolution. The location of each mutated amino acid residue is marked in red (color figure online)
Discussion
Whole exome sequencing analysis has become an effective tool to identify disease-causing mutations in large families. In this study, we utilized whole exome sequencing to characterize a large Chinese family with HCM and AF and identified a missense mutation, R186Q, in the TNNI3 gene that causes AF and HCM in the family. TNNI3 mutation R186Q was first reported in a French patient with HCM (Richard et al. 2003) and later in two families with incomplete penetrance of HCM (Mogensen et al. 2004). In one Caucasian family, four of the seven R186Q carriers developed HCM, whereas in the other Asian family, one of the four mutation carriers developed the disease (Mogensen et al. 2004). Similarly, in the large Chinese family studied here, nine family members were found to carry the R186Q mutation, but three of them (III:2, III:9, and IV:7) were not affected with HCM or AF. This can be explained by reduced, age-dependent penetrance of the R186Q mutation. Since individual III:9 and IV:7 were still at a young age of 22 and 6 years, respectively, they may not reach the age of onset.
Mutation R186Q was associated with a particularly severe outcome. The proband of the Caucasian family with R186Q died suddenly at the age of 32 years (Mogensen et al. 2004). Individual III:1 in the Chinese family studied here (Fig. 2; Table 2) died suddenly at the age of 43 years. Identification of the responsible mutations in this family provides important information to facilitate appropriate medical management for mutation carriers and their offspring. Moreover, since the carriers with mutation R186Q are at a high risk of sudden death, living carriers in the Chinese family should be monitored closely.
Three mutation carriers in our Chinese family, including II-1 (obligate carrier), II-2, and III-1 (obligate carrier), were affected by both HCM and AF (Table 2; Fig. 2). In contrast, none of the previously reported 12 carriers of the R186Q mutation was affected with AF (Richard et al. 2003; Mogensen et al. 2004). It remains to be investigated whether the causal role of the R186Q mutation in troponin I in AF is limited to the Chinese population.
To further establish the association between troponin I mutations and AF, we sequenced all exons and exon–intron boundaries of the TNNI3 gene in 1127 AF patients. We identified four novel genomic variants (E64G, M154L, E187G, and D196G) in TNNI3. The four variants did not exist in 1583 non-AF controls. Moreover, we also screened all exons and exon–intron boundaries of the TNNI3 gene in 1583 non-AF controls, but no TNNI3 variants were identified in the controls. Thus, a significant association was found between TNNI3 mutations and AF (P = 0.03) by statistical genetic association analysis. Variants E64G, M154L, E187G, and D196G did not exist in the NHLBI 6500 Exome database, the ExAC Browser database with 60,706 exomes and other existing databases.
It is interesting to note that E64G is the first troponin I variant identified in the helices 1 and 2 region. All four variants occur at amino acid residues with a high degree of conservation across species, indicating that these residues were important for cardiac troponin I function. Bioinformatic analysis predicts that all four variants may be damaging to the function of troponin I.
Because E187G is next to the R186Q mutation, it is located in a functionally important domain, which provides support that E187G may be a pathogenic mutation for AF. For variant D196G, it is interesting that at the same amino acid residue 196, one missense mutation, R196N was previously reported in a French patient with HCM (Richard et al. 2003). Thus, R196G identified in a patient with AF in this study may be a pathogenic mutation.
The three-dimensional structure of the 52 kDa core domain of human cardiac troponin I was determined (Takeda et al. 2003). Helices 1 and 2 (residues 40–80 and 90–130, respectively), of troponin I interacts with troponin T and forms the “IT-arm”, which interacts with the C-lobe of troponin C (Fig. 4a) (Wang et al. 2012). The E64G mutation is located in the first critical region (32–79) in the first α-helix that interacts with troponin C. Amino acid residues 150–159 form the third α-helix that binds to the N-lobe of troponin C. The M154L mutation is located in the troponin I–C interaction domain. The fact that both E64G and M154L are located in the critical domain of cardiac troponin I that interacts with troponin C provides additional support that both variants may be pathogenic to AF. Moreover, based on the analysis, it is likely that E64G and M154L variants of cardiac troponin I may cause a functional effect on the interaction between troponin I and troponin C, leading to the development of AF. Together, these data and follow-up statistical analysis suggest that variants in TNNI3 in aggregate are associated with risk of AF (P = 0.03 by Fisher’s exact text). Our results are consistent with a gene expression profiling study of chronic AF, which showed that TNNI3 expression was regulated in AF and this may be one of the particularly characteristic of AF (Lamirault et al. 2006).
Previously reported disease-causing mutations were identified mostly in patients with lone AF, i.e. AF without structural heart disease and other diseases. However, it is well known that risk of AF is significantly increased by advancing age, male sex, hypertension, coronary artery disease, myocardial infarction, heart failure, congenital heart disease, valvular disease, diabetes mellitus, obesity, rheumatic heart disease, hyperthyroidism, and sleep apnea. In fact, the majority of AF cases occur in the context of other diseases. It is interesting to note that all four AF-associated TNNI3 variants, i.e. E64G, M154L, E187G, and D196G, were identified in patients with both AF and other common cardiovascular risk factors (Table 7). Variant E64G was identified in an AF patient with hypertension and diabetes mellitus, M154L was found in an AF patient with hypertension and heart failure, and E187G and D196G were identified in AF patients with coronary artery disease (Table 7). Therefore, troponin I mutations are associated with the typical, common form of AF. The underlying heart diseases may act as potential trigger or substrate for AF, which promote atrial remodeling of AF and increase risk of AF. The four missense variants in TNNI3 may account for stress-induced AF under the background of other cardiovascular diseases. However, the detailed molecular mechanism by which troponin I mutations cause AF in the context of other cardiovascular diseases should be investigated in detail in the future.
GWAS have identified genomic variants at more than 10 loci for AF. However, the TNNI3 locus was not among the loci identified by GWAS. GWAS examine relatively common variants for association with human diseases. Moreover, the TNNI3 variants identified in this study are all private mutations which do not exist in any public database, including the largest genomic variant database with 60,706 exomes. Due to the minor allele frequency of 0 %, no association can be performed with each single mutation alone with the population-based studies. Therefore, previous GWAS failed to identify the TNNI3 locus for AF. This highlights one of the unique advantages for next generation sequencing (either whole exome sequencing or whole genome sequencing), which can uncover rare and/or private genomic variants that are associated with a human disease. With regard to the methodology, we performed genome-wide linkage analysis for the Chinese AF/HCM family with the R186Q mutation using more than 400 markers which span the entire human genome by every 10 cM before the whole exome sequencing technology became available. We failed to identify a positive locus. No positive linkage was found with markers spanning the TNNI3 locus (LOD score = 0.20 and −3.42 at D19S418 and D19S210, respectively), or other candidate variants identified by whole exome sequencing listed in Table 4 (LOD scores = −4.41 to 0.89). The reason for the failure of linkage analysis is because two unaffected family members III-9 and IV-7 turned to be mutation carriers (Fig. 2), which dramatically reduced LOD scores. This again highlights a unique advantage of next generation sequencing, which can identify a potential pathogenic mutation in a family first, and then genotype–phenotype analysis in the family can identify the true causal mutation.
There are several limitations with the present study. First, one major limitation of this study is that unfortunately, family members declined further genetic analysis so that we were unable to perform co-segregation analysis in families for the four TNNI3 variants. Second, lack of functional studies is another limitation. Future functional studies are needed to show whether these variants have any functional effect on the function of troponin I. Third, the number of internal reference ancestry of 1583 controls is low. The significant association between aggregated TNNI3 variants and AF needs to be replicated in another independent population, ideally in a non-Chinese population, in the future. Fourth, although the Fisher’s exact test detected a significant association between TNNI3 variants in aggregate and risk of AF in the cohort (P = 0.03), SNP-set (Sequence) Kernel Association Test (SKAT) (Ionita-Laza et al. 2013) failed to yield a significant P value (0.23 before adjustment of covariates, 0.33 after adjusting for age and gender, and 0.46 after adjusting for sex, age, coronary artery disease, type 2 diabetes and hypertension). The discrepancy may be due to the minor allele frequency of 0 % for all four missense variants and the small sample size. Future studies with a much larger population may be needed to resolve the issue.
In conclusion, the novel finding of this study is that mutation R186Q in TNNI3 causes both AF and HCM in a large Chinese family. Moreover, four novel mutations in TNNI3 were identified in patients with AF alone, including E64G, M154L, E187G, and D196G. These data expand the clinical spectrum of TNNI3 mutations. Moreover, the data in this study revealed an unexpected finding that variants in troponin I involved in contraction–relaxation control of the heart are associated with risk of common AF, a disease with electrical defects in the heart, which may reveal a new biological pathway for the pathogenesis of AF. Finally, this study provides evidence to support the ‘rare variants, common disease’ hypothesis to explain missing heritability for common human disease like AF.
Abbreviations
- AF:
-
Atrial fibrillation
- HCM:
-
Hypertrophic cardiomyopathy
- TNNI3:
-
Cardiac troponin I
- SIFT:
-
Sorting intolerant from tolerant
- PROVEAN:
-
Protein variation effect analyzer
- SNPs:
-
Single-nucleotide polymorphisms
References
Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, Schnabel RB, Bis JC, Boerwinkle E, Sinner MF, Dehghan A, Lubitz SA, D’Agostino RB Sr, Lumley T, Ehret GB, Heeringa J, Aspelund T, Newton-Cheh C, Larson MG, Marciante KD, Soliman EZ, Rivadeneira F, Wang TJ, Eiriksdottir G, Levy D, Psaty BM, Li M, Chamberlain AM, Hofman A, Vasan RS, Harris TB, Rotter JI, Kao WHL, Agarwal SK, Stricker BHC, Wang K, Launer LJ, Smith NL, Chakravarti A, Uitterlinden AG, Wolf PA, Sotoodehnia N, Kottgen A, van Duijn CM, Meitinger T, Mueller M, Perz S, Steinbeck G, Wichmann HE, Lunetta KL, Heckbert SR, Gudnason V, Alonso A, Kaab S, Ellinor PT, Witteman JCM (2009) Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet 41:879–881
Bhavsar PK, Brand NJ, Yacoub MH, Barton PJ (1996) Isolation and characterization of the human cardiac troponin I gene (TNNI3). Genomics 35:11–23
Camm AJ, Lip GYH, De Caterina R, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Ž, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, Reviewers D, Vardas P, Al-Attar N, Alfieri O, Angelini A, Blömstrom-Lundqvist C, Colonna P, De Sutter J, Ernst S, Goette A, Gorenek B, Hatala R, Heidbüchel H, Heldal M, Kristensen SD, Le Heuzey J-Y, Mavrakis H, Mont L, Filardi PP, Ponikowski P, Prendergast B, Rutten FH, Schotten U, Van Gelder IC, Verheugt FWA (2012) 2012 focused update of the ESC guidelines for the management of atrial fibrillation: an update of the 2010 ESC guidelines for the management of atrial fibrillation developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 33:2719–2747
Chang X, Wang K (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49:433–436
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688
Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, Christensen K (2009) Familial aggregation of atrial fibrillation a study in Danish twins. Circ Arrhythm Electrophysiol 2:378–383
Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim Y-H, McAnulty JH, Zheng Z-J, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJL (2014) Worldwide epidemiology of atrial fibrillation: a global burden of disease 2010 study. Circulation 129:837–847
Du W, Bautista JF, Yang H, Diez-Sampedro A, You S-A, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK (2005) Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 37:733–738
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11:446–450
Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, Sinner MF, de Bakker PIW, Mueller M, Lubitz SA, Fox E, Darbar D, Smith NL, Smith JD, Schnabel RB, Soliman EZ, Rice KM, Van Wagoner DR, Beckmann B-M, van Noord C, Wang K, Ehret GB, Rotter JI, Hazen SL, Steinbeck G, Smith AV, Launer LJ, Harris TB, Makino S, Nelis M, Milan DJ, Perz S, Esko T, Kottgen A, Moebus S, Newton-Cheh C, Li M, Mohlenkamp S, Wang TJ, Linda Kao WH, Vasan RS, Nothen MM, MacRae CA, Ch Stricker BH, Hofman A, Uitterlinden AG, Levy D, Boerwinkle E, Metspalu A, Topol EJ, Chakravarti A, Gudnason V, Psaty BM, Roden DM, Meitinger T, Wichmann HE, Witteman JCM, Barnard J, Arking DE, Benjamin EJ, Heckbert SR, Kaab S (2010) Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 42:240–244
Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Muller-Nurasyid M, Krijthe BP, Lubitz SA, Bis JC, Chung MK, Dorr M, Ozaki K, Roberts JD, Smith JG, Pfeufer A, Sinner MF, Lohman K, Ding J, Smith NL, Smith JD, Rienstra M, Rice KM, Van Wagoner DR, Magnani JW, Wakili R, Clauss S, Rotter JI, Steinbeck G, Launer LJ, Davies RW, Borkovich M, Harris TB, Lin H, Volker U, Volzke H, Milan DJ, Hofman A, Boerwinkle E, Chen LY, Soliman EZ, Voight BF, Li G, Chakravarti A, Kubo M, Tedrow UB, Rose LM, Ridker PM, Conen D, Tsunoda T, Furukawa T, Sotoodehnia N, Xu S, Kamatani N, Levy D, Nakamura Y, Parvez B, Mahida S, Furie KL, Rosand J, Muhammad R, Psaty BM, Meitinger T, Perz S, Wichmann HE, Witteman JCM, Kao WHL, Kathiresan S, Roden DM, Uitterlinden AG, Rivadeneira F, McKnight B, Sjogren M, Newman AB, Liu Y, Gollob MH, Melander O, Tanaka T, Stricker BHC, Felix SB, Alonso A, Darbar D, Barnard J, Chasman DI, Heckbert SR, Benjamin EJ, Gudnason V, Kaab S (2012) Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 44:670–675
Farah C, Reinach F (1995) The troponin complex and regulation of muscle contraction. FASEB J 9:755–767
Fuster V, Rydén LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, Halperin JL, Le Heuzey J-Y, Kay GN, Lowe JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann S, Smith SC, Jacobs AK, Adams CD, Anderson JL, Antman EM, Hunt SA, Nishimura R, Ornato JP, Page RL, Riegel B, Priori SG, Blanc J-J, Budaj A, Camm AJ, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Zamorano JL (2006) ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace 8:651–745
Fye WB (2006) Tracing atrial fibrillation-100 years. New Engl J Med 355:1412
Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011) 2011 ACCF/AHA Guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 58:2703–2738
Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A, Thorleifsson G, Kristjansson K, Palsson A, Blondal T, Sulem P, Backman VM, Hardarson GA, Palsdottir E, Helgason A, Sigurjonsdottir R, Sverrisson JT, Kostulas K, Ng MCY, Baum L, So WY, Wong KS, Chan JCN, Furie KL, Greenberg SM, Sale M, Kelly P, MacRae CA, Smith EE, Rosand J, Hillert J, Ma RCW, Ellinor PT, Thorgeirsson G, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K (2007) Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448:353–357
Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, Gulcher J, Mathiesen EB, Njolstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Kucera G, Stubblefield T, Carter S, Roden D, Ng MCY, Baum L, So WY, Wong KS, Chan JCN, Gieger C, Wichmann HE, Gschwendtner A, Dichgans M, Kuhlenbaumer G, Berger K, Ringelstein EB, Bevan S, Markus HS, Kostulas K, Hillert J, Sveinbjornsdottir S, Valdimarsson EM, Lochen M-L, Ma RCW, Darbar D, Kong A, Arnar DO, Thorsteinsdottir U, Stefansson K (2009) A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet 41:876–878
Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett JC, Olson TM (2008) Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. New Engl J Med 359:158–165
Hu D, Sun Y (2008) Epidemiology, risk factors for stroke, and management of atrial fibrillation in China. J Am Coll Cardiol 52:865–868
Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X (2013) Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet 92:841–853
Kannel WB, Wolf PA, Benjamin EJ, Levy D (1998) Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 82:2N–9N
Koressaar T, Remm M (2007) Enhancements and modifications of primer design program Primer3. Bioinformatics 23:1289–1291
Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE (1995) The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 98:476–484
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081
Lamirault G, Gaborit N, Le Meur N, Chevalier C, Lande G, Demolombe S, Escande D, Nattel S, Léger JJ, Steenman M (2006) Gene expression profile associated with chronic atrial fibrillation and underlying valvular heart disease in man. J Mol Cell Cardiol 40:173–184
Lu Q, Wu X, Morimoto S (2013) Inherited cardiomyopathies caused by troponin mutations. J Geriatr Cardiol 10:91
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Mogensen J, Murphy RT, Kubo T, Bahl A, Moon JC, Klausen IC, Elliott PM, McKenna WJ (2004) Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:2315–2325
Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, Hedley PL, Christiansen M, Olesen S-P, Haunsø S, Schmitt N, Jespersen T, Svendsen JH (2012) High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet 5:450–459
Olesen MS, Nielsen MW, Haunso S, Svendsen JH (2014) Atrial fibrillation: the role of common and rare genetic variants. Eur J Hum Genet 22:297–306
Ren X, Xu C, Zhan C, Yang Y, Shi L, Wang F, Wang C, Xia Y, Yang B, Wu G, Wang P, Li X, Wang D, Xiong X, Liu J, Liu Y, Liu M, Liu J, Tu X, Wang QK (2010) Identification of NPPA variants associated with atrial fibrillation in a Chinese GeneID population. Clin Chim Acta 411:481–485
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet J-P, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M, For the EHFP (2003) Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107:2227–2232
Shi L, Li C, Wang C, Xia Y, Wu G, Wang F, Xu C, Wang P, Li X, Wang D, Xiong X, Bai Y, Liu M, Liu J, Ren X, Gao L, Wang B, Zeng Q, Yang B, Ma X, Yang Y, Tu X, Wang Q (2009) Assessment of association of rs2200733 on chromosome 4q25 with atrial fibrillation and ischemic stroke in a Chinese Han population. Hum Genet 126:843–849
Sinner MF, Tucker NR, Lunetta KL, Ozaki K, Smith JG, Trompet S, Bis JC, Lin H, Chung MK, Nielsen JB, Lubitz SA, Krijthe BP, Magnani JW, Ye J, Gollob MH, Tsunoda T, Müller-Nurasyid M, Lichtner P, Peters A, Dolmatova E, Kubo M, Smith JD, Psaty BM, Smith NL, Jukema JW, Chasman DI, Albert CM, Ebana Y, Furukawa T, Macfarlane PW, Harris TB, Darbar D, Dörr M, Holst AG, Svendsen JH, Hofman A, Uitterlinden AG, Gudnason V, Isobe M, Malik R, Dichgans M, Rosand J, Van Wagoner DR, Consortium M, Consortium A, Benjamin EJ, Milan DJ, Melander O, Heckbert SR, Ford I, Liu Y, Barnard J, Olesen MS, Stricker BHC, Tanaka T, Kääb S, Ellinor PT (2014) Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation 130:1225–1235
Takeda S, Yamashita A, Maeda K, Maéda Y (2003) Structure of the core domain of human cardiac troponin in the Ca (2+)-saturated form. Nature 424:35–41
Tian X-L, Kadaba R, You S-A, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q (2004) Identification of an angiogenic factor that when mutated causes susceptibility to Klippel–Trenaunay syndrome. Nature 427:640–645
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115–e115
Wang Q (2008) Atrial fibrillation genetic considerations: the basic scientist’s perspective. In: Natale A, Jalife J (eds) Atrial Fibrillation. Humana Press, Totowa, pp 133–144
Wang K, Li M, Hakonarson H (2010a) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164–e164
Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F, Liao Y, Fang F, Chen Q, Tu X, Wang QK (2010b) Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem Biophy Res Comm 398:98–104
Wang F, Xu C, He Q, Cai J, Li X, Wang D, Xiong X, Liao Y-H, Zeng Q, Yang Y, Cheng X, Li C, Yang R, Wang C, Wu G, Lu Q, Bai Y, Huang Y, Yin D, Yang Q, Wang X, Dai D, Zhang R, Wan J, Ren J, Li S, Zhao Y, Fu F, Huang Y, Li Q, Shi S, Lin N, Pan Z, Li Y, Yu B, Wu Y, Ke Y, Lei J, Wang N, Luo C, Ji L, Gao L, Li L, Liu H, Huang E, Cui J, Jia N, Ren X, Li H, Ke T, Zhang X, Liu J, Liu M, Xia H, Yang B, Shi L, Xia Y, Tu X, Wang QK (2011) Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet 43:345–349
Wang H, Chalovich JM, Marriott G (2012) Structural dynamics of troponin i during Ca2+-activation of cardiac thin filaments: a multi-site Förster resonance energy transfer study. PLoS One 7:e50420
Wolf PA, Abbott RD, Kannel WB (1991) Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 22:983–988
Zhang S (2009) Atrial fibrillation in mainland China: epidemiology and current management. Heart 95:1052–1055
Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R, Wang L, Chen Q, Wang QK (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135:1017–1027
Acknowledgments
We are grateful for study subjects for their participation of this project. We thank current and previous members of Wang laboratory for help and technical assistance in GeneID.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by the China National Natural Science Foundation Key Program (31430047), Chinese National Basic Research Programs (973 Programs 2013CB531101 and 2012CB517801), Hubei Province’s Outstanding Medical Academic Leader Program, Hubei Province Natural Science Key Program (2014CFA074), the China National Natural Science Foundation Grant (91439129, NSFC-J1103514), NIH/NHLBI Grants R01 HL121358 and R01 HL126729, Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education, and the “Innovative Development of New Drugs” Key Scientific Project (2011ZX09307-001-09). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
Chuchu Wang declares that she has no conflict of interest. Manman Wu declares that she has no conflict of interest. Jin Qian declares that he has no conflict of interest. Bin Li declares that he has no conflict of interest. Chengqi Xu declares that he has no conflict of interest. Sisi Li declares that she has no conflict of interest. Shanshan Chen declares that she has no conflict of interest. Yuanyuan Zhao declares that she has no conflict of interest. Yufeng Huang declares that she has no conflict of interest. Lisong Shi declares that he has no conflict of interest. Xiang Cheng declares that he has no conflict of interest. Yuhua Liao declares that he has no conflict of interest. Qiuyun Chen declares that he has no conflict of interest. Yunlong Xia declares that he has no conflict of interest. Wei Yao declares that he has no conflict of interest. Gang Wu declares that he has no conflict of interest. Mian Cheng declares that she has no conflict of interest. Qing K. Wang declares that he has no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of College of Life Science and Technology, Huazhong University of Science and Technology and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Communicated by S. Hohmann.
C. Wang, M. Wu, J. Qian, B. Li, X. Tu, and C. Xu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wang, C., Wu, M., Qian, J. et al. Identification of rare variants in TNNI3 with atrial fibrillation in a Chinese GeneID population. Mol Genet Genomics 291, 79–92 (2016). https://doi.org/10.1007/s00438-015-1090-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1090-y