
Abstract
The Lesion Simulating Disease (LSD) genes encode a family of zinc finger proteins that play a role in programmed cell death (PCD) and other biological processes, such as plant growth and photosynthesis. In the present study, we report the reconstruction of the evolutionary history of the LSD gene family in Viridiplantae. Phylogenetic analysis revealed that the monocot and eudicot genes were distributed along the phylogeny, indicating that the expansion of the family occurred prior to the diversification between these clades. Sequences encoding proteins that present one, two, or three LSD domains formed separate groups. The secondary structure of these different LSD proteins presented a similar composition, with the β-sheets being their main component. The evolution by gene duplication was identified only to the genes that contain three LSD domains, which generated proteins with equal structure. Moreover, genes encoding proteins with one or two LSD domains evolved as single-copy genes and did not result from loss or gain in LSD domains. These results were corroborated by synteny analysis among regions containing paralogous/orthologous genes in Glycine max and Populus trichocarpa. The Ka/Ks ratio between paralogous/orthologous genes revealed that a subfunctionalization process possibly could be occurring with the LSD genes, explaining the involvement of LSD members in different biological processes, in addition to the negative regulation of PCD. This study presents important novelty in the evolutionary history of the LSD family and provides a basis for future research on individual LSD genes and their involvement in important pathway networks in plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The sessile nature of plants means that they are able to adjust their metabolic processes according to environmental changes. During the signaling of stress responses, which trigger changes at the transcriptome, cellular, and physiological levels (Atkinson and Urwin 2012), one of the well-characterized reactions is known as the hypersensitive response (HR), which triggers the network of programmed cell death (PCD) (van Doorn et al. 2011). PCD regulates the growth and spread of lesions at the initial site of damage and in the cells that surround this area (Mur et al. 2008; Rusterucci et al. 2001). Efforts to elucidate the genes that mediate the PCD processes have resulted in the identification of the Lesion Simulating Disease (LSD) protein family. The LSD family was initially identified in Arabidopsis thaliana by the analysis of mutants that spontaneously form necrotic lesions in the absence of pathogenic infection (Dietrich et al. 1994). Genes belonging to this family negatively regulate PCD under stress conditions (Dietrich et al. 1997; Epple et al. 2003) and are exclusive to Viridiplantae (Cabreira et al. 2013). The A. thaliana LSD1 gene (AtLSD1) is the well-characterized member of this family and acts as a cellular hub that makes a central contribution to the oxidative stress response (Kaminaka et al. 2006). This gene enhances ROS (reactive oxygen species)-scavenging capacity in the signaling pathway that is related to oxidative stress (Jabs et al. 1996; Kliebenstein et al. 1999; Mateo et al. 2004; Mullineaux and Baker 2010). Together with EDS1 (Enhanced disease susceptibility1) and PAD4 (Phytoalexin deficient4), AtLSD1 acts as an ROS/ethylene homeostatic switch, controlling light acclimation and pathogen defense (Muhlenbock et al. 2008). Moreover, AtLSD1 regulates acclimation to excess excitation energy (Mateo et al. 2004), the response to cold stress (Huang et al. 2010), and lysigenous aerenchyma formation under hypoxic conditions (Muhlenbock et al. 2007). The role of AtLSD1 in the regulation of cellular signaling homeostasis, photosynthesis, water use efficiency, and seed yield has recently been reported (Wituszynska et al. 2013). Additionally, the gene OsLSD1 (Oryza sativa LSD1) regulates PCD and callus differentiation (Wang et al. 2005); OsLOL2 (Oryza sativa LSD1-like 2) is involved in rice growth and disease resistance (Bhatti et al. 2008; Xu and He 2007); BohLOL1 (Bambusa oldhamii LDS1-like 1) (Yeh et al. 2011) participates in growth and biotic stress response in bamboo, and GmLSD1 to GmLSD8 (Glycine max LSD) are modulated in response to fungi and dehydration stresses (Cabreira et al. 2013). These studies have demonstrated the involvement of LSD members in important biological processes in plants, especially responses to abiotic and biotic stresses.
The LSD proteins contain exclusively the zinc finger LSD domain CxxCRxxLMYxxGASxVxCxxC (Dietrich et al. 1997). This characteristic allows differentiation between the LSD gene family and the metacaspase gene family, since metacaspase genes present one zinc finger LSD domain and the peptidase_C14 domain. The statement about which domain(s) compose the proteins is important to distinguish between these gene families. The consensus sequence of LSD domains in LSD proteins shows broad conservation (Cabreira et al. 2013) and is required for protein–protein interactions (Coll et al. 2010; He et al. 2011a, b; Kaminaka et al. 2006; Li et al. 2013). The LSD domains contain conserved cysteine residues that allow the formation of the C2C2 arrangement (Cabreira et al. 2013), essential for the nuclear localization of the protein (He et al. 2011a). The changes in LSD protein conformation mediated by the presence of a different number of LSD domains and the evolution mechanism that is involved in the generation of LSD proteins (containing one, two, or three LSD domains) remain unexplored.
In the present study, we report the phylogenetic reconstruction of the LSD gene family in Viridiplantae considering the secondary structure of proteins, chromosomal location, duplication pattern, and synteny analysis. These results provide important insights about the evolutionary history of the LSD family. These insights could be useful in further studies of PCD mechanisms in plants.
Materials and methods
LSD gene annotation and structure analysis
Previously identified sequences (Cabreira et al. 2013) were used to reconstruct an LSD phylogenetic tree. These sequences correspond to genes identified in Manihot esculenta, Ricinus communis, Linum usitatissimum, Populus trichocarpa, Medicago truncatula, Lotus japonicus, Phaseolus vulgaris, Glycine max, Cucumis sativus, Prunus persica, Malus domestica, Arabidopsis thaliana, Arabidopsis lyrata, Capsella rubella, Brassica rapa, Thellungiella halophila, Carica papaya, Citrus sinensis, Citrus clementina, Eucalyptus grandis, Vitis vinifera, Mimulus guttatus, Aquilegia caerulea, Sorghum bicolor, Zea mays, Setaria italica, Oryza sativa, Brachypodium distachyon, Selaginella moellendorffii, Physcomitrella patens, Chlamydomonas reinhardtii, and Volvox carteri. In reference to our previous annotation, the following four corrections were performed according to the recent Phytozome update (v.9.1, http://phytozome.org): (a) the sequences previously annotated as Aco1 (Aquilegia caerulea 1) and Aco2 were removed from the analysis (Aco1 is unavailable in the Phytozome, and Aco2 corresponds to an alternative transcript of Aco3), and the sequences previously named Aco3 and Aco4 were renamed as Aco1 and Aco2, respectively; (b) the sequences annotated as Ppe4 (Prunus persica 4) and Bdi5 (Brachypodium distachyon 5) were also removed because they are unavailable in the Phytozome, precluding further analysis. The sequences used for alignments are provided in Supplementary Material 1.
We analyze the intron/exon structure and the intron phase distribution using the Gene Structure Display Server (GSDS) program, developed by the Center of Bioinformatics (CBI), Peking University (Guo et al. 2007). The sequences were accessed using the Phytozome database (June/2013).
Phylogenetic analysis of the LSD genes
The complete sequences from the 113 LSD genes were aligned using the Muscle algorithm as implemented in MEGA v.5.05 (Tamura et al. 2011). The sequences were edited manually and back-translated to nucleotide sequences for analyses at the DNA level. Contiguous insertion–deletion events (indels) that had more than one base pair (bp) were treated as single mutations (Simmons and Ochoterena 2000). A Bayesian inference (BI) was generated using BEAST v. 1.4.7 (Drummond and Rambaut 2007); a run of 207 chains was performed, and the trees were sampled every 1000 generations. The Yule tree prior, the HKY substitution model, and the uncorrelated log-normal relaxed clock were used in the BEAST analysis. The TRACER v.1.4 (http://beast.bio.ed.ac.uk/Tracer) was used to check the convergence of the Monte Carlo Markov chains (MCMCs) and for adequate effective sample sizes (EES >200) after the first 10 % of the generations had been deleted as burn-in. The final joint sample was used to estimate the maximum clade credibility tree with the TreeAnnotator program, which is part of the BEAST package. The statistical support for the clades was determined by accessing the Bayesian posterior probability (PP). The trees were visualized using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
Protein secondary structure prediction
We previously identified LSD proteins containing three, two, and one LSD domain(s) (Cabreira et al. 2013). To evaluate whether proteins with a distinct number of LSD domains adopt a different structure, we analyzed the secondary structure of the LSD proteins. The analysis of protein sequences of GmaLSD1, GmaLSD8, and OsaLSD5 (Oryza sativa LSD5), which have three, two, and one LSD domain(s), respectively, was conducted using the PSIPRED secondary structure prediction server (http://bioinf.cs.ucl.ac.uk/psipred/) (Buchan et al. 2010; McGuffin et al. 2000). We used the protein that does not belong to the LSD family as a negative control for this analysis. The AthMC1 (Arabidopsis thaliana metacaspase 1_At1g02170) contains the LSD1-like zinc finger domain (characteristic of the LSD family) and the peptidase_C14 domain (characteristic of the caspase family). We analyzed the positions of the LSD domains using the SMART database (http://smart.embl-heidelberg.de/).
Chromosomal localization, duplication events, synteny, and Ka/Ks substitution rate analysis
We analyzed the chromosomal localization and the mechanisms that are involved with the LSD genes evolution. The species previously studied (Cabreira et al. 2013) and with a genome available in the PLAZA v.2.5 (http://bioinformatics.psb.ugent.be/plaza/) database were analyzed using the WGmapping tool, search id HOM000593 (July/2013).
We analyzed 100 kb syntenic regions between orthologous genes in G. max LSD (GmaLSD) and Populus trichocarpa LSD (PtrLSD). In addition, the syntenic regions that contain GmaLSD and PtrLSD paralogous genes were considered. We performed the analyses in the Genome Duplication Database (PGDD) (http://chibba.agtec.uga.edu/duplication/) (Lee et al. 2013), which uses the BLASTP tool to search for potential anchors (E <1e−5, top 5 matches) among every possible chromosome pair in multiple genomes. The homolog pairs identified are used as the input in the multiple collinearity scan (MCsan) program, and an E value <1e−10 is used as a significant cutoff (June/2013). We search the putative classification of the genes identified in the Phytozome and the PLAZA databases (July/2013). In addition, we accessed the protein similarity of duplicated genes in Phytozome (August/2013).
We analyze the ratio of the number of nonsynonymous substitutions per nonsynonymous site (Ka) to the number of synonymous substitutions per synonymous site (Ks) in the orthologous and paralogous GmaLSD and PtrLSD genes using the PGDD database (http://chibba.agtec.uga.edu/duplication/) (June/2013). In this database, the protein sequences were aligned using CLUSTALW as a guide to the CDS (coding DNA sequence) alignments by PAL2NAL (http://abacus.gene.ucl.ac.uk/software/paml.html). Finally, the Nei–Gojobori method was used to generate the Ka and Ks ratio.
Results
Genes with different LSD domain structures form separate groups along the LSD gene family evolution
The unrooted phylogenetic tree generated by Bayesian information (BI) was well supported, as show by posterior probability (PP) (Fig. 1). Two major clades of sequences were observed, the first comprising the sequences with one and two LSD domains and the second including the sequences with three LSD domains (Fig. 1). The phylogeny was structured according to the number of LSD domains encoded by the LSD genes. The Rco2 (Ricinus communis 2) gene was the unique exception to this pattern. It encodes two LSD domains and localized in the group of genes that encode proteins with three LSD domains. We do not identify clades comprising only monocot and eudicot genes. These genes were interspersed along the phylogenetic tree, forming several subclades. We found a subclade containing only LSD genes from chlorophyte, lycophyte, and moss.

Evolutionary relationships among the 113 LSD genes based on unrooted tree obtained by Bayesian inference. The PP is shown above the branches. The taxa terminologies are abbreviated using the first letter of the genus and the first two letters of the species named according to (Cabreira et al. 2013), (see Supplementary Table S2). The black shading indicates the sequences with three LSD domains. The blue shading indicates the sequences with two LSD domains. The red shading indicates the sequences with one LSD domain. The eudicot species (triple asterisk), the monocot species (double asterisk), and the basal organism (asterisk) are shown. The right side provides a detailed illustration of the relative intron/exon length and number in each gene. The intron/exon figures were generated using the GSDS program and redrawn. The orange square indicates the Rco2 sequence (color figure online)
We compare the exons/introns of the individual LSD genes (Fig. 1). Among the 113 sequences, the number of introns ranged from 0 (Ppa1 and Ppa2) to 9 (Vvi3). In general, phylogenetically related genes exhibited a similar gene intron/exon composition. In the 27 sequences of subclades A and C (two LSD domains; Fig. 1, blue genes), around 63 % contains three introns, except Sbi2, Mes4, and Csi3 (two introns); Ptr4, Lus7, Mes2, Cpa1, and Tha3 (four introns); Sit1 (six introns); and Mgu3 (seven introns). The subclade B genes (one LSD domain; Fig. 1, red genes) contain one to six introns. The subclade D (three LSD domains; Fig. 1, black genes) formed by monocots and chlorophyte, lycophyte, and moss comprised sequences that ranged from zero (Ppa1 and Ppa2) to six introns (Sit 3 and Smo1). In the 37 sequences of subclade E (three LSD domains; Fig. 1, black genes), 76 % contains five introns, except Gma6 (three introns); Bdi1, Gma7, and Aco1 (four introns); Csi1 and Lus5 (six introns); Sit5 (seven introns); and Vvi3 and Mtr2 (eight introns). In the 35 sequences of subclade F (three LSD domains; Fig. 1, black genes), around 72 % contained four introns and the sequences Csa3 (two introns); Rco2, Tha1, Bra5, Ath2, and Osa2 (three introns); Mgu2, Gma4, and Lus2 (five introns); and Vvi2 (six introns) were exceptions.
The product of RNA splicing results from the joining of a 5′ splicing site to a 3′ splicing site (Sharp 1981). The RNA splicing is an important mechanism, which can generate different intron phases and different reading frames. The reading frame is related to the position of the intron within or between codons. Introns between codons are designated phase 0, introns between the first and the second bases of a codon are designated phase 1, and introns between the second and the third bases of a codon are designated phase 2. A correlation between intron phases results in an excess of symmetric exons and symmetric exon sets were reported (Long et al. 1995). We observe that the intron phases were remarkably well conserved in the related genes. In general, considering the subclades that contain sequences with three LSD domains, subclade E had the first and second introns in phase 1, and subclade F had the first, second, and third introns in phase 1. We identified sequences with variable intron phase in subclade D (formed by monocot and chlorophyte, lycophyte, and moss). Among the sequences with two LSD domains, the majority possessed the first and second introns in phase 1.
Distinct number of LSD domains and the maintenance of secondary structure in LSD proteins
The prediction of secondary structure, which constitutes the second level of protein structure, provides the location of β-sheets or/and α-helices within a protein or protein family. To predict which structure (β-sheets or α-helices) is predominant in the chain of LSD proteins, we analyzed the amino acid sequences of three different LSD proteins. In general, the predictions were accurate and reliable, based on the confidence of prediction value represented in Fig. 2 (blue bars). We observed that β-sheets are the main component of the secondary structures of GmaLSD1 (three LSD domains), GmaLSD8 (two LSD domains), and OsaLSD5 (one LSD domains) (see Fig. 2 and Supplementary Material 2 for details). As exemplified by soybean and rice, these observations were confirmed in several species such as M. esculenta, R. communis, C. papaya, Z. mays, and S. bicolor (Supplementary Material 3). Therefore, the number of LSD domains did not alter the secondary structure of LSD proteins. AtMC1 (negative control) revealed a different protein structure than that of the LSD proteins. It has both β-sheets and α-helices, but the α-helices are the main component. We identified two β-sheets in regions that contain the zinc finger LSD domains in LSD proteins, but only one β-sheet was found in this region in the AtMC1 control.

Secondary structure of LSD proteins. GmaLSD1 (three LSD domains), GmaLSD8 (two LSD domains), and OsaLSD5 (one LSD domain) were analyzed using the PSIPRED program. AthMC1 was used as a control for the analysis. The orange line under the amino acid sequence highlights the LSD domains, and the blue line highlights the C14 domain (color figure online)
Genome distribution and expansion of LSD genes
Figure 3 illustrates the chromosomal location of LSD genes and the putative mechanism of their duplication. The complete results are presented in Supplementary Table S1. We found LSD genes distributed on various chromosomes throughout the genomes analyzed.

Analysis of chromosomal location and the mechanism of LSD gene generation in the available species using the WGmapping tool in the PLAZA database. For each species, the individual scale in Mb is shown. The black vertical line represents the chromosomes, scaffolds, or contigs and shows their number at the top. The black, red, and green horizontal lines represent the single-copy genes, the block, and the tandem-duplicated genes, respectively (color figure online)
Single LSD isoforms (without a corresponding paralogous gene) were more often found than genes subjected to block and tandem duplication processes. This was observed for the single LSD gene in Chlamydomonas reinhardtii and for the two copies of the LSD gene in chromosome 7 and 4 for S. bicolor and Z. mays, respectively. Nevertheless, we observed that the block and tandem duplication contributed substantially to LSD family expansion in species such as P. trichocarpa and G. max. These species have passed through polyploidization events.
We identified that the duplication processes were restricted to genes with three LSD domains (Supplementary Table S1). All genes that contained two or one LSD domains did not have a matching paralogous gene.
Although some gene copies were generated from tandem or block duplication, we found that some genes appeared as part of both processes, as the genes in chromosome 3 and 12 of O. sativa ssp. japonica. These data indicate that these genes are included in a set of tandem-duplicated genes that are present in a block-duplicated region.
Syntenic analysis of LSD orthologous/paralogous genes in Glycine max and Populus trichocarpa
We analyze a 100-kb syntenic region between LSD orthologous/paralogous genes. Supplementary Table S2 shows the protein family classification for all genes per syntenic region based on the Phytozome and PLAZA databases. In addition, we analyze the Ka/Ks values.
We chose two related species (soybean and poplar) with the entire genomes assembled and annotated and with a relatively large number of LSD members. Poplar and soybean have experienced different numbers of genome duplications: poplar underwent one complete genome duplication after the genome triplication event (γ) and soybean passed through two duplication events (Lin and Paterson 2011). Therefore, this analysis allows a comparison between syntenic regions after a different number of genome duplications.
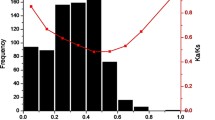
Figure 4 illustrates the syntenic region between orthologous genes in soybean (GmaLSD) compared with poplar (PtrLSD) genes. Poplar and soybean frequently contain the same protein families distributed in their syntenic regions, indicating a common origin for the chromosomes. We observed Ka/Ks ratios in orthologous genes that suggest a negative selection. Only one exception to this pattern was observed in the syntenic regions between GmaLSD3 and PtrLSD1 and GmaLSD3 and PtrLSD2. In these regions, the Ka/Ks ratio of Glyma15g22100 and Ptrichoc_0011s15820 and Glyma15g22100 and Ptrichoc001s43290 (belonging to the nitrate, formate, and iron dehydrogenase family) suggests neutral selection.

Syntenic regions between GmaLSD and PtrLSD orthologous genes analyzed in the PGDD database. The green horizontal line represents the chromosome. The blue and red vertical line and arrows represent the duplicated orthologous genes; the red line represents the sequence that was used as the search query (indicated at the top of each of the syntenic regions). The white arrow indicates that there were no duplicated genes. The gene code that was used in the PGDD is located next to the arrows. The e value for each syntenic region is shown (color figure online)
We observed three different types of block duplication between poplar and soybean (Supplementary Table S2). A small duplication block was found in the syntenic regions that contain GmaLSD1 and PtrLSD1 (78.0 % protein similarity), GmaLSD1 and PtrLSD2 (75.1 % protein similarity), GmaLSD2 and PtrLSD1 (69.3 % protein similarity), GmaLSD3 and PtrLSD1 (67.2 % protein similarity), GmaLSD3 and PtrLSD2 (59.9 % protein similarity), GmaLSD4 and PtrLSD1 (75.3 % protein similarity), GmaLSD4 and PtrLSD2 (73.0 % protein similarity), GmaLSD5 and PtrLSD3 (82.8 % protein similarity), and GmaLSD5 and PtrLSD5 (82.1 % protein similarity). A large duplication block was observed in the syntenic region that contains GmaLSD6 and PtrLSD3 (75.3 % protein similarity) and GmaLSD7 and PtrLSD3 (77.6 % protein similarity, data not shown). A huge duplication block was observed in the syntenic regions that possess GmaLSD6 and PtrLSD5 (75.3 % protein similarity), GmaLSD7 and PtrLSD5 (77.6 % protein similarity), and GmaLSD8 and PtrLSD4 (58.6 % protein similarity).
Figure 5 shows the syntenic regions between LSD paralogous genes in soybean and poplar. In accordance with the syntenic regions between the orthologous genes, we observed a high degree of conservation in the protein families. For the majority of the genes, the Ka/Ks ratio also suggested negative selection (Supplementary Table S2). Two exceptions were observed: in the syntenic region that contains GmaLSD3 and GmaLSD4 and in the region that contains Glyma09g09990 and Glyma15g22100 (nitrate, fromate, and iron dehydrogenase family). These exceptions could suggest neutral selection. The same pattern was observed for Glyma01g40750 and Glyma17g16560 (E3 ubiquitin ligase family) in the syntenic region that contains GmaLSD6 and GmaLSD7.

Syntenic regions between the GmaLSD and PtrLSD paralogous genes analyzed in the PGDD database. The green horizontal line represents the chromosome. The blue and red vertical line and arrows represent the duplicated paralogous genes; the red line represents the sequence that was used as the search query (indicated at the top of each of the syntenic regions). To illustrate the lack of duplication of GmaLSD8 and PtrLSD4, Glyma07g31590 and Ptrichoc0004s04280 were used as query sequences. The white arrow indicates that there were duplicated genes. The gene code that was used in PGDD is located next to the arrows. The e value for each syntenic region is shown. The black squares represent the lack of duplicated GmaLSD8 and PtrLSD4 genes (color figure online)
We verified two different types of duplication block in LSD paralogous genes (Supplementary Table S2). A huge duplication block was found between GmaLSD1 and GmaLSD2 (99.4 % protein similarity), GmaLSD3 and GmaLSD4 (87.8 % protein similarity), GmaLSD5 and GmaLSD6 (86.9 % protein similarity), GmaLSD6 and GmaLSD7 (78.9 % protein similarity), and PtrLSD3 and PtrLSD5 (78.2 % protein similarity). A large duplication block was observed in the syntenic region that contains GmaLSD5 and GmaLSD7 (80 % protein similarity) and PtrLSD1 and PtrLSD2 (88.3 % protein similarity).
We observed that the number of LSD domain was maintained between paralogous/orthologous genes (Figs. 4, 5). Therefore, the syntenic regions showed that duplicated LSD genes originate proteins with equal structure of domains.
Discussion
LSD gene diversification occurred before the separation of the monocots and eudicots
The phylogenetic analysis performed in this study shows that the distribution of the LSD genes among the Viridiplantae species reflects the number of LSD domains that they encode and that this distribution is well supported (Fig. 1). The proposed scenario based on the unrooted tree suggests that all genes evolved during the same time and emphasizes the idea that the common ancestor of each of these genes preceded the separation of monocots and eudicots. Additionally, the incongruence observed in the localization of Rco2 (with two LSD domains) could be explained by the finding that while other proteins with two LSD domains comprise the second and third domains (i.e., the core and C-terminal domains), Rco2 is exclusively formed by the first and third LSD domains (i.e., the N- and C-terminal domains) (data not shown). Thus, despite the absence of the second domain, Rco2 is closer to proteins that show three LSD domains.
Structural divergence between exon/intron within gene families is also a mechanism for their evolution (Li et al. 2009). The differences observed in the exon–intron structure of paralogous/orthologous genes could be explained by several mechanisms, such as exon/intron gain/loss, point mutation, insertion/deletion, exonization (a process in which an intronic or intergenic sequence becomes exonic), and pseudoexonization (the opposite process of exonization) in a DNA sequence (Xu et al. 2011). Although related paralogous/orthologous genes contain exon/intron that is conserved, events of exon/intron modifications might have occurred during the evolution of the LSD family. We observed modifications in the number of intron to specific genes, as observed in the subcluster that is contained in subclade E (between the Tha2 and Gma6 genes) (Fig. 1). The genes that belong to this subcluster contain five introns, except Mtr2, Gma7, and Gma6.
Genes that showed modifications in their intron number had also alterations in their intron phase. This pattern is remarkable in the subclade that is formed between the Tha2 and Gma6 genes (contained in subclade E). In these, Mtr2 and Gma7 showed modifications, but remained clustered with related genes. We conclude that, despite the variation, they still resemble their corresponding paralogous/orthologous genes.
In summary, we verify that the analysis of intron/exon structure of LSD genes corroborates the phylogenetic analysis. Therefore, the combination of these results suggests that all the LSD genes might have a common ancestor.
β-sheet composition in LSD proteins is maintained independent of the number of LSD domains
Protein secondary structure carries information about the structural arrangements that occur in a protein. To date, data involving the proteins of the LSD family are associated with protein–protein interactions (Coll et al. 2010; He et al. 2011b; Kaminaka et al. 2006; Li et al. 2013), the role of the zinc finger LSD domains (He et al. 2011a), and their conservation in protein sequences (Cabreira et al. 2013). These studies revealed that zinc finger LSD domains are fundamental for the functionality of these proteins.
β-sheets are formed by the pairing of multiple β-strands that are held together by typical patterns of hydrogen bonds that run in parallel or antiparallel (essential to maintain protein stability), which involve interactions between residues that are often separated by large distances along the primary sequence of proteins (Cheng and Baldi 2005). We showed that β-sheets are the main component in the assembly of these LSD proteins (Fig. 2; see also Supplementary Material 2 and Supplementary Material 3 to an overview of these results). Three sequential β-sheets (indicated by the yellow arrow) generally overlap the LSD domain position (indicated by the orange line) (Fig. 2). Regardless of the number of LSD domains (exemplified as GmLSD1, GmLSD8, and OsaLSD5), the structures of β-sheets are maintained. Thus, the high conservation of LSD domains (Cabreira et al. 2013) and β-sheets that overlap with their positions points to a possible similar protein secondary structure in all the LSD family. The results of previous studies and the present data suggest an important role for LSD domains in LSD protein structure and activity.
The majority of LSD genes are single-copy genes
In gene families, gene duplication events are fundamental for the families’ evolution because duplicated genes provide the raw materials for the generation of new gene functions (Yin et al. 2013). Duplication of genomic content can occur by many independent mechanisms, such as tandem duplication (local duplications that involve one or two genes), block/segmental duplications (duplications of subchromosomal-length regions), and whole-genome duplications (WGD) originated by polyploidy events (Flagel and Wendel 2009).
The analysis of the distribution of LSD genes reveals that they are located on various chromosomes, and several members were found on duplicated chromosomal segments (Fig. 3). Similar results were observed for the annexin gene family in Viridiplantae (Jami et al. 2012).
Block duplication process is more frequent in plants because most of them are polyploids, and numerous duplicated chromosomal blocks are maintained within their genomes (Yin et al. 2013). Several plant species, such as A. thaliana (Simillion et al. 2002), rice (Yu et al. 2005), and soybean (Schmutz et al. 2010), experienced WGD during their evolution. This phenomenon is particularly prominent in eudicot plants, which share a γ event (Lin and Paterson 2011).
The genes originated by the tandem duplication process do not present a matching paralog (Fig. 3; Supplementary Table S1), suggesting that the duplicated copies are quickly lost. The exceptions to this scenario are the tandem-duplicated genes in poplar (PtrLSD1 and PtrLSD2) and soybean (GmaLSD3 and GmaLSD4), and species that had undergone genome duplications during their evolutionary histories (Lin and Paterson 2011). Therefore, we suggested that the majority of the LSD genes are single-copy genes that were duplicated, but maintained throughout evolution in an ancestral genome. These events resulted in a current scenario that consists of a large number of orthologous LSD genes.
A common origin of LSD genes is particularly interesting for sequences that possess one and two LSD domains. Although certain sequences with three LSD domains are subject to duplications, the sequences with one or two LSD domains do not show duplicated genes (Supplementary Table S1). Synteny analysis of GmaLSD8 and PtrLSD4 (both of which present two LSD domains) supports this result (Fig. 5). These genes are located in a very large duplication block and do not have matching paralogs. This result shows that they are descendants from an ancestral genome and that their duplication did not occur after the split between these genomes or that the duplicated copy was lost during evolution.
Synteny analysis demonstrates high conservation between the related species poplar (Populus trichocarpa) and soybean (Glycine max)
Synteny is an inference that two or more chromosomes or segments are derived from a common ancestor (Lyons et al. 2008). The analysis of physical synteny may be used to explain the events that a genome has undergone prior to achieving its current structural form. These genomic events can include the evolutionary repositioning of genes responsible for shared phenotypes between two related species (McClean et al. 2010).
Although several breakpoints occur in the syntenic regions between poplar and soybean, a significant synteny is still maintained (Fig. 4). These breakpoints can be explained by the different number of duplications in these species. Thus, syntenic regions containing LSD genes identified as intraspecies (GmaLSD × GmaLSD and PtrLSD × PtrLSD) and interspecies (GmaLSD × PtrLSD) emphasize that the LSD genes originated from a common ancestor.
LSD genes with the same number of LSD domains formed syntenic regions (Figs. 4, 5). As exemplified by soybean and poplar, this observation was confirmed in several species such as M. esculenta, R. communis, C. papaya, and S. bicolor (data not shown). Therefore, these results show that the LSD genes that encode proteins with one, two, and three LSD domains are not the result of loss or gain in LSD domains. We conclude that the duplicated copies retain the original number of domains.
Although duplicated genes can undergo neofunctionalization (when one copy acquires a novel function) or subfunctionalization (in which both copies are mutated and adopt complementary functions), pseudogenization is the most common fate of the copies after the duplication process (Cagliari et al. 2011). The Ka/Ks ratio generates insights into the processes that drive the changes at the molecular level. A Ka/Ks ratio >1 shows that neofunctionalization through positive selection acted during the sequence divergence, and a Ka/Ks ratio <1 suggests that subfunctionalization through negative selection operated during the divergence (Fan et al. 2013; Zhang et al. 2002).
The results for the homologous Rxp regions in soybean suggest that smaller Ks values (0.16, 0.17) are associated with recent duplication regions and that high Ks values (0.62–0.67) are associated with ancient polyploidy events (Kim et al. 2009). Based on these data, we observed that the syntenic regions with GmaLSD1 and GmaLSD2, GmLSD3 and GmaLSD4, and GmaLSD5 and GmaLSD6 can be associated with recent duplication regions (low Ks values). Moreover, the syntenic regions with GmaLSD6 and GmaLSD7 and GmaLSD5 and GmaLSD7 generally exhibit high Ks values, suggesting an association with an ancient polyploidy event. Therefore, these results suggest that the GmaLSD genes could have been derived from different duplication events.
Because gene duplication makes a major contribution to the evolution of new gene functions, duplicated genes can experience a substantial relaxation of selection compared with unduplicated genes (Kondrashov et al. 2002). An evolutionary model of gene duplication proposed by Kondrashov et al. (2002) holds that an apparent early phase of relaxed constraint or even near-neutrality that is followed by a negative selection period occurs at the first stages after gene duplication. The model proposed that the later stages of this process most likely provide a long-term advantage by enabling the creation of new functions. In terms of the syntenic regions analyzed, the Ka/Ks ratio shows that both paralogous and orthologous sequences appear to be evolving under similar levels of negative selection (Supplementary Table S2). This finding indicates that a subfunctionalization process probably could be occurring in LSD genes. In fact, we observed different expression patterns of GmaLSD genes in various organs and plants subjected to Phakopsora pachyrhizi infection and dehydration (Cabreira et al. 2013). Thus, the differential expression together with the low Ka/Ks supports a possible subfunctionalization of these genes. Previous studies have shown that the prevalence of variable expression in soybean after polyploidization has revealed that several paralogous genes undergo subfunctionalization (Fan et al. 2013; Roulin et al. 2013). Accordingly, our results reinforce the theory of the retention of duplicate genes through subfunctionalization, which may constitute a transitional step to neofunctionalization (Roulin et al. 2013).
Figure 6 summarizes and provides a comprehensive overview of the results. In summary, we state that the diversification of the LSD family precedes the separation of monocots and eudicots. We observed that the different number of LSD domains does not alter the protein secondary structure, which suggests a similar conformation in all LSD proteins. Proteins that are composed of one, two, and three domains are not a result of a loss or gain in LSD domains. Duplicated LSD genes generated copies with equal numbers of LSD domains. The majority of the LSD genes are maintained as a single copy in the different species and are orthologous in several genomes. Together, these results suggest the existence of a common ancestor of the LSD proteins. Finally, although the majority of members are related to the negative regulation of PCD, LSD members probably could be involved in other biological processes, due the occurrence of functionalization processes.

Model of possible LSD family evolution. The rectangles represent genes encoding proteins with three LSD domains (black), two LSD domains (blue), and one LSD domain (red). The dark pink and green rectangles represent the mutated copy with complementary functions. The black rectangle shows the copy that keeps the original function, and the brown rectangle indicates the copy that acquires a novel function (color figure online)
References
Atkinson NJ, Urwin PE (2012) The interaction of the plant biotic and abiotic stresses: from genes to the field. J Exp Bot 63:3523–3543
Bhatti KH, Xu C, Wu J, He C (2008) Overexpression of rice OsLOL2 gene confers disease resistance in tobacco to Pseudomonas syringae pv. tabaci. Prog Nat Sci 18:807–812
Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT (2010) Protein annotation and modelling servers at University College London. Nucleic Acids Res 38:W563–W568
Cabreira C, Cagliari A, Bucker-Neto L, Wiebke-Strohm B, de Freitas LB, Marcelino-Guimaraes FC, Nepomuceno AL, Margis-Pinheiro MM, Bodanese-Zanettini MH (2013) The Lesion Simulating Disease (LSD) gene family as a variable in soybean response to Phakopsora pachyrhizi infection and dehydration. Funct Integr Genomics 13(3):323–338
Cagliari A, Turchetto-Zolet AC, Maraschin FS, Loss G, Margis R, Margis-Pinheiro M (2011) The evolutionary history of CBF transcription factors: gene duplication of CCAAT–binding factors NF-Y in plants In: Felix F (ed) Gene duplication, vol 1. InTech, Rijeka, pp 1–27
Cheng J, Baldi P (2005) Three-stage prediction of protein beta-sheets by neural networks, alignments and graph algorithms. Bioinformatics 21(Suppl 1):i75–i84
Coll NS, Vercammen D, Smidler A, Clover C, Van Breusegem F, Dangl JL, Epple P (2010) Arabidopsis type I metacaspases control cell death. Science 330(6009):1393–1397
Dietrich RA, Delaney TP, Uknes SJ, Ward ER, Ryals JA, Dangl JL (1994) Arabidopsis mutants simulating disease resistance response. Cell 77(4):565–577
Dietrich RA, Richberg MH, Schmidt R, Dean C, Dangl JL (1997) A novel zinc finger protein is encoded by the Arabidopsis LSD1 gene and functions as a negative regulator of plant cell death. Cell 88(5):685–694
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Epple P, Mack AA, Morris VR, Dangl JL (2003) Antagonistic control of oxidative stress-induced cell death in Arabidopsis by two related, plant-specific zinc finger proteins. Proc Natl Acad Sci USA 100(11):6831–6836
Fan C, Wang X, Hu R, Wang Y, Xiao C, Jiang Y, Zhang X, Zheng C, Fu YF (2013) The pattern of Phosphate transporter 1 genes evolutionary divergence in Glycine max L. BMC Plant Biol 13:48
Flagel LE, Wendel JF (2009) Gene duplication and evolutionary novelty in plants. New Phytol 183(3):557–564
Guo AY, Zhu QH, Chen X, Luo JC (2007) GSDS: a gene structure display server. Yi Chuan 29(8):1023–1026
He S, Huang K, Zhang X, Yu X, Huang P, An C (2011a) The LSD1-type zinc finger motifs of Pisum sativa LSD1 are a novel nuclear localization signal and interact with importin alpha. PLoS One 6(7):e22131
He S, Tan G, Liu Q, Huang K, Ren J, Zhang X, Yu X, Huang P, An C (2011b) The LSD1-interacting protein GILP is a LITAF domain protein that negatively regulates hypersensitive cell death in Arabidopsis. PLoS One 6(4):e18750
Huang X, Li Y, Zhang X, Zuo J, Yang S (2010) The Arabidopsis LSD1 gene plays an important role in the regulation of low temperature-dependent cell death. New Phytol 187(2):301–312
Jabs T, Dietrich RA, Dangl JL (1996) Initiation of runaway cell death in an Arabidopsis mutant by extracellular superoxide. Science 273(5283):1853–1856
Jami SK, Clark GB, Ayele BT, Ashe P, Kirti PB (2012) Genome-wide comparative analysis of annexin superfamily in plants. PLoS One 7(11):e47801
Kaminaka H, Nake C, Epple P, Dittgen J, Schutze K, Chaban C, Holt BF III, Merkle T, Schafer E, Harter K, Dangl JL (2006) bZIP10-LSD1 antagonism modulates basal defense and cell death in Arabidopsis following infection. EMBO J 25(18):4400–4411
Kim KD, Shin JH, Van K, Kim DH, Lee SH (2009) Dynamic rearrangements determine genome organization and useful traits in soybean. Plant Physiol 151(3):1066–1076
Kliebenstein DJ, Dietrich RA, Martin AC, Last RL, Dangl JL (1999) LSD1 regulates salicylic acid induction of copper zinc superoxide dismutase in Arabidopsis thaliana. Mol Plant Microbe Interact 12(11):1022–1026
Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV (2002) Selection in the evolution of gene duplications. Genome Biol 3(2):RESEARCH0008
Lee TH, Tang H, Wang X, Paterson AH (2013) PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res 41:D1152–D1158
Li W, Liu B, Yu L, Feng D, Wang H, Wang J (2009) Phylogenetic analysis, structural evolution and functional divergence of the 12-oxo-phytodienoate acid reductase gene family in plants. BMC Evol Biol 9:90
Li Y, Chen L, Mu J, Zuo J (2013) Lesion Simulating Disease1 interacts with catalases to regulate hypersensitive cell death in Arabidopsis. Plant Physiol 163(2):1059–1070
Lin L, Paterson AH (2011) Size variation in homologous segments across divergent plant genomes. Mob Genet Elements 1(2):92–96
Long M, de Souza SJ, Gilbert W (1995) Intron phase correlations and the evolution of intron/exon structure of genes. Proc Natl Acad Sci USA 92:1249512499
Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Wang X, Bowers J, Paterson A, Lisch DMF (2008) Finding and comparing syntenic regions among Arabidopsis and the Outgroups Papaya, Poplar, and Grape: CoGe with Rosids. Bioinformatics 148:1772–1781
Mateo A, Muhlenbock P, Rusterucci C, Chang CC, Miszalski Z, Karpinska B, Parker JE, Mullineaux PM, Karpinski S (2004) Lesion Simulating Disease1 is required for acclimation to conditions that promote excess excitation energy. Plant Physiol 136(1):2818–2830
McClean PE, Mamidi S, McConnell M, Chikara S, Lee R (2010) Synteny mapping between common bean and soybean reveals extensive blocks of shared loci. BMC Genom 11:184
McGuffin LJ, Bryson K, Jones D (2000) The PSIPRED protein structure prediction server. Bioinformatics 16(4):404–405
Muhlenbock P, Plaszczyca M, Mellerowicz E, Karpinski S (2007) Lysigenous aerenchyma formation in Arabidopsis is controlled by Lesion Simulating Disease1. Plant Cell 19(11):3819–3830
Muhlenbock P, Szechynska-Hebda M, Plaszczyca M, Baudo M, Mateo A, Mullineaux PM, Parker JE, Karpinska B, Karpinski S (2008) Chloroplast signaling and Lesion Simulating Disease1 regulate crosstalk between light acclimation and immunity in Arabidopsis. Plant Cell 20(9):2339–2356
Mur LA, Kenton P, Lloyd AJ, Ougham H, Prats E (2008) The hypersensitive response; the centenary is upon us but how much do we know? J Exp Bot 59(3):501–520
Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, Stacey G, Doerge RW, Jackson SA (2013) The fate of duplicated genes in a polyploid plant genome. Plant J 73:143–153
Rusterucci C, Aviv DH, Holt BF III, Dangl JL, Parker JE (2001) The disease resistance signaling components EDS1 and PAD4 are essential regulators of the cell death pathway controlled by LSD1 in Arabidopsis. Plant Cell 13(10):2211–2224
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 463(7278):178–183
Sharp PA (1981) Speculations on RNA splicing. Cell 23:643–646
Simillion C, Vandepoele K, Van Montagu MC, Zabeau M, Van de Peer Y (2002) The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci USA 99(21):13627–13632
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Syst Biol 49(2):369–381
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
van Doorn WG, Beers EP, Dangl JL, Franklin-Tong VE, Gallois P, Hara-Nishimura I, Jones AM, Kawai-Yamada M, Lam E, Mundy J, Mur LAJ, Petersen M, Smertenko A, Taliansky M, Van Breusegem F, Wolpert T, Woltering E, Zhivotovsky B, Bozhkov PV (2011) Morphological classification of plant cell deaths. Cell Death Differ 18:1241–1246
Wang L, Pei Z, Tian Y, He C (2005) OsLSD1, a rice zinc finger protein, regulates programmed cell death and callus differentiation. Mol Plant Microbe Interact 18(5):375–384
Wituszynska W, Slesak I, Vanderauwera S, Szechynska-Hebda M, Kornas A, Van Der Kelen K, Muhlenbock P, Karpinska B, Mackowski S, Van Breusegem F, Karpinski S (2013) Lesion Simulating Disease1, enhanced disease susceptibility1, and phytoalexin deficient 4 conditionally regulate cellular signaling homeostasis, photosynthesis, water use efficiency, and seed yield in Arabidopsis. Plant Physiol 161(4):1795–1805
Xu C, He C (2007) The rice OsLOL2 gene encodes a zinc finger protein involved in rice growth and disease resistance. Mol Genet Genomics 278(1):85–94
Xu G, Guo C, Shan H, Kong H (2011) Divergence of duplicate genes in exon-intron structure. Proc Natl Acad Sci USA 109(4):187–1192
Yin G, Xu H, Xiao S, Qin Y, Li Y, Yan Y, Hu Y (2013) The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol 13(1):148
Yu J, Wang J, Lin W, Li S, Li H, Zhou J, Ni P, Dong W, Hu S, Zeng C, Zhang J, Zhang Y, Li R, Xu Z, Li X, Zheng H, Cong L, Lin L, Yin J, Geng J, Li G, Shi J, Liu J, Lv H, Li J, Deng Y, Ran L, Shi X, Wang X, Wu Q, Li C, Ren X, Li D, Liu D, Zhang X, Ji Z, Zhao W, Sun Y, Zhang Z, Bao J, Han Y, Dong L, Ji J, Chen P, Wu S, Xiao Y, Bu D, Tan J, Yang L, Ye C, Xu J, Zhou Y, Yu Y, Zhang B, Zhuang S, Wei H, Liu B, Lei M, Yu H, Li Y, Xu H, Wei S, He X, Fang L, Huang X, Su Z, Tong W, Tong Z, Ye J, Wang L, Lei T, Chen C, Chen H, Huang H, Zhang F, Li N, Zhao C, Huang Y, Li L, Xi Y, Qi Q, Li W, Hu W, Tian X, Jiao Y, Liang X, Jin J, Gao L, Zheng W, Hao B, Liu S, Wang W, Yuan L, Cao M, McDermott J, Samudrala R, Wong GK, Yang H (2005) The genomes of Oryza sativa: a history of duplications. PLoS Biol 3(2):e38
Zhang L, Vision TJ, Gaut BS (2002) Patterns of nucleotide substitution among simultaneously duplicated gene pairs in Arabidopsis thaliana. Mol Biol Evol 19(9):464–1473
Acknowledgment
This study was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Programa de Pós-Graduação em Genética e Biologia Molecular da Universidade Federal do Rio Grande do Sul (PPGBM/UFRGS), GENOSOJA/CNPq, and BIOTECSUR II/MCT.
Conflict of interest
The authors declare that they have no conflicts of interest.
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by B. Yang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cabreira, C., Cagliari, A., Bücker-Neto, L. et al. The phylogeny and evolutionary history of the Lesion Simulating Disease (LSD) gene family in Viridiplantae. Mol Genet Genomics 290, 2107–2119 (2015). https://doi.org/10.1007/s00438-015-1060-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1060-4