
Abstract
Ligon lintless-1 (Li1) is a Gossypium hirsutum mutant that is controlled by a dominant gene that arrests the development of cotton fiber after anthesis. Two F2 mapping populations were developed from mutant (Li1 × H7124) F1 plants in 2012 and 2013; each was composed of 142 and 1024 plants, respectively. Using these populations, Li1 was mapped to a 0.3-cM region in which nine single-strand conformation polymorphism markers co-segregated with the Li1 locus. In the published G. raimondii genome, these markers were mapped to a region of about 1.2 Mb (the Li1 region) and were separated by markers that flanked the Li1 locus in the genetic map, dividing the Li1 region into three segments. Thirty-six genes were annotated by the gene prediction software FGENESH (Softberry) in the Li1 region. Twelve genes were candidates of Li1, while the remaining 24 genes were identified as transposable elements, DNA/RNA polymerase superfamily or unknown function genes. Among the 12 candidate genes, those encoding ribosomal protein s10, actin protein, ATP synthase, and beta-tubulin 5 were the most-promising candidates of the Li1 mutant because the function of these genes is closely related to fiber development. High-throughput RNA sequencing and quantitative PCR revealed that these candidate genes had obvious differential gene expression between mutant and wild-type plants at the fiber elongation stage, strengthening the inference that they could be the most likely candidate gene of the Li1 mutant phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cotton fiber is an important raw material for the textile industry. Pursuing a high-fiber yield of good quality is the ultimate goal of conventional cotton breeding (Arpat et al. 2004; Lee et al. 2007). However, this is also a challenging job for scientific research because of the complex genetic foundation governing fiber development. According to length, cotton fiber can be divided into two categories: fuzzy fiber (<5 mm) and lint fiber (>10 mm). Fuzzy fiber is not only very short but also tightly attached to the surface of seeds. Lint fiber can be further divided into short fiber (<21 mm), medium-length fiber (28–32 mm), and long fiber (>35 mm) (Rong et al. 2005). The growth and development of cotton fiber, which originates from a single epidermal cell that protrudes outward and extends, can be artificially divided into four stages: fiber initiation (day 0–5), fiber elongation and formation of primary cell wall (day 0–25), formation of secondary cell wall (day 20–45), and fiber maturation (day 40–50, varies with cotton varieties) (Arpat et al. 2004; Lee et al. 2006, 2007). Only 25–30 % of seed epidermal cells (about 20,000 per ovule) could normally protrude and thus form mature fiber (Guan et al. 2011; Qin and Zhu 2011). Lint fiber cells usually begin to protrude before anthesis or on anthesis day, whereas fuzzy fiber generally delays several days (Lee et al. 2007). The number of fibers on the surface of the seed depends on how many epidemic cells protrude outward, which is closely related to cotton fiber yield. Cotton fiber elongation and protrusion occur almost simultaneously. The fastest elongation stage of the fiber cells and the most active period of various biochemical reactions occur during the period of 5–25 days after anthesis (DPA). The fiber length is largely determined during this period, which is also the key period affecting fiber quality (Qin and Zhu 2011).
So far, the genes that regulate fiber development were inferred by analyzing the expression of genes from different organs at different times by northern blot, reverse transcription (RT)-PCR, in situ hybridization, and RNA interference, or by analyzing gene expression and phenotype variation of transgenic cottons (Lee et al. 2007; Qin and Zhu 2011; Shangguan et al. 2010). The possible functions of some genes regulating fiber initiation and development have been revealed through extensive investigations and summarized in several reviews (Lee et al. 2007; Qin and Zhu 2011; Shangguan et al. 2010). However, we know little about the molecular mechanism of cotton fiber development, which finally determines the fiber quality.
Mutants are enormously appropriate materials to study cotton gene functions related to fiber development. Cotton fiber mutants can be divided into three categories: fiberless seeds (such as Fbl and Sma-4(ha)), seeds with very short-lint fiber (such as Li1, Li2, and Lix) (Cai et al. 2013; Karaca et al. 2002; Kohel et al. 1992), and fuzzless seeds with lint fiber (such as N1, sma-4 (fz), n2, xu142, and more than 100 recently reported naked mutants) (Rong et al. 2005; Sun et al. 2012; Turley and Kloth 2002). Because a single gene mutation in some of these mutants can affect the initiation and development of cotton fibers (Rong et al. 2005; Sun et al. 2012; Kohel et al. 2002; Turley and Kloth 2008; Turley et al. 2007), the isolation of these genes is critical to elucidate the molecular basis of cotton fiber development. Therefore, much effort has been expended in this research area. Until now, only seven fiber mutants have been roughly mapped (Karaca et al. 2002; Rong et al. 2005).
The Ligon lintless-1 (Li1) mutant was first discovered by Griffee and Ligon in 1929 (Griffee and Ligon 1929). The phenotype of the mutant showed twisted stems, curled leaves, and seeds without a long staple. Kohel and others argued that Li1 was a mutant controlled by a dominant gene that arrested the development of cotton fiber 5–7 DPA (Kohel 1972; Narbuth and Kohel 1990). The fiber elongation factor was inferred to be the most important element in determining the final phenotype of the seeds without a long staple (Kohel et al. 1992). Kohel et al. also determined that the fiber cell wall of Li1 appeared to be thicker than that of other normal cotton. Additionally, the survival rate of homozygous Li1 plants was quite low (Kohel et al. 1993; Narbuth and Kohel 1990). Recently, a mutant with a similar phenotype controlled by a dominant gene was created and mapped to a region of chromosome (Chr.) 04(A), which was thought to be homoeologous to the region of Li1 on Chr.22(D) (Cai et al. 2013). Because the Li1 mutant is a model for studying fiber elongation and both primary and secondary cell wall thickening processes, several groups have comprehensively explored gene alterations in the Li1 mutant. For example, gene expression in Li1 mutant ovules and young fibers was analyzed by fluorescence quantitative PCR and high-throughput RNA sequence analysis in an attempt to explain why the Li1 mutant had no lint fibers (Ding et al. 2014; Gilbert et al. 2013, 2014; Liu et al. 2012). Only a small number of genes in Li1 ovules showed differential expression during the cotton fiber initiation stage compared with that of wild-type ovules. Simultaneously, differential expression of several secondary cell wall development-related genes such as expansin, tubulin, and sucrose synthase was identified in the young fiber of Li1 and wild-type plants during the fiber elongation stage, probably explaining the phenotype of the Li1 mutant (Bolton et al. 2009; Gilbert et al. 2013, 2014). In addition to the preliminary study of the mutant at the transcriptional level, researchers conducted an expression analysis at the protein level in the Li1 mutant during fiber elongation using the protein two-dimensional electrophoresis (2-DE) technique (Zhao et al. 2010). The results indicated that the expression of many proteins was quite low in Li1 plants compared with normal plants. These proteins included those that were responsible for protein folding and stability; protein transportation among the cytoplasm, nucleus, and vesicles; and signal transduction. Zhao et al.’s (2010) work also revealed that the expression of many cytoskeletal proteins was significantly down-regulated during cotton fiber development, which resulted in cytoskeletal disorganization during the process of fiber development and thus affected fiber elongation due to the very low content of cytoskeletal protein in the fiber of the Li1 mutant.
Fine mapping of the mutant is a critical step to clone the candidate genes. The Li1 locus has been mapped to Chr.22(D) by several laboratories (Gilbert et al. 2013; Karaca et al. 2002; Rong et al. 2005). Kahara et al. identified a simple sequence repeat (SSR) marker (MP4030) 12.83 cM away from the Li1 locus (Karaca et al. 2002). Comparatively, Rong et al. mapped the Li1 mutation to a narrower location with a relatively larger F2 population (Rong et al. 2005). In this research, the Li1 locus was flanked by restriction fragment length polymorphism (RFLP) markers Gate4CA09 and Coau1J04, which were 2.7 and 1.3 cM away, respectively. Recently, Li1 was further mapped to Chr.22 adjacent to the SSR marker TMB2500 with a distance of 0.8 cM (Gilbert et al. 2013). Nevertheless, much progress needs to be achieved in order to clone the mutant gene. Here, we report the fine mapping of the Li1 locus in a region of about 0.3 cM and 1.2 Mb, which was a gene-sparse region; 36 genes were annotated. The expression of the candidate genes was investigated, and the most-promising candidate genes were further deduced based on map position information and gene expression.
Materials and methods
Development of mapping populations
F1 plants were developed from crosses between the heterozygous Li1 mutant and G. barbadense variety H7124. The G. barbadense variety was used in this research to add the polymorphism. F1 plants with a mutant-type phenotype were used to produce F2 populations. Two mapping populations consisting of 142 and 1024 F2 plants were developed in 2012 and 2013 for rough and fine mapping, respectively. All plants were planted in our university farm in Linan, Zhejiang Province, China.
Association analysis between phenotype and genotype in F2 mapping populations
The plant morphology of the Li1 mutant and its near-isogenic wild-type plant was observed, and the association between phenotype and genotype was subsequently analyzed in the selfed offspring of the Li1 mutant and F2 populations derived from F1 plants with mutant phenotypes including plant height, node number, and boll number. These phenotypes were measured on September 16, 2013, at the stage of later flower season. Plant height was measured from the cotyledon node to the top of the plant. Once the Li1 gene was fine mapped, the mutant types of F2 plants were classified as heterozygote and homozygote based on the genotype of markers that co-segregated with the phenotype of the Li1 mutant. The phenotype of the two groups of plants was analyzed, and the statistical significance of the difference was calculated to determine if there was a close association between phenotype and genotype.
DNA extraction and marker development
DNA extraction
Young leaf buds (2–3) of each sample were chosen and placed in a 2-mL eppendorf tube for DNA extraction. DNA was extracted by the procedure reported previously (Zhou et al. 2010).
Marker development
The Li1 mutant gene was mapped previously to the middle region of Chr.22(D) (Rong et al. 2005). In this research, the published mapping information was used as the initial point to fine map the Li1 gene. The sequences of RFLP markers flanking the Li1 gene in the published paper were searched using the basic local alignment search tool (BLAST) against the G. raimondii genome sequence (Paterson et al. 2012) to limit the region where the Li1 gene was probably located. First, the sequences corresponding to two end markers of the targeted region were selected to design the primers for the single-strand conformation polymorphism (SSCP) screening. Once the Li1 gene was confirmed to be in the region, more sequences were selected at positions every 10 kb until the candidate gene was determined. Primers were designed considering several factors, such as fragment length and coverage of introns, as reported previously (He et al. 2013).
SSCP procedure
PCR was performed as the protocol reported previously (He et al. 2013), and PCR conditions were modified until 1–2 clear bands of both parents were detected for a particular pair of primers. PCR products were checked by agarose gel electrophoresis. SSCP analysis, including heat denaturation, electrophoresis, and silver staining, was performed according to the procedure that we reported previously (He et al. 2013).
Genetic mapping
F2 individuals from F1 plants with a mutant phenotype segregated into wild and mutant type, as found in the selfed offspring of Li1 mutant plants. The mutant phenotype was scored as a dominant marker. Mapping of the Li1 mutant gene was carried out in two stages. The genetic map was first constructed using the data from a small population consisting of 142 plants that were planted in 2012. In 2013, a large mapping population consisting of 1024 F2 plants was used for fine mapping. Phenotype data, together with the scoring data of SSCP markers, were used to construct the genetic map by running MAPMAKER/EXP 3.0 (Lander et al. 1987). The Kosambi mapping function, logarithm of odds score of >3.0, and a maximum distance of 40 cM were used to declare the linkage of the markers.
Gene annotation
The sequences between two markers, P95 and P98, were first extracted from the G. raimondii complete genome sequence (Paterson et al. 2012). The sequence corresponding to about 2.57 Mb was then used to predict cotton genes by the software FGENSH with the default settings. The annotation process was conducted by uploading all of the predicted sequences into a local Blast2GO system where the software translated BLAST with the non-redundant database was installed.
RNA extraction and gene expression analysis
Total RNA was isolated from various tissue samples of wild and mutant plants from self pollination of a mutant plant using the cetyltrimethylammonium bromide–sour phenol extraction method as described by Jiang and Zhang (Jiang and Zhang 2003). RNA samples were treated with DNase I (TaKaRa, Japan) to eliminate contaminating genomic DNA. To conduct RT-PCR, approximately 2 μg total RNA from each sample was used to synthesize first-strand cDNA in a 20-μL reaction solution using an M-MLV reverse transcription kit (TaKaRa, Japan). Moreover, the synthesized cDNAs were utilized as templates in the following semi-quantitative PCR analysis (semi-qPCR) and real-time quantitative PCR (qPCR) reactions.
PCR amplification was performed using Takara Taq polymerase. The PCR was conducted in a heated-lid thermal cycler (Eppendorf, Germany) with the following procedure: 95 °C for 3 min, followed by 28–30 cycles of 95 °C for 30 s, 52–55 °C for 30 s, and 72 °C for 90 s, and ending with an elongation step of 72 °C for 10 min. A cotton constitutive gene, UBQ7, was used as an internal standard to adjust the concentration of the cDNA of each sample used in the semi-qPCR analysis. The amplified fragments were examined by electrophoresis on a 1.5 % (w/v) agarose gel.
The cDNAs used to detect the gene expression were the same as those employed for qPCR analysis. The reaction was conducted on the Applied Biosystems 7300 Real-Time PCR System using the SYBER premix ExTaq kit (TaKaRa, Japan) according to the manufacturer’s instructions. The amplification of the target genes was monitored by SYBR-Green fluorescence signal every cycle. The cycle threshold value was used for gene expression analysis.
Results
Fine mapping of the Li1 gene
In the published map (Rong et al. 2005), the Li1 locus co-segregated with Coau1J04 and was flanked by Gate4CA09 and pAR0078. When the sequences of these RFLP markers were searched against the complete genome sequence of G. raimondii, they were found to be 14.41 Mb (14,760,834–29,172,417 bp) apart in the middle of D12 of G. raimondii (Fig. 1c, d). A total of 185 pairs of primers (first stage) were designed after several rounds of mapping efforts (Supplementary Table 1) for rough mapping. Among these, 103 primers produced 1–2 clear bands in both parents and were then used to screen the polymorphism between them. As a result, 44 polymorphic primers were detected and used to build the map. Three linkage groups were constructed using 142 F2 plants developed from the cross between Li1 and H7124 in 2012. The Li1 mutant was first roughly mapped to a linkage group consisting of 17 SSCP markers and a Li1 gene, which spanned 40.3 cM (Fig. 1a). The markers were not evenly distributed on the linkage group and were divided into three segments. The Li1 gene was mapped to the middle segment and was tightly flanked by P95 and P98 which were 0.3 and 0 cM away from Li1, respectively. In the published G. raimondii sequence (Paterson et al. 2012), P95 and P98 were 2.57 Mb apart. The sequence between the two markers was extracted and predicted by the gene prediction software FGENESH. As a result, 323 genes were identified in the region with potentially complete gene structures.

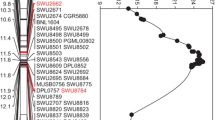
Genetic and physical maps of the Li1 mutant. a The rough map of the Li1 gene. b The fine map of the Li1 gene. c The physical map of the Li1 gene. Two bars at the right side of the map represent two segments containing the DNA sequences that co-segregated with the Li1 locus. d The map of the Li1 gene from Rong et al. (2005). The right map was the genetic reference map reported by Rong et al. (2004). The lines connect the same DNA sequence in different maps
To narrow the region including the Li1, a larger F2 population including 1024 plants was adopted to further map the targeted gene. More SSCP markers (88) were developed in the region between P95 and P98, where the Li1 gene was probably located; this was determined in the above rough mapping. A linkage group covering 35.84 cM and consisting of 27 markers (26 SSCPs and the Li1 gene) was built using this population (Fig. 1b). The Li1 gene was delimited to a region of 0.3 cM and was flanked by P214 and P223. There were also three recombinants found in 1024 F2 plants between P214 and P223. More markers were further used in this region to identify the exact location of these three recombinants, resulting in nine SSCP markers that co-segregated with Li1. Finally, two recombination events were detected between Li1 and P214, and one recombination event was detected between Li1 and P233.
Because all SSCP markers on the above genetic map were developed using the published G. raimondii genome sequence, it was very easy to discern locations on the diploid D genome sequence. After comparing the marker order on the genetic map of tetraploid cotton and the physical map of the diploid D genome sequence, it was determined that the maps retained good synteny except for the middle region of the genetic map (between P200 and P095), where the Li1 gene was located. In G. raimondii, P200 and P095 were 1.69 Mb apart. In the middle region of the genetic map, Li1 co-segregated with nine SSCP markers, whereas in the physical map of G. raimondii, they were separated into two segments by four markers (P257, P251, P191, and P223; Fig. 1c). Segment I was between P257 and P200, which spanned 740 kb, and segment II was between P214 and P223, which spanned 233 kb. Four recombinants were identified between P257/P251 and P191/P233 in 1024 F2 plants; these recombination events were mapped on both sides of the Li1 gene region in tetraploid cotton, but there was only 291 kb between P257 and P223 in the diploid D genome. This result implied that the D genome of either diploid or tetraploid cotton experienced chromosome structural changes in this region.
Gene annotation of the Li1 region
The above analysis indicated that Li1 candidate genes probably existed in a region of chromosome D12 that included three segments (segment I, II, and interval) in the diploid D genome, which is called the Li1 region hereafter. The sequences in this region were annotated, and 36 genes were identified by FGENESH, which included 21 genes in segment I, 5 genes in segment II, and 10 genes in interval (Tables 1, 2). Most of the inferred genes (27/36) belonged to three main groups: known function (12 genes), transposable elements (8 genes), and DNA/RNA polymerase superfamily (7 genes). The remaining 9 genes are unknown function genes. The level of gene density was estimated as the gene number divided by the total base pairs of the segment. Higher gene enrichment was found in segment I than in segment II (2.84 vs. 2.15 genes/100 kb, respectively). In addition, the gene type also varied substantially between the two segments. For example, six transposable elements were identified in segment I but not in segment II. Moreover, many more genes that have known functions were found in segment I than in segment II (9 vs. 1). In addition to the genes in segments I and II, ten annotated genes in the interval region between them are also listed in Table 1 and summarized in Table 2. Similarly, genes encoding proteins with no annotation (3), DNA/RNA polymerase superfamily proteins (2) and transposable element proteins (3) were the main types of genes in the interval region.
Expression analysis of candidate genes
Transcript expression profiling of the RNA extracted from leaves was compared between mutant and wild-type leaves in our laboratory (Ding et al. 2014). Differentially expressed genes in leaf tissues of mutant versus wild-type plants were identified, and the underlying pathways and potential genes that control leaf and fiber development were subsequently inferred. All predicated genes in the Li1 region and their leaf profile data are presented in Table 1. Most of the genes in this region were not detected, or they were expressed at a very low level. Six genes in segment I and one gene in interval showed high gene expression (reads per kilobase of gene model per million mapped reads [RPKM] > 150) in both mutant and wild-type plants, but only three genes (beta-tubulin 5 [P254], actin [P258], and ribosomal protein s10 gene [P272]) showed about a twofold difference between mutant and wild-type plants.
To validate the expression data from RNA-seq analysis, seven genes mentioned above were first analyzed together, and four genes displayed very low expression (P254, P260, P262, and P263) by semi-qPCR in stem and 0 DPA ovules (Fig. 2) in both wild and mutant type. Most results were consistent with those from RNA-seq analysis, except P254 and P260, which showed high expression in RNA-seq analysis of leaf but did not demonstrate good amplification in either wild-type or mutant plants (Fig. 2). The five genes (P258, P269, P272, P273, and P274), showing higher expression in both RNA-seq and semi-qPCR, were analyzed for their expression in more tissues, including stems, leaves, and different stages of ovules and fibers by semi-qPCR and qRT-PCR (Figs. 3, 4). Overall, the results of the two methods were well correlated. Different genes proved to have different expression patterns in these tissues. As observed in stems and ovules at 0 PDA, P274 showed very high expression, whereas P272 had no or low gene expression in most of the tested tissues (Fig. 3). When the expression patterns were compared between the wild-type and mutant tissues, the pattern was very complicated and displayed the opposite expression among different tissues. A gene that exhibited higher expression in mutant than in the wild-type tissue sometimes had the opposite relative expression in other tissues. For example, P258 showed higher gene expression in the stem of the wild-type plant than in the mutant, whereas in 10-PDA fiber, the mutant demonstrated much higher gene expression than the wild-type plant.

Expression patterns of the candidate genes in the Li1 region were analyzed by reverse transcription-PCR in stem and 0 days post anthesis (DPA) ovules of wild-type and mutant plants. M DNA ladder (Takara, Japan)

Expression patterns of the candidate genes in the Li1 region were analyzed by semi-quantitative PCR of stem, leaf, ovules, and fiber at different developmental stages of wild-type (W) and mutant (M) plants. DPA days post anthesis, Fib fiber

Expression patterns of the candidate genes in the Li1 region were analyzed by quantitative PCR in stem (S), leaf (L), ovules, and fiber at different developmental stages of wild-type and mutant plants
Morphology variation of mutant plants in the F2 population and its association with the genotype of markers tightly linked to Li1.
A total of 1024 F2 plants were observed for their phenotype, and all plants could be easily divided into wild and mutant types (Fig. 5). The mutant plants were very weak, and their stems and leaves twisted as reported (Narbuth and Kohel 1990). The ratio of mutant to wild is 2:1, which significantly deviated from the one gene segregation model (3:1) as that found previously (Rong et al. 2005). The failure of homozygote mutant individuals to survive during embryogenesis was believed to be the main reason for the low number of mutant plants in the F2 offspring (Narbuth and Kohel 1990; Rong et al. 2005). In the field, mutant plants varied considerably in vitality, including plant height, boll number, and node number (Table 3).

Wild-type and Li1 mutant plants. Wild-type (right), heterozygote Li1 mutant (middle), and homozygote Li1 mutant (left) plants
As observed in the above mapping efforts of the Li1 gene, nine SSCP markers co-segregated with the Li1 gene. Segregation analysis of these markers indicated that 134 plants were homozygotes of Gossypium hirsutum (Li1, genotype 1), 558 were heterozygotes (genotype 2), and 332 were homozygotes of G. barbadense (genotype 3), which deviated significantly from the expected ratio of 1:2:1 (χ 2 value 3.8E−19) (Fig. 6). The plant number of genotype 1 was much less than that of genotype 3 (134 vs. 332). When the F2 plants were classified into three groups according to the genotypes of the markers co-segregating with the Li1 gene, they exhibited obviously different growth vigor. The plants with genotype 1 and 2 displayed the mutant phenotype, whereas the plants of genotype 3 had a normal plant phenotype. Of the plants with a mutant phenotype, the plants of genotype 1 exhibited much weaker growth than heterozygote plants (genotype 2) in several aspects, such as plant height, branch number, and boll number (Table 3; Fig. 5), even when all plants displayed the mutant phenotype. Although the range of each measured trait overlapped among different groups, the three groups of plants showed a significant difference in almost all of their measured phenotypes on average (Table 3). For example, the main stem of the plants that were homozygotes of G. hirsutum was 78.7 cm in height, which was significantly shorter than that of the heterozygote plants (132.1 cm), while the latter was also significantly shorter than that of the plants that were homozygotes of G. barbadense (180.1 cm).

Number of plants with different heights across three genotypes of the markers co-segregating with the Li1 phenotype in the F2 population derived from a cross between Li1 mutant and H7124
Discussion
Difficulties in cloning the Li1 gene
Genetic mapping is the first step to clone the targeted gene for further function investigation. Several efforts have been made to delimit the Li1 gene in the last 10 years through genetic mapping with DNA markers. Two laboratories have mapped the Li1 gene to a rough region of Chr.22(D) (Gilbert et al. 2013; Karaca et al. 2002). Both of the parents that they utilized were G. hirsutum varieties, which resulted in a low level of polymorphism, and that was probably the main reason why they could not map the gene to a fine position. Another reason was that the whole genome sequence was not available at that time, which made it difficult to develop new markers to further approach the targeted gene. Comparatively, Rong et al. (2005) provided a much narrower position of the Li1 gene because two cotton species (G. barbadense and G. hirsutum) were used as the parents. In that research, the Li1 gene co-segregated with Coau1J04 and mapped to a 3.2-cM region that was flanked by Gate4CA09 and pAR0078, 1.3 and 1.9 cM from Li1, respectively. Nevertheless, it was quite difficult to further delimit the targeted gene without the whole cotton genome reference sequence. In addition, as indicated by Rong et al. (2005), the region containing Li1 shows unusual high marker density in the reference map, and it has tentatively been suggested to include the centromere (Rong et al. 2004). The flanking markers (Gate4CA09 and pAR0078) were 1.3 and 1.9 cM from the Li1 locus, respectively, but they spanned 14.41 Mb, which was 4.5 Mb/cM, much higher than the average of 606 kb/cM (Rong et al. 2004). These regions normally contain more heterochromatin than other regions; a characteristic that inhibits recombination. The interspecific crosses between G. hirsutum and G. barbadense used to produce the mapping populations in this study and by Rong et al. (2005) probably were another reason causing the low recombination in this region detected in these researches, because the large divergence between two homoelogous chromosomes will normally inhibit their crossover.
We started the allocation of the mutant gene based on the map reported by Rong et al. (2005) with the aid of the published diploid D genome reference sequence (Paterson et al. 2012). Using a small mapping population, we first roughly mapped the Li1 locus to the same region that was reported by Rong et al. (2005), confirming that the Li1 gene truly existed in the region as reported (Rong et al. 2005). We compared the marker order of Chr.22(D) (Fig. 1a) and its physical position on the counterpart of the diploid D genome (Fig. 1c), we determined that both chromosomes retained very good synteny. In our rough map, Li1 co-segregated with P098, which was at a position of 23,687,483 bp on the published sequence of G. raimondii Chr.12. In the map published by Rong et al. (2005), Coau1J04, which co-segregated with Li1, was at the position of 22,634,985 bp, 1.052 Mb away from the P098 marker. These mapping results confirmed that both maps were correct and that the Li1 gene was located in the region around these two markers, which facilitated further marker development to fine map the gene. Using a relatively larger mapping population, the Li1 gene was further mapped to a region between P223 and P214, where nine SSCP markers co-segregated with Li1. It was obvious that some markers mapped to the same position because physically they were so close in the heterochromatin region that recombination rarely occurred and could not be detected using a relatively large population (Fig. 1b, c). Comparing the order of these markers on Chr.22(D) of tetraploid cotton to D12 of diploid cotton indicated that the orders were not the same, suggesting that either tetraploid or diploid cotton experienced chromosome structural changes. Moreover, the markers flanking the region of the Li1 gene on Chr.22(D) were neighbors on D12 of G. raimondii. The middle region of D12 could be divided into two segments according to their position on Chr.22(D): segment I (740.1 kb) and segment II (233.9 kb), which had markers that co-segregated with Li1, and the interim segment (291.2 kb), which included the markers flanking the Li1 locus on the genetic map. These chromosome structure changes make it difficult to infer the location of the Li1 candidate gene.
In this research, 1.27 Mb of DNA sequence (between P200 and P214) only contained 12 genes (0.011 genes/kb) excluding transposable elements, DNA/RNA polymerases, and those without an annotated function in the National Center for Biotechnology Information. The gene density in this region was much lower than the average of 0.056 genes/kb, which is another typical characteristic of a region with heterochromatin, in addition to the low number of recombination events. Although three recombination events were observed between P214 and P223 in this fine-mapping study, it is challenging to determine their location because of the low number of recombinants and the varied chromosome structure of this region in tetraploid cotton and the reference DNA sequence of G. raimondii, as discussed above. We tried 66 pairs of primers to screen the polymorphism in this region (between P214 and P200) and achieved the current mapping results.
Candidate genes and possible mechanism causing abnormal growth of the Li1 mutant
In addition to the mapping efforts, transcript profile analysis was adopted in several laboratories to explore the candidate genes of the Li1 locus (Bolton et al. 2009; Ding et al. 2014; Gilbert et al. 2013, 2014; Liu et al. 2012; Naoumkina et al. 2015). Most of these studies focused primarily on gene expression alterations between mutant and wild-type plants during the fiber elongation stages (3–24 DPA), and they agreed that many genes belonging to several metabolic pathways were critical for fiber growth and development. However, each lab proposed quite different conclusions. For example, Bolton et al. (2009) thought that three candidate gene families—expansin, sucrose synthase, and tubulin—were probably the candidate genes responsible for the decreased growth of fibers in Li1, while Liu et al. (2012) indicated that the auxin-signaling and sugar-signaling pathways might play major roles in the modulation of early fiber elongation. Gilbert et al. (2013) believed that the sampling time (either the late elongation stage or the early secondary cell wall synthesis stage) explained why they “failed to identify significant numbers of differentially expressed transcripts”, and no consistent conclusion was obtained. Even the results from Fang’s laboratory that were published at a different time varied considerably (Gilbert et al. 2013, 2014; Naoumkina et al. 2015). They explained that the differing conclusions between their first (Gilbert et al. 2013) and second paper (Gilbert et al. 2014) were probably caused by the environmental conditions of the plant growth, and the differences between their second paper and third paper (Naoumkina et al. 2015) were “due to limitations of microarray technology” used in the second paper. This indicated that the cotton fiber elongation process is a very complicated process that involves many signaling pathways. Li1 is a dominant mutant that affects the reproductive growth and vegetative growth throughout the cotton lifecycle. Therefore, we inferred that the Li1 mutant phenotype was caused by a critical gene, a mutation in which influences many other genes directly and indirectly involved in cotton energy metabolism, plant growth, development, and morphogenesis. As a result of the Li1 mutation, many genes that are highly affected by Li1 would display complex expression patterns in different growth stages and organs. According to this hypothesis, it probably will be easy to explain why the same gene exhibited higher expression in the mutant than in the wild-type plant in one tissue but expressed an opposite trend in other tissues, which was observed in this study. Actually, this weird gene expression pattern was also noticed for some deliberately selected genes (cellulose synthase) at different time points of the fiber elongation stage, although some of them did not reach statistical significance when they were re-checked by qPCR (Gilbert et al. 2013).
Gilbert et al. (2013) delimited Li1 to a 17-Mb region between the best hits of the flanking SSR markers TMB2500 and DPL0489, which included the Li1 region flanked by P200 and P214 identified in this research. Using Affymetrix microarrays, 24 genes in this interval showed altered expression in the Li1 mutant compared with the wild-type plants, but none of them was located in the 1.27-Mb Li1 region in this study. In addition, 2-DE combined with local expressed sequence tag database-assisted tandem mass spectrometry analysis revealed 81 differentially expressed proteins from Li1 fibers that were assigned to different functional categories, which included 54 proteins that were down-regulated and 27 that were up-regulated (Zhao et al. 2010). When our previous leaf RNA-seq data were analyzed (Ding et al. 2014), most (7) of the genes predicated to have function (12, category 4) in the Li1 region showed high expression in both wild-type and mutant plants (RPKM > 150, Table 1); one marker (P254) was in the interval and six were in segment I. However, only two genes (ribosomal protein s10 gene [Rps10] and actin gene) had over twofold altered gene expression in mutant compared to wild-type plants; Rps10 was down-regulated and actin was up-regulated. Four genes (beta-tubulin 5 gene [Bt5], flap endonuclease 1-like gene, tbcc domain-containing protein 1-like gene, and ATP synthase gene [ATPS]) showed obvious differential gene expression in the Li1 mutant compared to the wild-type plant (Table 1). According to our mapping position, gene expression data, and a gene function related to fiber development, we inferred that Rps10, actin, ATPS, and Bt5 are the promising candidate genes of the Li1 locus.
The Rps10 encodes the S10 protein, which is part of the small subunit of mitochondrial ribosomes. An abnormal phenotype similar to that of the Li1 mutant was observed when this gene was silenced in Arabidopsis (Majewski et al. 2009; Schippers and Mueller-Roeber 2010). Homozygous plants presented developmentally early onset of silencing, a very efficient decrease in the level of Rps10 transcripts, and a severe and uniform phenotype, called P1. A wide variety of morphological and developmental abnormalities was also observed within the hemizygous transformants, allowing their classification into three categories: P2, P3, and P4 (Majewski et al. 2009; Schippers and Mueller-Roeber 2010). In A. thaliana, several ribosomal protein loss-of-function mutations, which affect cell division or cell expansion and consequently result in deformed leaf size and shape, have been identified (Schippers and Mueller-Roeber 2010). In our previous leaf RNA-seq analysis (Ding et al. 2014), Rps10 had about 6.5 times higher gene expression in wild-type plants than in mutant plants (RPKM: 24,513 vs. 3773), which was consistent with the results of our stem and 15 DPA fiber semi-qPCR analysis. However, in the 0 and 15 DPA ovules and 18 DPA fibers, this gene was expressed at much higher levels in mutant plants than in wild-type plants. On the basis of the altered gene expression and similar phenotype change when this gene is interrupted in Arabidopsis, Rps10 was a promising candidate of the Li1 mutant locus.
Actin protein is the raw material that is used to construct the cell skeleton (Janda et al. 2014). To investigate the role of the actin cytoskeleton during fiber development, 15 G. hirsutum actin (GhACT) cDNA clones were characterized by RNA gel blot and qPCR (Li et al. 2005). GhACT1 was expressed predominantly in fiber cells. RNA interference of GhACT1 caused a significant decrease in its mRNA and protein levels, and it disrupted the actin cytoskeleton network in fibers. These results suggested that GhACT1 played an important role in fiber elongation. In another study, a number of cytoskeleton-related proteins showed a remarkable decrease in protein abundance in the Li1 fibers through a comparative proteomic analysis between the Li1 mutant and its wild-type counterpart (Zhao et al. 2010). Accordingly, the architecture of the actin cytoskeleton was severely deformed, the microtubule organization was moderately altered, and there was a dramatic disruption of vesicle trafficking (Zhao et al. 2010). In our study, a previously unidentified Gossypium actin gene was identified. Previous RNA-seq data and semi-qPCR demonstrated that this gene was significantly up-regulated in the Li1 mutant leaves and 10 and 18 DPA fibers; in contrast, this gene was obviously down-regulated in Li1 mutant stem and 18-DPA ovule. We believed that the actin gene contributes to the housekeeping functions in plants, and that any unusual expression in the mutant would lead to a failed cell skeleton organization. Therefore, the abnormal expression of this gene could cause the Li1 mutant’s phenotype in leaf, stem, and fibers.
F1F0-ATP synthase catalyzes the synthesis of ATP from ADP and phosphate in mitochondria (Stock et al. 1999). The δ1 subunit of mitochondrial ATP synthase was significantly up-regulated during cotton fiber cell elongation according to microarray and qPCR (Pang et al. 2010). Both the relative level and duration of GhATPδ1 transcripts correlated positively with the final fiber length of different cotton germplasms. Elongating fiber cells had a significantly elevated ATP to ADP ratio, suggesting that a higher energy input is required for primary fiber cell wall formation and elongation (Pang et al. 2010). The ATPS is mainly down-regulated in several Li1 mutant tissues such as stem, ovules at 10 DPA and 18 DPA, and fibers at 15 DPA compared to the wild-type tissues. Although altered gene expression did not reach a twofold difference, this result together with its mapping location suggests that ATPS might be related to the Li1 candidate locus.
Microtubules play an important role in plant cell morphology and development (Liew et al. 2015). For example, microtubule depolymerization by a specific antagonist causes the loss of directionality of root hair growth (Bibikova et al. 1999). Microtubule reorientation is the key in changing the growth orientation, leading to Arabidopsis trichome branching (Mathur and Chua 2000). Root hair and trichome are tissues that develop from epidermal cells-like cotton fibers. Transcription profiling revealed that nine GhTUBs, including GhTUB1, were highly expressed in elongating fiber cells compared to their expression in fuzzless–lintless mutant ovules (He et al. 2008). In addition, seven of nine TUAs were highly expressed in developing fibers (Li et al. 2007). Bt5 was in the interval segment, and it was up-regulated nearly twofold in the Li1 mutant leaf compared to the wild-type leaf according to RNA-seq analysis. These results suggested that this gene could be a Li1 mutant candidate gene.
Lethal function of Li1
Li1 has several diverse functions that affect the normal growth and development of almost all cotton organs, including leaf, stem, petal, and boll (Griffee and Ligon 1929; Kohel et al. 1992). The Li1 homozygous mutation also prohibits the regular development of embryos, resulting in incompletely developed embryos that are believed to be lethal. In this study, we found that the mutant plants that were homozygous for the markers that co-segregated with Li1 generally grew much slower than the mutant plants with co-segregating heterozygous markers. Therefore, we deduced that not all homozygous embryos were lethal and about 14 % of mutant F2 plants (26/186, Table 3) were thought to have the homozygous genotype (LiLi). The homozygous and heterozygous Li1 mutant plants that were identified using co-segregating markers can be explored to clone the candidate gene by comparing the sequences of the two types of mutant plants. These plants also will be very useful to study the difference in gene expression and plant tissue structure, which will facilitate the candidate gene identification and functional confirmation.
References
Arpat AB, Waugh M, Sullivan JP, Gonzales M, Frisch D, Main D, Wood T, Leslie A, Wing RA, Wilkins TA (2004) Functional genomics of cell elongation in developing cotton fibers. Plant Mol Biol 54:911–929
Bibikova TN, Blancaflor EB, Gilroy S (1999) Microtubules regulate tip growth and orientation in root hairs of Arabidopsis thaliana. Plant J 17:657–665
Bolton JJ, Soliman KM, Wilkins TA, Jenkins JN (2009) Aberrant expression of critical genes during secondary cell wall biogenesis in a cotton mutant, Ligon lintless-1 (Li-1). Comp Funct Genomics. doi:10.1155/2009/659301
Cai C, Tong X, Liu F, Lv F, Wang H, Zhang T, Guo W (2013) Discovery and identification of a novel Ligon lintless-like mutant (Lix) similar to the Ligon lintless (Li1) in allotetraploid cotton. Theor Appl Genet 126:963–970
Ding M, Jiang Y, Cao Y, Lin L, He S, Zhou W, Rong J (2014) Gene expression profile analysis of Ligon lintless-1 (Li1) mutant reveals important genes and pathways in cotton leaf and fiber development. Gene 535:273–285
Gilbert MK, Turley RB, Kim HJ, Li P, Thyssen G, Tang Y, Delhom CD, Naoumkina M, Fang DD (2013) Transcript profiling by microarray and marker analysis of the short cotton (Gossypium hirsutum L.) fiber mutant Ligon lintless-1 (Li1). BMC Genom 14:403
Gilbert MK, Kim HJ, Tang Y, Naoumkina M, Fang DD (2014) Comparative transcriptome analysis of short fiber mutants Ligon-lintless 1 and 2 reveals common mechanisms pertinent to fiber elongation in cotton (Gossypium hirsutum L.). PLoS ONE 9:e95554
Griffee F, Ligon LL (1929) Occurrence of lintless cotton plants and the inheritance of the character lintless. Agron J 21:711–717
Guan X, Lee JJ, Pang M, Shi X, Stelly DM, Chen ZJ (2011) Activation of Arabidopsis seed hair development by cotton fiber-related genes. PLoS One 6:e21301
He XC, Qin YM, Xu Y, Hu CY, Zhu YX (2008) Molecular cloning, expression profiling, and yeast complementation of 19 β-tubulin cDNAs from developing cotton ovules. J Exp Bot 59:2687–2695
He S, Zheng Y, Chen A, Ding M, Lin L, Cao Y, Zhou W, Rong J, Jenkins J (2013) Converting restriction fragment length polymorphism to single-strand conformation polymorphism markers and its application in the fine mapping of a trichome gene in cotton. Plant Breed 132:337–343
Janda M, Matoušková J, Burketová L, Valentová O (2014) Interconnection between action cytoskeleton and plant defense signaling. Plant Signal Behav 9:e976486
Jiang J, Zhang T (2003) Extraction of total RNA in cotton tissues with CTAB-acidic phenolic method. Cotton Sci 15:166–167
Karaca M, Saha S, Jenkins JN, Zipf A, Kohel R, Stelly DM (2002) Simple sequence repeat (SSR) markers linked to the Ligon lintless (Li1) mutant in cotton. J Hered 93:221–224
Kohel RJ (1972) Linkage tests in upland cotton, Gossypium hirsutum L. Crop Sci 12:66–69
Kohel RJ, Narbuth EV, Benedict CR (1992) Fiber development of Ligon lintless-2 mutant of cotton. Crop Sci 32:733–735
Kohel RJ, Benedict CR, Jividen GM (1993) Incorporation of [14C] glucose into crystalline cellulose in aberrant fibers of a cotton mutant. Crop Sci 33:1036–1040
Kohel RJ, Stelly DM, Yu J (2002) Tests of six cotton (Gossypium hirsutum L.) mutants for association with aneuploids. J Hered 93:130–132
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lee JJ, Hassan OS, Gao W, Wei NE, Kohel RJ, Chen XY, Payton P, Sze SH, Stelly DM, Chen ZJ (2006) Developmental and gene expression analyses of a cotton naked seed mutant. Planta 223:418–432
Lee JJ, Woodward AW, Chen ZJ (2007) Gene expression changes and early events in cotton fibre development. Ann Bot 100:1391–1401
Li XB, Fan XP, Wang XL, Cai L, Yang WC (2005) The cotton ACTIN1 gene is functionally expressed in fibers and participates in fiber elongation. Plant Cell 17:859–875
Li L, Wang XL, Huang GQ, Li XB (2007) Molecular characterization of cotton GhTUA9 gene specifically expressed in fibre and involved in cell elongation. J Exp Bot 58:3227–3238
Liew KM, Xiang P, Zhang LW (2015) Mechanical properties and characteristics of microtubules: a review. Compos Struct 124:98–108
Liu K, Sun J, Yao L, Yuan Y (2012) Transcriptome analysis reveals critical genes and key pathways for early cotton fiber elongation in Ligon lintless-1 mutant. Genomics 100:42–50
Majewski P, Wołoszyńska M, Jańska H (2009) Developmentally early and late onset of Rps10 silencing in Arabidopsis thaliana: genetic and environmental regulation. J Exp Bot 60:1163–1178
Mathur J, Chua NH (2000) Microtubule stabilization leads to growth reorientation in Arabidopsis trichomes. Plant Cell 12:465–477
Naoumkina M, Thyssen GN, Fang DD (2015) RNA-seq analysis of short fiber mutants Ligon-lintless-1 (Li 1) and -2 (Li 2) revealed important role of aquaporins in cotton (Gossypium hirsutum L.) fiber elongation. BMC Plant Biol 15(1):65
Narbuth EV, Kohel RJ (1990) Inheritance and linkage analysis of a new fiber mutant in cotton. J Hered 81:131–133
Pang Y, Wang H, Song WQ, Zhu YX (2010) The cotton ATP synthase δ1 subunit is required to maintain a higher ATP/ADP ratio that facilitates rapid fibre cell elongation. Plant Biol 12:903–909
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, Llewellyn D, Showmaker KC, Shu S, Udall J, Yoo MJ, Byers R, Chen W, Doron-Faigenboim A, Duke MV, Gong L, Grimwood J, Grover C, Grupp K, Hu G, Lee TH, Li J, Lin L, Liu T, Marler BS, Page JT, Roberts AW, Romanel E, Sanders WS, Szadkowski E, Tan X, Tang H, Xu C, Wang J, Wang Z, Zhang D, Zhang L, Ashrafi H, Bedon F, Bowers JE, Brubaker CL, Chee PW, Das S, Gingle AR, Haigler CH, Harker D, Hoffmann LV, Hovav R, Jones DC, Lemke C, Mansoor S, ur Rahman M, Rainville LN, Rambani A, Reddy UK, Rong JK, Saranga Y, Scheffler BE, Scheffler JA, Stelly DM, Triplett BA, Van Deynze A, Vaslin MF, Waghmare VN, Walford SA, Wright RJ, Zaki EA, Zhang T, Dennis ES, Mayer KF, Peterson DG, Rokhsar DS, Wang X, Schmutz J (2012) Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492:423–427
Qin YM, Zhu YX (2011) How cotton fibers elongate: a tale of linear cell-growth mode. Curr Opin Plant Biol 14:106–111
Rong JK, Abbey C, Bowers JE, Brubaker CL, Chang C, Chee PW, Delmonte TA, Ding XL, Garza JJ, Marler BS, Park CH, Pierce GJ, Rainey KM, Rastogi VK, Schulze SR, Trolinder NL, Wendel JF, Wilkins TA, Williams-Coplin TD, Wing RA, Wright RJ, Zhao XP, Zhu LH, Paterson AH (2004) A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166:389–417
Rong J, Pierce GJ, Waghmare VN, Rogers CJ, Desai A, Chee PW, May OL, Gannaway JR, Wendel JF, Wilkins TA, Patersn AH (2005) Genetic mapping and comparative analysis of seven mutants related to seed fiber development in cotton. Theor Appl Genet 111:1137–1146
Schippers JHM, Mueller-Roeber B (2010) Ribosomal composition and control of leaf development. Plant Sci 179:307–315
Shangguan X, Yu N, Wang L, Chen X (2010) Recent advances in molecular biology research on cotton fiber development cotton. Springer, Berlin, pp 161–175
Stock D, Leslie AG, Walker JE (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286:1700–1705
Sun YL, Jia YH, He SP, Zhou ZL, Sun JL, Pang BY, Du XM (2012) Genetic analysis of fuzzless in cotton germplasm. Hereditas (Beijing) 34:1073–1078
Turley RB, Kloth RH (2002) Identification of a third fuzzless seed locus in upland cotton (Gossypium hirsutum L.). J Hered 93:359–364
Turley RB, Kloth RH (2008) The inheritance model for the fiberless trait in upland cotton (Gossypium hirsutum L.) line SL1-7-1: variation on a theme. Euphytica 164:123–132
Turley R, Vaughn K, Scheffler J (2007) Lint development and properties of fifteen fuzzless seed lines of Upland cotton (Gossypium hirsutum L.). Euphytica 156:57–65
Zhao PM, Wang LL, Han LB, Wang J, Yao Y, Wang HY, Du XM, Luo YM, Xia GX (2010) Proteomic identification of differentially expressed proteins in the Ligon lintless mutant of upland cotton (Gossypium hirsutum L.). J Proteome Res 9:1076–1087
Zhou W, Huang C, Gong Y, Feng Q, Gao F (2010) Molecular cloning and expression analysis of an ANS gene encoding anthocyanidin synthase from purple-fleshed sweet potato [Ipomoea batatas (L.) Lam]. Plant Mol Biol Rep 28:112–121
Acknowledgments
Dr. Xiongming Du of the Cotton Research Institute, Chinese Academy of Agricultural Sciences (CRI-CAAS) at Anyang, China, kindly provided all cotton materials including the Li1 mutant. We sincerely express our thanks to an anonymous reviewer for his/her helpful comments, suggestions, and corrections.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This study was funded jointly by the National High Technology Research and Development Program of China (863 Program, Grant No. 2011AA100202), the National Natural Science Foundation of China (Grant Nos. 31200909 and 31301372), and the State Key Laboratory of Cotton Biology Open Fund (CB2014A06) to Yurong Jiang.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Zhang
Y. Jiang and M. Ding have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jiang, Y., Ding, M., Cao, Y. et al. Genetic fine mapping and candidate gene analysis of the Gossypium hirsutum Ligon lintless-1 (Li1) mutant on chromosome 22(D). Mol Genet Genomics 290, 2199–2211 (2015). https://doi.org/10.1007/s00438-015-1070-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1070-2