
Abstract
Objective
Lung cancer (LC) remains a threatening health issue worldwide. Methyltransferase-like protein 3 (METTL3) is imperative in carcinogenesis via m6A modification of microRNAs (miRNAs). This study estimated the effect of METTL3 in LC by regulating m6A methylation-mediated pri-miR-663 processing.
Methods
miR-663 expression in 4 LC cell lines and normal HBE cells was determined using RT-qPCR. A549 and PC9 LC cells selected for in vitro studies were transfected with miR-663 mimics or inhibitor. Cell viability, migration, invasion, proliferation, and apoptosis were detected by CCK-8, Transwell, EdU, and flow cytometry assays. The downstream target genes and binding sites of miR-663 were predicted via Starbase database and validated by dual-luciferase assay. LC cells were delivered with oe-METTL3/sh-METTL3. Crosslinking between METTL3 and DGCR8 was verified by co-immunoprecipitation. Levels of m6A, miR-663, and pri-miR-663 were measured by m6A dot blot assay and RT-qPCR. m6A modification of pri-miR-663 was verified by Me-RIP assay. Finally, the effects of METTL3 in vivo were ascertained by tumor xenograft in nude mice.
Results
miR-663 was upregulated in LC cells, and miR-663 overexpression promoted cell proliferation, migration, invasion, and inhibited apoptosis, but miR-663 knockdown exerted the opposite effects. miR-663 repressed SOCS6 expression. SOCS6 overexpression annulled the promotion of miR-663 on LC cell growth. METTL3 bound to DGCR8, and METTL3 silencing elevated the levels of pri-miR-663 and m6A methylation-modified pri-miR-663, and suppressed miR-663 maturation and miR-663 expression. METTL3 facilitated tumor growth in mice through the miR-663/SOCS6 axis.
Conclusion
METTL3 promotes LC progression by accelerating m6A methylation-mediated pri-miR-663 processing and repressing SOCS6.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer (LC) ranks the most prevailing and fatal malignancy, accounting for 11.6% morbidity and 18.4% mortality (Chen et al. 2020a; Xu et al. 2020b). Smoking, environmental, and occupational factors are primary contributors to the occurrence of LC (Liu et al. 2021). Despite recent advancements in treatment, the 5-year overall survival is merely estimated to be around 17% (Puri et al. 2017). Herein, extensive research is necessitated to explore biological characteristics and nosogenesis of LC and to find molecular markers.
microRNAs (miRNAs) can mediate numerous intracellular pathways in cancers by functioning as tumor-suppressive or carcinogenic factors (Seo et al. 2019). In particular, miR-663 can maintain non-small cell lung carcinoma (NSCLC) by repressing mitochondrial outer membrane permeabilization via PUMA/BBC3 and BTG2 (Fiori et al. 2018). The regulatory mechanism of its participation in LC remains to be explored. Moreover, miRNAs exert vital biological functions by impeding expressions of their target genes (Li et al. 2019). miR-1260b modulates NSCLC cell cycle and growth by targeting suppressor of cytokine signaling 6 (SOCS6) (Xia et al. 2019). In addition, Starbase bioinformatics predicted a potential binding between miR-663 and SOCS6. Therefore, we proposed the hypothesis that miR-663 may mediate SOCS6 in LC to further elucidate the mechanism by which miR-663 regulates LC.
N6-methyladenosine (m6A) methylation is considered the most copious modification existing in eukaryotic mRNAs (Barbieri et al. 2017; Wang et al. 2020). The dysregulation of m6A methylation modification may affect the mRNA processing, degradation, and translation, contributing to the activation of oncogenes and inactivation of anti-oncogenes, which is tightly associated with the development of malignant tumors and drug resistance (Zeng et al. 2020). Methyltransferase-like 3 (METTL3) is conceived as the RNA methyltransferase involved in mRNA biosynthesis, decay, and translation control via m6A modification (Wei et al. 2019). METTL3 imposes a promotional action in gastric cancer by mediating m6A modification of HDGF mRNA (Wang et al. 2020) and manipulates epithelial–mesenchymal transition (EMT), migration, and invasion of breast cancer through m6A modification of lncRNA MALAT1 (Zhao et al. 2021). METTL3 is unveiled to be upregulated in LC tissues (Chen et al. 2020b). Moreover, METTL3 and m6A RNA modification are enhanced during TGF-β-induced EMT of LC cells (Wanna-Udom et al. 2020).
m6A modification of diverse miRNAs is implicated in cancer stemness and tumorigenesis (Wu 2020). Specifically, m6A-elicited miR-143-3p reinforces the brain metastasis of LC through the regulation of vasohibin-1 (Wang et al. 2019a). Furthermore, METTL3 facilitates the maturation of miR-126-5p by regulating m6A modification of pri-miR-126-5p (Bi et al. 2021). METTL3 exerts a carcinogenic role by interacting with DGCR8 and strengthening pri-miR221/222 processing in the m6A-dependent manner (Han et al. 2019). Intriguingly, the regulation of METTL3 is also elucidated in miR-663b processing (Alarcon et al. 2015). Based on the aforementioned findings, this study postulated the involvement of METTL3 in LC by regulating m6A methylation of pri-miR-663 and SOCS6, which lent insight into novel therapeutic targets for LC.
Materials and methods
Ethics statement
All animal experiments were approved by the laboratory animal welfare and ethics committee of Hainan Hospital of PLA General Hospital. Numerous efforts were made to minimize animal number and pain.
Cell culture and treatment
Human bronchial epithelial (HBE) cells and 4 LC cell lines (A549, H1299, H358, and PC9) were provided by ATCC (Manassas, VA, USA). HBE cells were cultured in Dulbecco’s modified Eagle medium, while LC cells were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) at 37 °C in an incubator with 5% CO2. Both media contained 10% fetal bovine serum (FBS, Invitrogen) and 1% P/S (100 U/mL penicillin and 100 μg/mL streptomycin).
Cell transfection and grouping
A549 LC cells were assigned to the following nine groups: blank group, inhi-negative control (NC) (transfected with inhi-NC for 24 h), miR-inhi (delivered with miR-663 inhibitor for 24 h), miR-inhi + sh-NC (co-transfected with miR-663 inhibitor and sh-NC for 24 h), miR-inhi + sh-SOCS6 (co-delivered with miR-663 inhibitor and sh-SOCS6 for 24 h), sh-NC (introduced with sh-NC for 24 h), sh-METTL3 (manipulated with sh-METTL3 for 24 h), sh-METTL3 + mimics NC (co-introduced with sh-METTL3 and mimics NC for 24 h), and sh-METTL3 + miR-663 mimics (co-manipulated with sh-METTL3 and miR-663 for 24 h).
PC9 LC cells were assigned to the following nine groups: blank group, mimics NC (delivered with mimics NC for 24 h), miR-663 mimics (transfected with miR-663 mimics for 24 h), miR-663 mimics + oe-NC (co-manipulated with miR-663 mimics and oe-NC for 24 h), miR-663 mimics + oe-SOCS6 (co-introduced with miR-663 mimics and oe-SOCS6 for 24 h), oe-NC (manipulated with oe-NC for 24 h), oe-METTL3 (introduced with oe-METTL3 for 24 h), oe-METTL3 + inhi-NC (co-transfected with oe-METTL3 and inhi-NC for 24 h), and oe-METTL3 + miR-inhi (co-delivered with oe-METTL3 and miR-inhi for 24 h).
Among them, miR-663 mimics/miR-663 inhibitor and their NC, SOCS6/METTL3 overexpression plasmid vectors and their NC, shRNAs, and sh-NC of SOCS6/METTL3 provided by GenePharma (Shanghai, China) were transfected into LC cells at 50 nM using Lipofectamine™ 2000 (Invitrogen).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA content was extracted from LC tissues and cells using the TRIzol reagent (Invitrogen). RNA concentration and purity were measured by an ultraviolet (UV) spectrophotometer (UV-1800, Shimadzu, Kyoto, Japan). PrimeScript™ RT reagent kits (Invitrogen) were employed to reversely transcribe 1 μg total RNA into cDNA. PCR amplification reactions were performed using Dy NAmo™ SYBR® Green qPCR kits (Finnzymes, Espoo, Finland) on a Bio-Rad CFX96 fluorescence qPCR instrument (ABI, Foster City, CA, USA). U6 acted as the internal control for miR-663. Data were evaluated using the 2−ΔΔCt method. Primers are exhibited in Table 1.
Cell counting kit-8 (CCK-8) assay
Transfected LC cells at 1 × 104/mL were seeded in 96-well plates with 100 μL cell suspension per well. After respective culture of 24 h, 48 h, and 72 h, 10 μL CCK-8 solution (Beyotime, Shanghai, China) was supplemented to each well, followed by another 2 h incubation. Thereafter, the optical density at 450 nm was determined through a microplate reader.
Transwell assays
Cell migration and invasion were detected using Transwell chambers (Millipore, Billerica, MA, USA). Transfected cells at 1 × 104/mL were placed in the apical chamber containing 200 μL serum-free medium, with FBS and culture medium added to the basolateral chamber. When detecting cell invasion, Matrigel was supplemented to Transwell chambers, but without Matrigel in the migration assay. Following 24 h culture, cells were fixed with methanol for 20 min, stained with 0.1% crystal violet solution, and analyzed under a microscope to, respectively, count the migrated and invaded cells in the basolateral chamber (Mao et al. 2021).
5-Ethynyl-2′-deoxyuridine (EdU) assay
Cell proliferation was estimated using EdU labeling kits (RiboBio, Guangzhou, Guangdong, China). Cells placed on 12-well plates were transfected and incubated with EdU labeling medium for 2 h. After paraformaldehyde and Triton X-100 treatment, Apollo Reaction Reagent was added to the cells and then reacted in the dark for 30 min. Then, 4′,6-diamidino-2-phenylindole was employed for nuclear staining. EdU-positive cells were photographed and percentages were computed.
Flow cytometry
Apoptosis was detected using flow cytometry. Following 24 h transfection, cells were rinsed twice with phosphate-buffered saline (PBS), detached with 0.25% ethylene diamine tetraacetic acid-free trypsin, centrifuged, then collected into flow tubes, and later centrifuged, with the supernatant discarded. After washing thrice with PBS, cells were resuspended in 70% ethanol solution and fixed overnight at − 20 °C. Subsequently, cells were incubated with Annexin V-fluorescein isothiocyanate and propidium iodide (PI) in the dark at room temperature for 10–15 min, and apoptosis was detected by a flow cytometer (Aceabio, San Diego, CA, USA) within 1 h. Early apoptotic cells were expressed as Annexin V+ and PI−. Late apoptotic cells were displayed as Annexin V+ and PI+. The early and late apoptotic cells were counted together.
Western blotting (WB)
Tissues and LC cells were treated with radio-immunoprecipitation assay lysate containing protease inhibitors (Beyotime) at 4 °C for 20 min and centrifuged at 3000g for 20 min with the supernatant removed, followed by determination of supernatant protein concentration using bicinchoninic acid kits (Beyotime). Proteins were separated with 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, and the separated proteins were electrotransferred to polyvinylidene fluoride membranes (Millipore). Next, membranes were transferred into 5% skim milk configured by Tris-buffered saline-tween (TBST), followed by shaking and blocking for 1 h at room temperature to impede non-specific binding. After that, they were incubated with primary antibodies anti-SOCS6 (1:300, ab197335, Abcam, Cambridge, UK) and anti-METTL3 (1:1000, ab195352, Abcam) overnight at 4 °C. Following washing twice with TBST, membranes were reacted for 1 h with secondary antibody horseradish peroxidase (HRP)-labeled goat anti-rabbit immunoglobulin G (IgG) at room temperature. Enhanced chemiluminescence working solution (Millipore) was applied for color development and photography. The density of protein bands was detected using ImageJ software 1.48 (NIH, Bethesda, MD, USA), with GAPDH as the internal reference.
RNA m6A dot blot assay
Poly (A) + RNAs were first heated at 65 °C for 5 min to be denatured, and then, they were transferred onto nitrocellulose membranes (Amersham, GE Healthcare, Boston, MA, USA) using the BioDot apparatus (Bio-Rad, Hercules, CA, USA). After UV crosslinking and blocking, membranes were incubated with primary antibody m6A antibody (1:1000, ab208577, Abcam) overnight at 4 °C, followed by incubation with secondary antibody HRP-labeled goat anti-mouse IgG (1:2000, ab150113, Abcam) and observation on a chemiluminescence system. Membranes were stained with 0.3 M sodium acetate solution (pH 5.2) containing 0.02% methylene blue to ensure consistency between different treatment groups.
Dual-luciferase reporter assay
Starbase database (http://starbase.sysu.edu.cn/) was applied for predicting the binding sites between miR-663 and SOCS6. The complementary sequences of miR-663 and SOCS6 and their mutation sequences were amplified and cloned into the pmir-GLO vectors (GenePharma) to construct pGL-SOCS6-wild-type (WT) and pGL-SOCS6-mutant (MUT) plasmids. Later, HEK293T cells (Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China) were, respectively, co-transfected with the constructed plasmids and mimic NC/miR-663 mimics (GenePharma) using Lipofectamine™ 2000 (Invitrogen), followed by detection of luciferase activity using a dual-luciferase assay kit (Promega, Madison, WI, USA).
m6A RNA immunoprecipitation (Me-RIP) assay
RIP assays were conducted using Magna RIP kits (Millipore). After UV radiation, cells were lysed by sonication at 48 °C. Thereafter, immunoprecipitation of endogenous DiGeorge syndrome critical region 8 (DGCR8) was performed utilizing an anti-DGCR8 antibody (1:1000, ab191875, Abcam) overnight at 4 °C. After two washes, RNA was extracted and subjected to RT-qPCR assay utilizing primers for primary miRNAs (pri-miRNAs) and normalized to input.
Similar to the above experiments, Me-RIP assay identified the binding of m6A RNA. Following digestion with DNase I (Sigma-Aldrich, St Louis, MO, USA), cells were lysed for 10 s by sonication for RNA interference. The magnetic beads were incubated with rabbit m6A antibody (1:500, ab151230, Abcam) for 1 h at room temperature, and then, the antibody-bead complex and RNA immunoprecipitation buffer (Magna RIP kit, Millipore, MA) were mixed thoroughly with RNA fragments, followed by overnight incubation at 4 °C. Subsequently, after digestion of the protein-RNA complex by proteinase K, RNA was extracted using phenol: chloroform: isoamyl alcohol (125:24:1), and subjected to RT-qPCR for detecting the pri-miR-663 expression using IgG antibody as blank control and normalized to input.
Co-immunoprecipitation (Co-IP) assay
Immunoprecipitation experiments were performed in A549 cells utilizing Pierce™ Co-IP kits (Thermo Fisher Scientific, Waltham, MA, USA). Immunoprecipitation of METTL3 was conducted utilizing an anti-METTL3 antibody (ab195352, 1:1000, Abcam) overnight at 4 °C. Following rinsing, the immunoprecipitated complex was reacted with RNase A or RNase inhibitor (Sigma-Aldrich) for 5 min at 37 °C, and subsequently, anti-DGCR8 antibody (ab191875, 1:1000, Abcam) was employed for WB analysis.
Xenograft tumor in nude mice
BALB/C female mice (aged 4–6 weeks, weighing 18–22 g) were provided by Charles River (SCXK (Beijing) 2016-0008, China). A549 cells transfected with sh-NC/sh-METTL3 and PC9 cells delivered with oe-NC/oe-METTL3 (1 × 107 cells) were subcutaneously injected into the armpit of each mouse using a sterile syringe, with five mice per group. After injection, mice were returned to the cage for continuous feeding. The tumor size was measured every 4 days, and tumor volume was calculated according to the formula [tumor volume (mm3)] = ab2/2, (a) the long diameter of tumor; (b) the short diameter of tumor. After 4 weeks, mice were euthanized, and then, tumors were removed and weighed (Liu et al. 2020).
Statistical analysis
Statistical analyses and plotting of data were processed using SPSS 21.0 (IBM Corp. Armonk, NY, USA) and GraphPad Prism 8.01 (GraphPad Software, San Diego, CA, USA). Measurement data were displayed as mean ± standard deviation (SD). Independent sample t test was adopted for comparisons between two groups. One-way analysis of variance (ANOVA) was employed for comparisons between multiple groups and Tukey's multiple comparisons test was applied for post hoc test. The p < 0.05 indicated significant differences.
Results
miR-663 promoted proliferation, migration, and invasion of LC cells
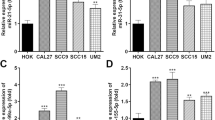
To probe into the mechanism of miR-663 in LC, we detected the expression of miR-663 in LC cell lines (A549, H1299, H358, and PC9) and normal HBE cells by RT-qPCR, which revealed prominently higher miR-663 in LC cells than that in normal cells (p < 0.01) (Fig. 1a). We selected A549 cells with relatively high miR-663 expression and PC9 cells with relatively low miR-663 expression for subsequent studies. Afterward, cells were, respectively, transfected with miR-663 inhibitor or miR-663 mimics to inhibit miR-663 expression in A549 cells (p < 0.05) (Fig. 1b) and to overexpress miR-663 in PC9 cells (p < 0.05) (Fig. 1c). CCK-8 assay showed that miR-663 overexpression significantly enhanced LC cell proliferation, while miR-663 inhibition reduced LC cell viability (all p < 0.01) (Fig. 1d, e). Transwell assays suggested that the migration and invasion of LC cells were remarkably increased in the miR-663 mimics group, whereas decreased after inhibition of miR-663 (all p < 0.01) (Fig. 1f, g). Additionally, EdU assay illustrated that miR-663 markedly promoted LC cell proliferation and miR-663 knockdown suppressed cell proliferation (Fig. 1h, i). Meanwhile, we detected apoptosis by flow cytometry and found that the apoptosis rate of the miR-663 mimics group was noticeably diminished relative to the blank group, and LC cell apoptosis was raised after inhibition of miR-663 (all p < 0.01) (Fig. 1j, k). Altogether, miR-663 facilitated LC cell proliferation, migration, and invasion, and repressed apoptosis, and miR-663 inhibition exerted opposite effects.

miR-663 promoted proliferation, migration, and invasion of LC cells. a–c miR-663 expression detected using RT-qPCR; d, e viability of A549 and PC9 cells assessed by CCK-8 assay; f, g migration and invasion of A549 and PC9 cells evaluated through Transwell assays; h, i cell proliferation analyzed via EdU method; j, k cell apoptosis detected by flow cytometry. Cell experiment was conducted three times and data were expressed as mean ± SD. One-way ANOVA was used for comparisons among multiple groups, followed by Tukey's test. **p < 0.01, ***p < 0.001
miR-663 inhibited SOCS6 expression
SOCS6 is weakly expressed in LC tissues (Xia et al. 2019). The Starbase database revealed that the 5′ terminal region of miR-663 was complementary to the 3′ untranslated region of SOCS6 (Fig. 2a). Dual-luciferase assay was designed according to the binding sites and then indicated that cells co-transfected with miR-663 mimics and SOCS6-WT showed conspicuously lower luciferase activity than those co-delivered with mimics NC and SOCS6-WT (p < 0.01), while cells transfected with SOCS6-MUT exhibited no obvious changes in luciferase activity (p > 0.05) (Fig. 2b). Furthermore, WB detection unveiled reduced SOCS6 levels in A549 and PC9 cells compared with HBE cells (all p < 0.01) (Fig. 2c). SOCS6 was notably increased after inhibition of miR-663 in A549 cells while decreased after overexpression of miR-663 in PC9 cells (all p < 0.01) (Fig. 2d, e). Overall, miR-663 targeted and suppressed SOCS6 expression.

miR-663 inhibited SOCS6 expression. a Starbase database predicted the binding relationship between miR-663 and SOCS6; b dual-luciferase assay verified the targeted binding of miR-663 and SOCS6; c–e WB measured SOCS6 expression. Cell experimentation was conducted three times, and data were exhibited as mean ± SD. One-way ANOVA was employed for comparisons among multiple groups, followed by Tukey's test. **p < 0.01, ***p < 0.001, ns indicated p > 0.05
Overexpression of SOCS6 partially annulled the promotion of miR-663 on LC cell growth
To investigate the function of SOCS6 on the promotion of miR-663 in LC cell growth, we simultaneously overexpressed miR-663 and SOCS6 in PC9 cells (p < 0.01) (Fig. 3a), and inhibited SOCS6 and miR-663 in A549 cells (p < 0.01) (Fig. 3b) for combined experiments. Subsequently, we found that the promoting effect of miR-663 on LC cell proliferation, migration, and invasion was reversed after overexpression of SOCS6 in PC9 cells, while the inhibitory effect of downregulated-miR-663 on LC cell growth was partially weakened after inhibition of SOCS6 in A549 cells (all p < 0.01) (Fig. 3c–i). Altogether, miR-663 could facilitate the proliferation, migration, and invasion of LC cells, and inhibit apoptosis by regulating SOCS6 expression.

Overexpression of SOCS6 partially annulled the promotion of miR-663 on LC cell growth. a, b SOCS6 expression was determined using WB; c cell viability detected using CCK-8 method; d, e cell migration and invasion estimated by Transwell assays; f, g cell proliferation assessed via EdU assay; h, i cell apoptosis analyzed through flow cytometry. Cell experiment was conducted three times, and data were presented as mean ± SD. One-way ANOVA was employed for comparisons among multiple groups, followed by Tukey's test. *p < 0.05, **p < 0.01, ***p < 0.001
METTL3-mediated m6A methylation regulated the maturation of miR-663 in LC cells
METTL3 represents a pivotal protein in the m6A-methyltransferase complex, which can modify pri-miRNAs through m6A in ovarian and bladder cancers, thus stimulating miRNA maturation and ultimately promoting the malignant progression of tumors (Bi et al. 2021; Han et al. 2019). Hence, we hypothesized that METTL3-mediated m6A methylation can modulate the maturation of miR-663 in LC cells. First, WB detection unraveled that METTL3 in A549 and PC9 cells was prominently higher than that in HBE cells, with the highest in A549 cells (all p < 0.05) (Fig. 4a). DGCR8, the critical partner of type III RNase Drosha, is vital in miRNA biogenesis generally by interacting with pri-miRNA and recruiting Drosha to cleave pri-miRNA at the correct site into pre-miRNA, and then, the pre-miRNA is cleaved by Dicer RNase to mature miRNA (Bi et al. 2021; Hang et al. 2021). Moreover, METTL3 interacts with DGCR8 and positively regulates the pri-miRNA process in an m6A-dependent manner (Wang et al. 2019b). Subsequently, Co-IP assay revealed the binding of METTL3 and DGCR8 and the waning of the crosslinking between METTL3 and DGCR8 after RNase treatment (Fig. 4b), indicating that this crosslinking may be partially mediated by miRNAs. Additionally, overexpression of METTL3 potentiated the crosslinking between METTL3 and DGCR8 (Fig. 4c), further confirming that METTL3 could influence the binding of DGCR8 to m6A-methylated miRNAs. Next, we determined RNA m6A levels in cells using the m6A dot blot assay and found that RNA m6A levels were increased in A549 and PC9 cells compared with HBE cells, and diminished after knockdown of METTL3, while raised after overexpression of METTL3 (Fig. 4d).

METTL3-mediated m6A methylation regulated the maturation of miR-663 in LC cells. A549 and PC9 cells were, respectively, transfected with sh-METTL3 or oe-METTL3. a WB assay was employed to detect METTL3 expression; b, c Co-IP was used to verify the crosslinking between METTL3 and DGCR8; d m6A dot blot assay was conducted to determine m6A level, with MB stain as loading control; e, f WB assay measured METTL3 expression; g, h RT-qPCR determined levels of miR-663 and pri-miR-663; i, j RIP and Me-RIP assays were introduced to verify the m6A modification of pri-miR-663. Cell experimentation was conducted three times and data were displayed as mean ± SD. One-way ANOVA was adopted for comparisons among multiple groups, followed by Tukey's test. **p < 0.01, ***p < 0.001, nsp > 0.05. Compared with A549 cells, #p < 0.05
To further explore whether METTL3 exerts an oncogenic property in LC by regulating miR-663, we overexpressed or silenced METTL3 expression in PC9 and A549 cells, respectively, and METTL3 level was detected by WB. The results unveiled that METTL3 expression was remarkably elevated in PC9 cells and reduced in A549 cells (Fig. 4e, f). We subsequently measured the levels of miR-663 and pri-miR-663 in LC cells by RT-qPCR, which demonstrated that miR-663 expression was prominently enhanced upon METTL3 overexpression in PC9 cells and lowered upon METTL3 silencing in A549 cells (all p < 0.01) (Fig. 4g). In addition, pri-miR-663 level was increased in METTL3-downregulated cells but decreased in METTL3-upregulated cells (all p < 0.01) (Fig. 4h). Finally, RIP and Me-RIP experiments revealed that METTL3 overexpression predominantly elevated the levels of pri-miR-663 crosslinked by DGCR8 immunoprecipitation and m6A modification of pri-miR-663 (all p < 0.01), while METTL3 silencing reduced the level of pri-miR-663 precipitated by DGCR8 and m6A antibodies (Fig. 4i, j). In short, METTL3 could regulate miR-663 maturation by mediating m6A methylation of pri-miR-663.
METTL3 facilitated LC cell growth by regulating miR-663
To further identify the function of METTL3 in LC, we inhibited METTL3 expression in A549 cells and overexpressed METTL3 in PC9 cells (all p < 0.05) (Fig. 5a, b). We observed that overexpression of METTL3 significantly stimulated LC proliferation, migration, and invasion, and inhibition of METTL3 impeded LC cell growth and promoted cell apoptosis (all p < 0.01) (Fig. 5c–j). miR-663 overexpression weakened the inhibition of METTL3 silencing on A549 cell growth and miR-663 knockdown annulled the promotion of METTL3 overexpression on PC9 cell growth (all p < 0.05) (Fig. 5c–j). To sum up, METTL3 facilitated LC cell growth by mediating miR-663.

METTL3 facilitated LC cell growth by regulating miR-663. a, b METTL3 expression was measured using WB; c, d cell viability detected using CCK-8 method; e, f cell migration and invasion assessed by Transwell assays; g, h cell proliferation evaluated via EdU assay; i, j cell apoptosis analyzed through flow cytometry. Cell experiment was conducted three times and data were expressed as mean ± SD. One-way ANOVA was employed for comparisons among multiple groups, followed by Tukey's test.*p < 0.05, **p < 0.01, ***p < 0.001
METTL3 promoted tumorigenesis and growth in mice via the miR-663/SOCS6 axis
Furthermore, we performed the tumor formation assay in nude mice to investigate the role of METTL3 in vivo. Nude mice were injected with sh-METTL3-transfected A549 cells and oe-METTL3-transfected PC9 cells, and then, we measured tumor size every 4 days and collected tumors on the 28th day (Fig. 6a). WB detection showed that relative to the LC group, METTL3 expression in LC tissues was markedly diminished in mice injected with A549 cells, whereas elevated in mice injected with PC9 cells (all p < 0.01) (Fig. 6b, c). Moreover, after downregulating METTL3 expression in LC tissues, miR-663 expression was noticeably decreased and SOCS6 was increased; after upregulating METTL3, miR-663 was raised and SOCS6 was reduced (all p < 0.01) (Fig. 6d–g). Additionally, inhibiting METTL3 prominently lowered the tumor formation ability of A549 cells and reduced tumor volume and weight, but overexpressing METTL3 raised the tumor formation ability of PC9 cells and augmented tumor volume and weight (Fig. 6h–k). Briefly, METTL3 could facilitate tumor growth in mice through the miR-663/SOCS6 axis.

METTL3 promoted tumorigenesis and growth in mice via the miR-663/SOCS6 axis. a Schematic diagram of tumor formation assay in vivo; b, c WB detected METTL3 expression; d, e RT-qPCR determined miR-663 expression; f, g WB measured SOCS6 expression; h, i changes of tumor volume was recorded; j, k representative images and weight of tumors. Data were exhibited as mean ± SD. One-way ANOVA was performed for comparisons among multiple groups, followed by Tukey's test. **p < 0.01, ***p < 0.001
Discussion
LC, the most general cancer and leading source of cancer-associated deaths, causes remarkable burdens on the world and 1.6 million deaths annually (Ho and Leung 2018; Manafi-Farid et al. 2021). miRNAs exert an essential impact on the onset and survival of LC (Fang et al. 2017). METTL3 is implicated in RNA biosynthesis, which dominants mRNA translation, alternative splicing, and miRNA maturation through m6A-dependent methylation (Xiao et al. 2020). m6A modification can affect the physiological and pathological processes of LC (Wu et al. 2021a). This study validated the mechanism of METTL3 in LC via the m6A methylation-mediated pri-miR-663 processing and SOCS6 expression (Fig. 7).

Mechanism of METTL3 in LC development by regulating the miR-663/SOCS6 axis
Current evidence unravels the substantial roles of miRNAs in controlling invasion, migration, and proliferation in LC (Tang et al. 2021). Aberrantly expressed miRNAs are common biomarkers of LC onset and advance (Chen et al. 2017). Intriguingly, miR-663 is upregulated in diverse cancers, including colorectal cancer (Yilmaz et al. 2020), nasopharyngeal carcinoma (Liang et al. 2017), ovarian cancer (Xie et al. 2019), and prostate cancer (Wang et al. 2017). Hence, we detected miR-663 expression and observed an elevated miR-663 in LC cells. After interference of miR-663 expression in A549 and PC9 cells, we noted that overexpression of miR-663 could promote LC cell proliferation, migration, and invasion and inhibit apoptosis, and downregulation of miR-663 owned the contrary effects. Consistently, the oncogenic role of miR-663 has been demonstrated in multifarious cancers, such as colorectal cancer, LC, nasopharyngeal carcinoma, and prostate cancer (Wang et al. 2019c). The involvement of miR-663 in NSCLC is also documented, and inhibition of miR-663 expression impedes NSCLC cell proliferation and tumor growth (Fiori et al. 2018). In mechanism, miRNAs could regulate gene expression by repressing mRNA translation and weakening mRNA stability (Lai et al. 2019). SOCS6 mainly engages in the negative modulation of tumor growth (Lawrenson et al. 2017; Sun et al. 2021) and is essential for normal cell growth by acting as a neoplasm suppressor in varied cancers (Wang et al. 2016; Yuan et al. 2018). Next, we identified that miR-663 targeted SOCS6. SOCS6 was under-expressed in LC cells and miR-663 inversely regulated SOCS6 expression. Besides, SOCS6 overexpression partially abrogated the catalytic role of miR-663 on LC cell growth. Our results are broadly in line with preceding findings that SOCS6 is downregulated in NSCLC cells and tissues, and decreased SOCS6 poses a promotion impact on cell proliferation and invasion, thereby accelerating NSCLC progression (Xia et al. 2019; Ye et al. 2021). SOCS6 loss is interrelated to poor survival of primary lung squamous cell carcinoma (Sriram et al. 2012). Similarly, miR-200b-3p affects LC development by negatively regulating SOCS6 expression (Chi et al. 2021). Collectively, miR-663 facilitated LC cell growth and suppressed apoptosis by inhibiting SOCS6 expression.
In eukaryotes, m6A is crucial in numerous biological processes and disorders such as cancer (Wang et al. 2021). m6A modification manipulates the fate and action of RNA, including mRNA stability, translation, RNA–protein interactions, and pri-miRNA processing (Wu et al. 2021b). METTL3, a determined methyltransferase for m6A methylation, is reported to be upregulated in numerous cancers (Gu et al. 2021; Peng et al. 2019). Therefore, we subsequently explored this mechanism of METTL3 in LC. Our results revealed upregulated METTL3 and RNA m6A levels in LC cells. METTL3 could bind to DGCR8 and positively mediate RNA m6A level. Moreover, METTL3 overexpression raised miR-663 expression, but decreased pri-miR-663 level, whereas METTL3 knockdown exerted the inverse functions. METTL3 advances the maturation of miRNAs (including miR-663b) by interacting with DGCR8 (Alarcon et al. 2015). Furthermore, METTL3 is upregulated in lung adenocarcinoma and METTL3 silencing lowers m6A level in PC9 and H3255 cells (Gao et al. 2021). Conjointly, METTL3 accelerated miR-663 maturation by promoting m6A methylation of pri-miR-663.
METTL3 is conceived as a paramount post-transcriptional regulator in miRNA biogenesis by enhancing levels of mature miRNAs through m6A modification of pri-miRNA (Diao et al. 2021). Subsequently, we clarified the effects of METTL3 on LC progression and found that overexpressing METTL3 gave an impetus to LC cell growth, which was nullified by miR-663 silencing. On the contrary, downregulating METTL3 prevented cell growth, which was counteracted by miR-663 upregulating. Our results are consistent with the preceding discovery that METTL3 could intensify LC cell growth by promoting oncogene translation and oncoprotein expression (Du et al. 2017; Lin et al. 2016). Furthermore, we verified the role of METTL3 in LC in vivo. Our results discovered that METTL3 stimulated tumor initiation and growth in mice by positively regulating miR-663 and negatively mediating SOCS6. METTL3 impacts tumor formation by regulating m6A modification in the mRNAs of pivotal oncogenes or tumor suppressors (Han et al. 2019). METTL3 knockdown elevates expressions of the SOCS family, including SOCS6, SOCS5, SOCS4, SOCS2, and SOCS2 (Song et al. 2021). Similarly, METTL3 maintains cancer tumorigenicity by suppressing SOCS2 to promote cell proliferation, including colon cancer (Xu et al. 2020a), gastric cancer (Jiang et al. 2020), and liver cancer (Chen et al. 2018). Briefly, METTL3 facilitated LC progression via the miR-663/SOCS6 axis.
To conclude, the study underlined that METTL3 accelerated m6A methylation-mediated pri-miR-663 processing, thereby inhibiting SOCS6 expression, and ultimately facilitating LC development. However, only two LC cell lines were selected for relevant studies, and more cell lines are warranted for in-depth study. Another limitation was derived from the lack of research on the function of METTL3 and miR-663 at the clinical level. Furthermore, we shall explore the downstream pathway of SOCS6 in mediating LC and the mechanism of m6A methylation modification of pri-miR-663 at the molecular level. Meanwhile, the correlation between aberrant expression of METTL3 and abnormal upregulation of miRNA deserves further investigation.
Availability of data and materials
All the data generated or analyzed during this study are included in this published article.
References
Alarcon CR, Lee H, Goodarzi H et al (2015) N6-methyladenosine marks primary microRNAs for processing. Nature 519:482–485
Barbieri I, Tzelepis K, Pandolfini L et al (2017) Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552:126–131
Bi X, Lv X, Liu D et al (2021) METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther 28:335–349
Chen Y, Min L, Ren C et al (2017) miRNA-148a serves as a prognostic factor and suppresses migration and invasion through Wnt1 in non-small cell lung cancer. PLoS ONE 12:e0171751
Chen M, Wei L, Law CT et al (2018) RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 67:2254–2270
Chen M, Zhu LL, Su JL et al (2020a) Prucalopride inhibits lung cancer cell proliferation, invasion, and migration through blocking of the PI3K/AKT/mTor signaling pathway. Hum Exp Toxicol 39:173–181
Chen WW, Qi JW, Hang Y et al (2020b) Simvastatin is beneficial to lung cancer progression by inducing METTL3-induced m6A modification on EZH2 mRNA. Eur Rev Med Pharmacol Sci 24:4263–4270
Chi Y, Zheng W, Bao G et al (2021) Circular RNA circ_103820 suppresses lung cancer tumorigenesis by sponging miR-200b-3p to release LATS2 and SOCS6. Cell Death Dis 12:185
Diao LT, Xie SJ, Lei H et al (2021) METTL3 regulates skeletal muscle specific miRNAs at both transcriptional and post-transcriptional levels. Biochem Biophys Res Commun 552:52–58
Du M, Zhang Y, Mao Y et al (2017) MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun 482:582–589
Fang C, Li XP, Gong WJ et al (2017) Age-related common miRNA polymorphism associated with severe toxicity in lung cancer patients treated with platinum-based chemotherapy. Clin Exp Pharmacol Physiol 44(Suppl 1):21–29
Fiori ME, Villanova L, Barbini C et al (2018) miR-663 sustains NSCLC by inhibiting mitochondrial outer membrane permeabilization (MOMP) through PUMA/BBC3 and BTG2. Cell Death Dis 9:49
Gao F, Wang Q, Zhang C et al (2021) RNA methyltransferase METTL3 induces intrinsic resistance to gefitinib by combining with MET to regulate PI3K/AKT pathway in lung adenocarcinoma. J Cell Mol Med 25:2418–2425
Gu Y, Chu X, Morgan JA et al (2021) Upregulation of METTL3 expression and m6A RNA methylation in placental trophoblasts in preeclampsia. Placenta 103:43–49
Han J, Wang JZ, Yang X et al (2019) METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer 18:110
Hang Q, Zeng L, Wang L et al (2021) Non-canonical function of DGCR8 in DNA double-strand break repair signaling and tumor radioresistance. Nat Commun 12:4033
Ho JC, Leung CC (2018) Management of co-existent tuberculosis and lung cancer. Lung Cancer 122:83–87
Jiang L, Chen T, Xiong L et al (2020) Knockdown of m6A methyltransferase METTL3 in gastric cancer cells results in suppression of cell proliferation. Oncol Lett 20:2191–2198
Lai X, Eberhardt M, Schmitz U et al (2019) Systems biology-based investigation of cooperating microRNAs as monotherapy or adjuvant therapy in cancer. Nucleic Acids Res 47:7753–7766
Lawrenson ID, Krebs DL, Linossi EM et al (2017) Cortical layer inversion and deregulation of Reelin signaling in the absence of SOCS6 and SOCS7. Cereb Cortex 27:576–588
Li P, Chen Y, Juma CA et al (2019) Differential inhibition of target gene expression by human microRNAs. Cells 8:791
Liang S, Zhang N, Deng Y et al (2017) miR-663 promotes NPC cell proliferation by directly targeting CDKN2A. Mol Med Rep 16:4863–4870
Lin S, Choe J, Du P et al (2016) The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 62:335–345
Liu S, Yang N, Wang L et al (2020) lncRNA SNHG11 promotes lung cancer cell proliferation and migration via activation of Wnt/beta-catenin signaling pathway. J Cell Physiol 235:7541–7553
Liu X, Jiang F, Wang Z et al (2021) Hypoxic bone marrow mesenchymal cell-extracellular vesicles containing miR-328-3p promote lung cancer progression via the NF2-mediated Hippo axis. J Cell Mol Med 25:96–109
Manafi-Farid R, Karamzade-Ziarati N, Vali R et al (2021) 2-[(18)F]FDG PET/CT radiomics in lung cancer: an overview of the technical aspect and its emerging role in management of the disease. Methods 188:84–97
Mao Y, He JX, Zhu M et al (2021) Correction to: Circ0001320 inhibits lung cancer cell growth and invasion by regulating TNFAIP1 and TPM1 expression through sponging miR558. Hum Cell 34:1287
Peng W, Li J, Chen R et al (2019) Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res 38:393
Puri S, Chatwal M, Gray JE (2017) Anti PD-L1 combined with other agents in non-small cell lung cancer: combinations with non-immuno-oncology agents. Expert Rev Respir Med 11:791–805
Seo HA, Moeng S, Sim S et al (2019) MicroRNa-based combinatorial cancer therapy: effects of micrornas on the efficacy of anti-cancer therapies. Cells 9:29
Song RH, Du P, Gao CQ et al (2021) METTL3 is involved in the development of Graves’ disease by inducing SOCS mRNA m6A modification. Front Endocrinol (lausanne) 12:666393
Sriram KB, Larsen JE, Savarimuthu Francis SM et al (2012) Array-comparative genomic hybridization reveals loss of SOCS6 is associated with poor prognosis in primary lung squamous cell carcinoma. PLoS ONE 7:e30398
Sun X, Sun Y, Li J et al (2021) SOCS6 promotes radiosensitivity and decreases cancer cell stemness in esophageal squamous cell carcinoma by regulating c-Kit ubiquitylation. Cancer Cell Int 21:165
Tang S, Li S, Liu T et al (2021) MicroRNAs: emerging oncogenic and tumor-suppressive regulators, biomarkers and therapeutic targets in lung cancer. Cancer Lett 502:71–83
Wang XJ, Zhang DL, Fu C et al (2016) MiR-183 modulates multi-drug resistance in hepatocellular cancer (HCC) cells via miR-183-IDH2/SOCS6-HIF-1alpha feedback loop. Eur Rev Med Pharmacol Sci 20:2020–2027
Wang S, Liu J, Li C et al (2017) Research of the effect of miR-663 on the proliferation of prostate cancer cells, and the correlations of miR-663 with pathological grade and clinical stage. J BUON 22:1011–1016
Wang H, Deng Q, Lv Z et al (2019a) N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol Cancer 18:181
Wang J, Ishfaq M, Xu L et al (2019b) METTL3/m(6)A/miRNA-873-5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating Keap1/Nrf2 pathway. Front Pharmacol 10:517
Wang N, Zeng L, Li Z et al (2019c) Serum miR-663 expression and the diagnostic value in colorectal cancer. Artif Cells Nanomed Biotechnol 47:2650–2653
Wang Q, Chen C, Ding Q et al (2020) METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 69:1193–1205
Wang H, Song X, Song C et al (2021) m(6)A-seq analysis of microRNAs reveals that the N6-methyladenosine modification of miR-21-5p affects its target expression. Arch Biochem Biophys 711:109023
Wanna-Udom S, Terashima M, Lyu H et al (2020) The m6A methyltransferase METTL3 contributes to transforming growth factor-beta-induced epithelial–mesenchymal transition of lung cancer cells through the regulation of JUNB. Biochem Biophys Res Commun 524:150–155
Wei W, Huo B, Shi X (2019) miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag Res 11:1177–1187
Wu KJ (2020) The role of miRNA biogenesis and DDX17 in tumorigenesis and cancer stemness. Biomed J 43:107–114
Wu H, Li F, Zhu R (2021a) miR-338-5p inhibits cell growth and migration via inhibition of the METTL3/m6A/c-Myc pathway in lung cancer. Acta Biochim Biophys Sin (shanghai) 53:304–316
Wu H, Xu H, Jia D et al (2021b) METTL3-induced UCK2 m(6)A hypermethylation promotes melanoma cancer cell metastasis via the WNT/beta-catenin pathway. Ann Transl Med 9:1155
Xia Y, Wei K, Yang FM et al (2019) miR-1260b, mediated by YY1, activates KIT signaling by targeting SOCS6 to regulate cell proliferation and apoptosis in NSCLC. Cell Death Dis 10:112
Xiao L, Zhao Q, Hu B et al (2020) METTL3 promotes IL-1beta-induced degeneration of endplate chondrocytes by driving m6A-dependent maturation of miR-126-5p. J Cell Mol Med 24:14013–14025
Xie HH, Huan WT, Han JQ et al (2019) MicroRNA-663 facilitates the growth, migration and invasion of ovarian cancer cell by inhibiting TUSC2. Biol Res 52:18
Xu J, Chen Q, Tian K et al (2020a) m6A methyltransferase METTL3 maintains colon cancer tumorigenicity by suppressing SOCS2 to promote cell proliferation. Oncol Rep 44:973–986
Xu Y, Jiang T, Wu C et al (2020b) CircAKT3 inhibits glycolysis balance in lung cancer cells by regulating miR-516b-5p/STAT3 to inhibit cisplatin sensitivity. Biotechnol Lett 42:1123–1135
Ye Y, Wu X, Long F et al (2021) Circular RNA _0015278 inhibits the progression of non-small cell lung cancer through regulating the microRNA 1278/SOCS6 gene axis. Ann Transl Med 9:1255
Yilmaz N, Yilmaz U, Tanbek K et al (2020) The role of miRNAs targeting K-ras and APC genes in colorectal cancer. Bratisl Lek Listy 121:554–557
Yuan D, Wang W, Su J et al (2018) SOCS6 functions as a tumor suppressor by inducing apoptosis and inhibiting angiogenesis in human prostate cancer. Curr Cancer Drug Targets 18:894–904
Zeng C, Huang W, Li Y et al (2020) Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol 13:117
Zhao C, Ling X, Xia Y et al (2021) The m6A methyltransferase METTL3 controls epithelial-mesenchymal transition, migration and invasion of breast cancer through the MALAT1/miR-26b/HMGA2 axis. Cancer Cell Int 21:441
Funding
The authors of this manuscript received no funding for the work reported in their manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All animal experiments were approved by the laboratory animal welfare and ethics committee of Hainan Hospital of PLA General Hospital. Numerous efforts were made to minimize animal number and pain.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, S., Lu, X., Zheng, D. et al. Methyltransferase-like 3 facilitates lung cancer progression by accelerating m6A methylation-mediated primary miR-663 processing and impeding SOCS6 expression. J Cancer Res Clin Oncol 148, 3485–3499 (2022). https://doi.org/10.1007/s00432-022-04128-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-022-04128-5