
Abstract
Cancer immunotherapy is a major breakthrough in tumor therapy and has been used in monotherapy or combination therapy. However, it has been associated with poor immune tolerance in some patients or immune-related adverse events. Therefore, ideal and reliable tumor elimination strategies are urgently needed to overcome these shortcomings. Phosphatidylserine (PS) is a negatively charged phospholipid, usually present in the inner lobules of eukaryotic cell membranes. Under certain physiological or pathological conditions, PS may be exposed on the outer leaflets of apoptotic cells serving as recognition signals by phagocytes and modulating the immune response. On the contrary, increased exposure of PS in the tumor microenvironment can significantly antagonize the body’s anti-tumor immunity, thereby promoting tumor growth and metastasis. During radiotherapy and chemotherapy, PS-mediated immunosuppression increases the PS levels in necrotic tissue in the tumor microenvironment, further suppressing tumor immunity. PS-targeted therapy is a promising strategy in cancer immunotherapy. It inhibits tumor growth and improves the anti-tumor activity of immune checkpoint inhibitors. A comprehensive understanding of the mechanism of PS-targeted therapy opens up a new perspective for future cancer immunotherapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Cancer is a serious public health challenge associated with high morbidity and mortality. Researchers have continuously explored new tumor targets in the past few decades. In recent years, immunotherapy has become the focus of new treatment approaches and has been shown to achieve remarkable results. However, the benefits achieved with monotherapy using immune checkpoint inhibitors (ICIs) were not ideal (Barrueto et al. 2020). Therefore, a clinical decision was made to use simultaneous blockade of CTLA-4 and PD-1. The possibility of this therapy inducing high-grade immune-related adverse events should thus be considered (Tang et al. 2020; Zhou et al. 2020). Further, immunotherapy may affect the tumor microenvironment (TME) to a large extent (Lei et al. 2020). Accumulating evidence reveals that TME plays a more prominent role in tumor immunity than ICIs (Kamal et al. 2020). Other tumor-related immunomodulatory therapies targeting TME or new treatment strategies that combine existing therapies have also been explored. Researchers have also gained interest in phosphatidylserine (PS)-targeted therapy. Phospholipids in eukaryotic cells have an asymmetric distribution. The negatively charged phosphatidylserine is mainly located in the inner membrane leaflet. Researchers have shown that the exposed PS is recognized during cell apoptosis, triggering phagocytosis. Activation of the PS receptor may have immunosuppressive effects by attenuation of the immune response. PS exposure in the tumor microenvironment may lead to immunosuppression and facilitate tumor growth (Shurin et al. 2009; Zohar and Shoenfeld 2018). Therefore, the location of PS on the cell membrane affects cell viability, tumor invasion, and metastasis (Iida et al. 2015). Therefore, targeting exposed PS in the TME with ICIs may offer a new strategy for improving tumor treatment success.
The basic mechanism of targeting PS
PS is generally located on the inner leaflet of the plasma membrane. Under normal physiological conditions, PS exposure on the outer membrane releases an “eat me” signal, inducing macrophages to engulf the cell (Calianese and Birge 2020). Although PS can also be transiently exposed on the surface of some immune cells including B cells, T cells and dendritic cells (DC), they may have a higher PS exposure threshold to circumvent autoimmunity (Fischer et al. 2006). Externalized PS is a cell marker, mainly responsible for the recognition and uptake of apoptotic cells by phagocytes, and inhibits the potential autoimmune response (Ishii et al. 2005). However, in TME, apoptosis-related key factors, such as hypoxia, adenosine, lactate, vascular endothelial growth factor, activated caspase, Ca2+ influx, among others, promote the externalization of PS to the outer leaflet of the plasma membrane (Francis et al. 2013; Vaupel and Multhoff 1887). The exposed PS binds to PS receptors on immune cells and weakens the innate and adaptive immune response by activating the immunosuppressive pathway, conducive to tumor immune escape (Freeman et al. 2010). Furthermore, PS exposure often occurs on the premise of cell necrosis or apoptosis in TME (Li et al. 2019). However, this phenomenon has also been observed in endothelial cells or extracellular vesicles of tumor or mesenchymal origin (Fendl et al. 2018). What is more, PS is also exposed in a variety of infectious pathogens and on the surface of infected cells, producing a weaker non-inflammatory immune response, called “apoptosis mimicry” (Hosseini et al. 2015). Presently, a variety of PS receptors have been discovered and are involved in different signal transduction pathways. Further, most PS receptors are related to the anti-inflammatory response, with some contributing to the pro-inflammatory response.
Induction and immunosuppressive effects of PS exposure on tumor cells
PS is absent on the external surface of vascular endothelial cells in normal cells. However, in the tumor microenvironment, oxidative stress can expose PS to the surface of the vascular endothelium of cancer cells (Sharma and Kanwar 2018). Generally, PS recognizes receptors on DC, macrophages, and T cells to suppress immunity (Chang et al. 2020). PS-binding to the receptors promotes the polarization of macrophages from a pro-inflammatory M1-like phenotype to a pro-tumor M2-like phenotype and leads to the secretion of immunosuppressive factors, such as IL-10 and TGF-β (Serinkan et al. 2005; Quan et al. 2018). IL-10 and TGF-β inhibit T cell activation by suppressing tumor antigen presentation by DC and induction of regulatory Tregs (Stewart et al. 2013). Meanwhile, PS on tumor-derived micro-vesicles has also been shown to inhibit T cell activation (Park and Kang 2019). PS has also been shown as an essential in mediating the combination of IFN-γ and IL-12 and prolonged inflammation (Frey and Gaipl 2011). Therefore, exposure of PS in tumor cells also has anti-tumor effects by mediating chronic inflammation.
Targeting PS receptors and cancer therapy
Several receptors recognize PS, including TIM (T cells, immunoglobulin, and mucin) gene family, stabilin-1/2, and TAM (Tyro, Axl, and Mertk) gene family members in TEM to cause immunosuppression (Dayoub and Brekken 2020). The TIM gene family, BAI1, and stabilin-1/2, directly bind to PS. However, the TAM gene family binds to PS indirectly via binding to protein ligands (Hochreiter-Hufford et al. 2013; David et al. 2012; Wu et al. 2018). We summarize the main receptors, their related characteristics and presenting cells. (Table 1).
TIM
The human TIM gene family mainly encodes three receptors, TIM-1, TIM-3, and TIM-4, which regulate various immune responses, such as asthma and infection (Burstyn-Cohen and Maimon 2019). Studies have shown that the TIM receptor recognizes PS by interacting with the conserved n-terminal immunoglobulin-like extracellular domain (Burstyn-Cohen and Maimon 2019). IM-1 is mainly expressed on CD4 + T cells and regulatory B cells. TIM-1 on Th2 cells is an effective costimulatory molecule for T cell activation. DC and macrophages highly express TIM-4 and participate in the inflammatory response (Kong et al. 2020).
TIM-3 is a type-I transmembrane protein, which is a specific marker for Th1 and Tc1 cells. TIM-3 is also expressed on cytotoxic CD8 + T cells, Th2 cells, Th17 cells, and regulatory T cells. It forms an interaction with PS through the extracellular IgV domain, which is a mucin stalk containing N- and o-chain glycosylation sites and an intracellular tail with a conserved tyrosine residue (Anderson 2014). In addition, TIM-3 is associated with poor prognosis in various cancers, including cervical cancer, gastric cancer, melanoma, and lung cancer. So far, Galectin-9, Carcinoembryonic Antigen Cell Adhesion Molecule-1 (Ceacam-1), High Mobility Group Histone B1 (HMGB1), and PS are the four ligands related to TIM-3 (Romero 2016; Du et al. 2017) (Fig. 1). The TIM-3/Galectin-9 pathway also promotes the production of myeloid-derived suppressor cells (MDSCs), which suppresses adaptive immunity, thus accelerating tumor growth and decrease autoimmunity (Zhou et al. 2019; Wang et al. 2014). As a molecule exposed on the surface of apoptotic cells, PS binds to TIM-3, promoting the clearance of apoptotic bodies (He et al. 2018). The number of apoptotic cells in the spleen of mice treated with TIM-3 mAb was shown to be increased, while the serum anti-dsdna antibodies were shown to be increased (Nakayama et al. 2009). The study also found that CD8 + DC expressing TIM-3 mediated phagocytosis of apoptotic cells and cross-presentation of related antigens to CD8 + T cells (Nakayama et al. 2009). In addition, allelic variants of TIM-3 have different binding affinities and phagocytic abilities to PS. The BALB/c alleles of TIM-3 have a higher binding to PS than HBA. DC and macrophages constitutively express TIM-3 and act as a negative immune regulator (Ocana-Guzman et al. 2016). An increase of the anti-inflammatory cytokines, such as IL-4 and TGF-β, polarizes the M1-like phenotype of macrophages to the M2-like phenotype, and increases the expression of TIM-3 (Ocana-Guzman et al. 2016). TIM-3+ phagocytes and PS expression mediate the uptake of apoptotic cells, thus initiating TIM-3-mediated inhibition (Sabatos-Peyton et al. 2018). A recent research revealed that TIM-3 promotes tumor growth by weakening immune activity. According to another study, TIM-3 was synergistically expressed with PD-1 in tumor-infiltrating lymphocytes (TIL) (Jie et al. 2017; Li et al. 2016). TIM-3+PD-1+ TILs showed a clear T cell failure phenotype, that was, unable to secrete interleukin 2 (IL-2), tumor necrosis factor (TNF), and interferon γ (IFN-γ). Another study revealed that PD-1 + CD8 + T cells in patients with melanoma also increased the expression of TIM-3 (Lu et al. 2017). The above results suggest that blocking TIM-3 and PD-1 may be beneficial in the recovery of T cell failure.

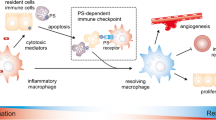
The PS receptor in immunosuppression. The four ligands related to TIM-3 are Galectin-9, Ceacam-1, HMGB1, and PS. Stabilin-1 and stabilin-2 directly bind to PS on the surface of apoptotic cells to activate Rac1. In the Gulp1-independent pathway, stabilin-2 acts on integrin β5 through FAS1, to activate Rac1 and induces cytoskeletal rearrangement. The TAM gene family (Tyro, Axl, and Mertk) binds to PS via Gas6 or protein S. The γ-carboxylated GLA domain of Gas6 and protein S directly bind to PS. Blocking vitamin K-dependent Gas6 γ-carboxylation can inhibit the activation of Axl on tumor cells and reduce tumor progression and metastasis
Stabilin-1 and stabilin-2
Stabilin-1 and stabilin-2 are mostly expressed in the liver, spleen, lymph nodes, bone marrow (stabilin-2), and adrenal cortex (stabilin-1) (Park and Kim 2019). Stabilin-1 and stabilin-2 directly bind to PS on the surface of apoptotic cells, activating Rac1 (Rac family small GTPase 1) (Park and Kim 2019). They reorganize the actin cytoskeleton through Gulp1 to phagocytose apoptotic cells. In the Gulp1-independent pathway, stabilin-2 acts on integrin β5 through its fascicle protein I (FAS1), activates Rac1 and induces cytoskeletal rearrangement (Park et al. 2016; Twarda-Clapa et al. 2018). Down-regulation of endogenous Gulp1 can attenuate phagocytosis of PS-exposed erythrocytes mediated by stabilin-1 and stabilin-2 (Park et al. 2010). On the contrary, overexpression of Gulp1 can enhance phagocytosis of PS-exposed red blood cells by stabilin-2. Stabilin-1 is also expressed in selectively activated macrophages (M2-like macrophages) (David et al. 2012). M2-like macrophages activate the body's repair response and promote tumor growth. Stabilin-1 was shown to be recruited in macrophages co-cultured with apoptotic cells and eliminated the dead cells via mediation with PS (Park et al. 2012). Immunosuppressive leukopenia in TME was observed after treatment with anti-Stabilin-1 showing that stabilin-1 in macrophages was involved in immune response against tumor cells. In addition, the growth of primary tumors in TME with stabilin-1-silenced macrophages was also reduced compared with the control group. TGF-β is an immunoregulatory factor related to the differentiation of immunosuppressive regulatory T cells. Park et al. showed that intervention with PS-exposed red blood cells or anti-stabilin-2 antibodies induced the secretion of TGF-β, indicating that stabilin-2 could regulate the inflammatory response after apoptosis (Park et al. 2008).
TAM
TAM genes are receptor tyrosine kinases (RTKs) expressed on various tumors. RTKs bind to PS via the γ-carboxylated bridging proteins Gas6 or protein S (Graham et al. 2014). The γ-carboxylated GLA domain of Gas6 and protein S directly binds to PS, and the receptor-binding domain is responsible for recognizing the TAM receptor (Wu et al. 2018; Meer and Poll 2014). The activation of RTKs on tumor cells is related to chemotherapy resistance. Blocking the vitamin K-dependent γ-carboxylation of Gas6 was shown to inhibit Axl activation on tumor cells and reduce tumor progression and metastasis in preclinical tumor models (Wu et al. 2018; Stasi et al. 2020). The combination of PS and TAM RTKs on tumor cells can also enhance the expression of PD-L1 on tumor cells (Kasikara et al. 2017). Therefore, anti-tumor immunity can be enhanced by blocking PS-mediated activation of TIM and TAM RTK pathways.
Targeting PS
Ran et al. covalently bound the monoclonal antibody of vascular cell adhesion molecule 1 (VCAM-1) to the extracellular domain of coagulation-related human tissue factor forming a ‘coagulant’ (Ran et al. 1998). The coagulant was selectively localized to tumor blood vessels expressing VCAM-1, thus promoting thrombus formation and delaying tumor growth. Interestingly, the coagulant was also located in the blood vessels expressing VCAM-1 in the heart and lungs of mice. However, these blood vessels did not show thrombosis (Lino et al. 2019). Further, they used immunohistochemistry to evaluate the distribution of monoclonal anti-phosphatidylserine (PS) antibodies. The results showed that VCAM-1 and PS were co-expressed in tumor blood vessels, but PS was not found on the external cell membrane of the heart and lungs (Gosk et al. 2008). They hypothesized that the difference in selectivity was due to the exposure of PS on the surface of blood vessel endothelial cells within TME, which was necessary for inducing blood coagulation (Ran et al. 1998). Ran et al. then developed a monoclonal antibody 9D2 against anionic phospholipids based on the exposure of PS on the outer leaflet of the cell membrane in tumor vasculature (Ran et al. 2005). 9D2 and Annexin V specifically localize to different epitopes in tumor blood vessels. The interaction of 9D2 and PS did not require Ca2+, unlike with Annexin V (Ran et al. 2002). Reactive oxygen species, hypoxia, and some inflammatory mediators, such as IL-1a and IFN-γ, in TME were shown to induce a moderate increase in PS exposure (Klein et al. 2021). Therefore, targeting exposed PS in tumor blood vessels may be an effective anti-cancer strategy.
The monoclonal antibody IgG (2aG4, 3G4, bavituximab, 1N11, and mch1N11) launched by Thorpe Laboratories against PS was shown to exert anti-tumor activity by destroying tumor blood vessels (Ran et al. 2005). The PS-targeting monoclonal antibody was shown to bind to PS via the serum cofactor β2 glycoprotein 1 (β2GP1). β2GP1 is an anionic phospholipid-binding protein in serum, which acts as a bridge between PS and monoclonal antibodies (Luster et al. 2006). PS-targeting monoclonal antibody binds to dimeric β2GP1 and PS on the plasma membrane with high affinity. However, β2GP1 could trigger the production of anti-phospholipid antibodies, causing autoimmune diseases such as systemic lupus erythematosus (Martinez-Flores et al. 2015). According to Mineo et al., monoclonal antibodies (1N11) could prevent the adverse events associated with anti-phospholipid antibodies.
Macrophages mediated the inhibition of tumor angiogenesis by 3G4, which was seen as reduced blood vessel density and plasma volume. This reveals that targeting PS can change the tumor microenvironment (Chang et al. 2020). In addition, targeting PS on tumor cells can prevent the conversion of the M1-like pro-inflammatory phenotype of macrophages to the M2-like pro-tumor phenotype (Dayoub and Brekken 2020). In orthotopic human breast xenotransplantation models, 40% of tumor blood vessels were combined with monoclonal antibodies. Preclinical studies showed that infected cells increased PS exposure when treated with mouse chimeric antibody 1N11 (mch1N11), thus increasing anti-tumor activity (Budhu et al. 2021). Human chimeric monoclonal antibodies may have anti-cancer effects mediated by macrophages through antibody-dependent cytotoxicity (ADCC) directed against PS+ tumor endothelial cells (Belzile et al. 2018). Meanwhile, the anti-cancer effect of monoclonal antibodies is amplified by chemotherapy. In short, increased PS exposure combined with PS-targeting antibody in tumor cell damage is beneficial to enhance anti-tumor immunity. The PS-targeting antibody enhances the killing effect through ADCC and reprogramming of immunosuppressive cells (Birge et al. 2016). At the same time, this process is accompanied by the maturation of DCs and the proliferation of effector T cells. Reprogramming is considered to be mediated by the blockade of the PS receptors of immunosuppressive cells and the interaction of antibodies with activated Fc receptors (FcRs) on DCs (Masuda et al. 2009; Naqvi et al. 2016). Reprogramming can be achieved by blocking the PS receptors of immunosuppressive cells or by binding antibodies to activated FcRs on DCs (Stavenhagen et al. 2007). The binding of antibodies to activated FcRs not only promotes DC maturation and antigen presentation ability, but also promotes the progress of adaptive immunity (Bajtay et al. 2006). Further studies have found that targeting apoptotic tumor cells to FcγR could provide effective DC vaccination against tumors (Murphy et al. 2014).
Targeting PS receptors
There has been an increased interest in PS-targeted therapy and the development of antibodies against PS receptors. TIM-3 antibody combined with anti-CTLA-4 and anti-PD-1 drugs was highly sensitive and well tolerated in patients with carcinogenic sarcoma (Ngiow et al. 2011). In addition, blockade of TIM-3 relieved immune suppression by reducing regulatory T cells in head and neck tumors (Liu et al. 2018). Several clinical trials targeting TIM-3 antibodies are summarized in Table 2. For example, NCT0368050 evaluated the effect of combined anti-TIM-3 antibody (TSR-022) and anti-PD-1 antibody in treating locally advanced or metastatic liver cancer. NCT03489343 was the first study to test the safety of Sym023 (Anti-TIM-3) in patients with metastatic solid malignancies or lymphomas without standard therapies. Unfortunately, most of these studies have not reported their findings. Harding et al. evaluated the effect of a new TIM-3 monoclonal antibody (LY3321367) alone or in combination with anti-PD-L1 antibodies in patients with advanced solid tumors (Harding et al. 2021). LY3321367 showed acceptable safety with good pharmacokinetics. However, it had moderate anti-tumor activity (Harding et al. 2021).
Antibodies targeting TAM have also attracted widespread attention. Tyro3 is considered an oncogene in various cancers, and it is specifically overexpressed in melanoma and colorectal cancer. Demarest et al. used monoclonal antibodies (mAbs) against Tyro3 to test their effect on the survival of melanoma cell lines. The results indicated that Tyro3 could increase the survival of melanoma cells and could be blocked by monoclonal antibodies (Demarest et al. 2013). Another study by Chien et al. found that targeting Tyro3 inhibited colon cancer epithelial–mesenchymal transition and increased drug sensitivity (Chien et al. 2016). Other studies developed monoclonal antibodies directed against Axl. AXL can activate oncogenic signaling pathways, leading to the migration of leukemic cancer cells. According to Duan et al., anti-AXL high-affinity antibody DAXL-88 could block the binding of AXL to its ligand GAS6 and inhibit GAS6-induced tumor cell invasion (Duan et al. 2019). In addition, tyrosine kinase of the AXL receptor is also considered a potential therapeutic target for NSCLC (Okimoto and Bivona 2015). BA3011 is also a monoclonal antibody that selectively binds to AXL in NSCLC and shows good efficacy (Samuel et al. 2007). At present, researchers have also developed a variety of drugs, such as ADC and CAB-AXL-ADC. Moreover, mouse models with MERTK-deficient tumors showed increased infiltration of leukocytes and CD8 + T lymphocytes (Cook et al. 2013). Targeting MerTK could enhance adaptive immunity following radiotherapy (Tormoen et al. 2020). Therefore, studies targeting PS receptors may provide a new perspective in tumor immunotherapy.
Combination of PS-targeted therapy and immune checkpoint inhibitors
In recent years, the clinical success of immunotherapy has drawn new attention to immuno-biology. Although immune checkpoint blockers have improved the anti-tumor response and survival rate, only a few patients benefit from them (Zhang et al. 2018). Freimark et al. revealed that PS-targeted treatment of melanoma in mice enhanced the anti-tumor activity of ICIs (Freimark et al. 2016). PS-targeting antibody combined with ICIs significantly inhibited tumor growth compared with a single agent. Combined therapy also increased the ratio of CD4+/CD8+ tumor-infiltrating lymphocytes (TILs) and induced the expression of pro-inflammatory mediators including IL-2, IFN-γ, and TNF-α (Freimark et al. 2016). IFN-γ can lead to chronic inhibition of T cell signaling pathway and the production of indoleamine 2,3-dioxygenase 1 (IDO), resulting in signal attenuation (Chinnadurai et al. 2014). However, another study revealed that IFN-γ-permitted MDSCs inhibited T cell effects through PD-1 ligands without IDO (Pistillo et al. 2020). Multiple immune checkpoints can be co-expressed on activated TILs. Several studies have shown that PD-L1 combined with TILs may be more accurate than a single-agent therapy with PD-L1 in predicting the survival of colorectal cancer (Wang et al. 2020). Moreover, the expression of PD-1 and PD-L1 on TILs effectively predicts the prognosis in small cell lung cancer (Sun et al. 2020). In addition, the ratio of CD8+ T cells to MDSCs and Tregs in the tumor microenvironment was also increased.
Gray et al. used single or combined therapy composed of PS-targeting and anti-PD-1 antibodies in mice carrying syngeneic EMT-6 or E0771 tumors (Gray et al. 2016). They found that treatment with PS-targeting antibodies could inhibit tumor growth and significantly enhance the anti-tumor activity of ICIs, with a statistically significant survival advantage in single-agent therapy (Gray et al. 2016). Secondary tumors in mice showed relative resistance to combination therapy. Further, the spleen cells were induced to produce large amounts of IFN-γ. An immune profile analysis showed enhanced expression of TIL and immune monitoring-related cytokines with PS-targeting and anti-PD-1 combined therapy, which was consistent with a previous study by Freimark et al. (2016). A recent study revealed that the combined therapy of PS-targeting mch1N11 and ICIs had a better anti-tumor effect than ICIs monotherapy (Freimark et al. 2016; Gray et al. 2016). The combination therapy based on mch1N11 effectively promoted the proliferation of CD4+/CD8+ TILs and induced the expression of pro-inflammatory cytokines, including IL-2, IFN-γ, and TNF-α. The ratio of MDSCs and Treg cells in tumor and spleen tissues was also up-regulated. The above results were comparable to studies showing that ICIs’ combined blockade was more effective than monotherapy (Colli et al. 2017). Lag3 is a negative regulatory target expressed on T cells (Long et al. 2018). The addition of anti-Lag-3 to mch1N11 and anti-PD-1 targeted therapy increased the tumor suppression effect to 99% and the tumor regression rate to 80% (Budhu et al. 2021; Belzile et al. 2018). Triple therapy in the animal model significantly enhanced antigen presentation and reduced tumor-promoting genes. What is more, radiotherapy and chemotherapy are inducers of PS externalization that also up-regulate the expression of PD-L1 in tumor cells.
So far, PS-targeted drugs have been launched, some of them undergoing clinical trials as combination therapy in multiple cancers. Bavituximab is an immunomodulatory chimeric monoclonal antibody that promotes immune response by inhibiting phosphatidylserine signaling. Table 3 summarizes relevant clinical trials of PS antibody (bavituximab) targeting in tumors. A single-arm phase II clinical trial evaluating combination therapy with bavituximab and sorafenib in the treatment of advanced hepatocellular carcinoma showed that the combined regimen did not aggravate the toxicity related to sorafenib (Mokdad et al. 2019). Another study evaluated the efficacy of the combined regimen in a mouse xenograft model of liver cancer (Stasi and Cappuzzo 2014). The results showed that the combined regimen was better than single-agent therapy, with sorafenib significantly increasing PS exposure in tumor blood vessels (Stasi and Cappuzzo 2014). The host immune response, such as antibody-dependent cellular cytotoxicity, is activated with the binding of bavituximab to PS. This leads to blood vessel destruction and enhancement of anti-tumor immunity.
Studies have also pointed out that combined therapy can significantly reduce the tumor micro-vessel density and levels of M2 macrophage and increase tumor endothelial cell apoptosis and the number of M1 macrophages (Gerber et al. 2015). Several preclinical studies have shown that the combination of bavituximab and chemotherapy can combat various solid tumors. Digumarti et al. evaluated the efficacy of bavituximab combined with paclitaxel and carboplatin in treating metastatic non-small cell lung cancer (NSCLC) (Digumarti et al. 2014). This combination showed tolerable safety and potential efficacy as a first-line treatment for advanced metastatic NSCLC (Digumarti et al. 2014). However, the potential risks of this combination need further evaluation. For example, blocking of MerTk (PS receptor) on T cells reduces the tumor-killing effect. In addition, the safety of combined anti-PS therapy and other anti-cancer drugs remains largely unknown.
The main limitations or risks of PS-targeted therapy
Although immunotherapy has achieved significant clinical results, only a few patients have benefited from ICIs. Moreover, immunotherapy is also associated with immune-related adverse events (Fan et al. 2021). Therefore, it is paramount to improve cancer immunotherapies. Drugs targeting PS have good tolerability and anti-tumor activity. However, there is a gap in their clinical applicability. First, the inhibition of PS receptors may reduce tumor-killing effects. Second, the safety of anti-PS therapy in combination with other ICIs is largely unknown. Third, the role of PS as a global checkpoint inhibitor in cancer needs to be further explored. Fourth, it is not known whether all PS receptors have immunosuppressive effects. Therefore, multiple studies are required to evaluate the different PS receptors in different tumors to develop new agents targeting the pathways.
Perspective and conclusion
Current cancer immunotherapy research aims to eliminate immunotherapy-related adverse events. To achieve this, research has employed a combination of PS-targeting antibodies with emerging therapies, such as radiotherapy and chemotherapy, immunotherapy, and oncolytic viruses, in various tumors. Thus, this review provides innovative and cross-cutting ideas and directions for developing PS-targeted drugs.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Abbreviations
- ADCC:
-
Antibody-dependent cytotoxicity
- Ceacam-1:
-
Carcinoembryonic Antigen Cell Adhesion Molecule-1
- DC:
-
Dendritic cells
- FAS1:
-
Fascicle protein I
- FcRs:
-
Fc receptors
- HMGB1:
-
High Mobility Group Histone B1
- ICIs:
-
Immune checkpoint inhibitors
- IDO:
-
Indoleamine 2,3-dioxygenase 1
- IFN-γ:
-
Interferon γ
- IL-2:
-
Interleukin 2
- 1N11:
-
Monoclonal antibodies
- mAbs:
-
Monoclonal antibodies
- MDSCs:
-
Myeloid-derived suppressor cells
- NSCLC:
-
Non-small cell lung cancer
- PS:
-
Phosphatidylserine
- Rac1:
-
Rac family small GTPase 1
- RTKs:
-
Receptor tyrosine kinases
- TAM:
-
Tyro, Axl, and Mertk
- TIM:
-
T cells, immunoglobulin, and mucin
- TME:
-
Tumor microenvironment
- TNF:
-
Tumor necrosis factor
- TIL:
-
Tumor-infiltrating lymphocytes
- VCAM-1:
-
Vascular cell adhesion molecule 1
- β2GP1:
-
β2 Glycoprotein 1
References
Anderson AC (2014) Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res 2:393–398
Bajtay Z, Csomor E, Sandor N, Erdei A (2006) Expression and role of Fc- and complement-receptors on human dendritic cells. Immunol Lett 104:46–52
Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR (2020) Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol 13:100738
Belzile O, Huang X, Gong J, Carlson J, Schroit AJ, Brekken RA, Freimark BD (2018) Antibody targeting of phosphatidylserine for the detection and immunotherapy of cancer. Immunotargets Ther 7:1–14
Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT et al (2016) Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ 23:962–978
Budhu S, Giese R, Gupta A, Fitzgerald K, Zappasodi R, Schad S, Hirschhorn D, Campesato LF, De Henau O, Gigoux M et al (2021) Targeting phosphatidylserine enhances the anti-tumor response to tumor-directed radiation therapy in a preclinical model of melanoma. Cell Rep 34:108620
Burstyn-Cohen T, Maimon A (2019) TAM receptors, phosphatidylserine, inflammation, and cancer. Cell Commun Signal 17:156
Calianese DC, Birge RB (2020) Biology of phosphatidylserine (PS): basic physiology and implications in immunology, infectious disease, and cancer. Cell Commun Signal 18:41
Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB, Shan JS, Stopeck AT (2015) A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med 4:1051–1059
Chang W, Fa H, Xiao D, Wang J (2020) Targeting phosphatidylserine for cancer therapy: prospects and challenges. Theranostics 10:9214–9229
Chien CW, Hou PC, Wu HC, Chang YL, Lin SC, Lin SC, Lin BW, Lee JC, Chang YJ, Sun HS, Tsai SJ (2016) Targeting TYRO3 inhibits epithelial-mesenchymal transition and increases drug sensitivity in colon cancer. Oncogene 35:5872–5881
Chinnadurai R, Copland IB, Patel SR, Galipeau J (2014) IDO-independent suppression of T cell effector function by IFN-gamma-licensed human mesenchymal stromal cells. J Immunol 192:1491–1501
Colli LM, Machiela MJ, Zhang H, Myers TA, Jessop L, Delattre O, Yu K, Chanock SJ (2017) Landscape of combination immunotherapy and targeted therapy to improve cancer management. Cancer Res 77:3666–3671
Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, Redente E, Sandahl M, Hunter DM, Strunk KE et al (2013) MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest 123:3231–3242
David C, Nance JP, Hubbard J, Hsu M, Binder D, Wilson EH (2012) Stabilin-1 expression in tumor associated macrophages. Brain Res 1481:71–78
Dayoub AS, Brekken RA (2020) TIMs, TAMs, and PS- antibody targeting: implications for cancer immunotherapy. Cell Commun Signal 18:29
Demarest SJ, Gardner J, Vendel MC, Ailor E, Szak S, Huang F, Doern A, Tan X, Yang W, Grueneberg DA et al (2013) Evaluation of Tyro3 expression, Gas6-mediated Akt phosphorylation, and the impact of anti-Tyro3 antibodies in melanoma cell lines. Biochemistry 52:3102–3118
Di Stasi R, De Rosa L, D’Andrea LD (2020) Therapeutic aspects of the Axl/Gas6 molecular system. Drug Discov Today 25:2130–2148
Digumarti R, Bapsy PP, Suresh AV, Bhattacharyya GS, Dasappa L, Shan JS, Gerber DE (2014) Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer 86:231–236
Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, Kane LP, Lu B (2017) TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int J Mol Sci 18:645
Duan Y, Luo L, Qiao C, Li X, Wang J, Liu H, Zhou T, Shen B, Lv M, Feng J (2019) A novel human anti-AXL monoclonal antibody attenuates tumour cell migration. Scand J Immunol 90:e12777
Fan Y, Geng Y, Shen L, Zhang Z (2021) Advances on immune-related adverse events associated with immune checkpoint inhibitors. Front Med 15:33–42
Fendl B, Eichhorn T, Weiss R, Tripisciano C, Spittler A, Fischer MB, Weber V (2018) Differential interaction of platelet-derived extracellular vesicles with circulating immune cells: roles of TAM receptors, CD11b, and phosphatidylserine. Front Immunol 9:2797
Fischer K, Voelkl S, Berger J, Andreesen R, Pomorski T, Mackensen A (2006) Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood 108:4094–4101
Francis RJ, Kotecha S, Hallett MB (2013) Ca2+ activation of cytosolic calpain induces the transition from apoptosis to necrosis in neutrophils with externalized phosphatidylserine. J Leukoc Biol 93:95–100
Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH (2010) TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 235:172–189
Freimark BD, Gong J, Ye D, Gray MJ, Nguyen V, Yin S, Hatch MM, Hughes CC, Schroit AJ, Hutchins JT et al (2016) Antibody-mediated phosphatidylserine blockade enhances the antitumor responses to CTLA-4 and PD-1 antibodies in melanoma. Cancer Immunol Res 4:531–540
Frey B, Gaipl US (2011) The immune functions of phosphatidylserine in membranes of dying cells and microvesicles. Semin Immunopathol 33:497–516
Gerber DE, Stopeck AT, Wong L, Rosen LS, Thorpe PE, Shan JS, Ibrahim NK (2011) Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res 17:6888–6896
Gerber DE, Hao G, Watkins L, Stafford JH, Anderson J, Holbein B, Oz OK, Mathews D, Thorpe PE, Hassan G et al (2015) Tumor-specific targeting by Bavituximab, a phosphatidylserine-targeting monoclonal antibody with vascular targeting and immune modulating properties, in lung cancer xenografts. Am J Nucl Med Mol Imaging 5:493–503
Gerber DE, Horn L, Boyer M, Sanborn R, Natale R, Palmero R, Bidoli P, Bondarenko I, Germonpre P, Ghizdavescu D et al (2018) Randomized phase III study of docetaxel plus bavituximab in previously treated advanced non-squamous non-small-cell lung cancer. Ann Oncol 29:1548–1553
Gosk S, Moos T, Gottstein C, Bendas G (2008) VCAM-1 directed immunoliposomes selectively target tumor vasculature in vivo. Biochim Biophys Acta 1778:854–863
Graham DK, DeRyckere D, Davies KD, Earp HS (2014) The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer 14:769–785
Gray MJ, Gong J, Hatch MM, Nguyen V, Hughes CC, Hutchins JT, Freimark BD (2016) Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res 18:50
Grilley-Olson JE, Weiss J, Ivanova A, Villaruz LC, Moore DT, Stinchcombe TE, Lee C, Shan JS, Socinski MA (2018) Phase Ib study of bavituximab with carboplatin and pemetrexed in chemotherapy-naive advanced nonsquamous non-small-cell lung cancer. Clin Lung Cancer 19:e481–e487
Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, Szpurka AM, Yamamoto N, Doi T, Fu S et al (2021) Blocking TIM-3 in treatment-refractory advanced solid tumors: a phase Ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin Cancer Res 27:2168–2178
He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR (2018) TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther 11:7005–7009
Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA, Klibanov AL, Yan Z, Mandell JW, Ravichandran KS (2013) Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497:263–267
Hosseini H, Li Y, Kanellakis P, Tay C, Cao A, Tipping P, Bobik A, Toh BH, Kyaw T (2015) Phosphatidylserine liposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive IgM producing B1a lymphocytes. Cardiovasc Res 106:443–452
Iida Y, Sunami E, Yamashita H, Hiyoshi M, Ishihara S, Yamaguchi H, Inoue A, Makide K, Tsuno NH, Aoki J et al (2015) Phosphatidylserine-specific phospholipase A1 (PS-PLA1) expression in colorectal cancer correlates with tumor invasion and hematogenous metastasis. Anticancer Res 35:1459–1464
Ishii H, Mori T, Shiratsuchi A, Nakai Y, Shimada Y, Ohno-Iwashita Y, Nakanishi Y (2005) Distinct localization of lipid rafts and externalized phosphatidylserine at the surface of apoptotic cells. Biochem Biophys Res Commun 327:94–99
Jie HB, Srivastava RM, Argiris A, Bauman JE, Kane LP, Ferris RL (2017) Increased PD-1(+) and TIM-3(+) TILs during cetuximab therapy inversely correlate with response in head and neck cancer patients. Cancer Immunol Res 5:408–416
Kamal Y, Schmit SL, Frost HR, Amos CI (2020) The tumor microenvironment of colorectal cancer metastases: opportunities in cancer immunotherapy. Immunotherapy 12:1083–1100
Kasikara C, Kumar S, Kimani S, Tsou WI, Geng K, Davra V, Sriram G, Devoe C, Nguyen KN, Antes A et al (2017) Phosphatidylserine sensing by TAM receptors regulates AKT-dependent chemoresistance and PD-L1 expression. Mol Cancer Res 15:753–764
Klein ME, Rieckmann M, Sedding D, Hause G, Meister A, Mader K, Lucas H (2021) Towards the development of long circulating phosphatidylserine (PS)- and phosphatidylglycerol (PG)-enriched anti-inflammatory liposomes: is PEGylation effective? Pharmaceutics 13:282
Kong X, Fu M, Niu X, Jiang H (2020) Comprehensive analysis of the expression, relationship to immune infiltration and prognosis of TIM-1 in cancer. Front Oncol 10:1086
Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, Li J, Li F, Tan HB (2020) Immune cells within the tumor microenvironment: biological functions and roles in cancer immunotherapy. Cancer Lett 470:126–133
Li J, Gray BD, Pak KY, Ng CK (2019) Targeting phosphatidylethanolamine and phosphatidylserine for imaging apoptosis in cancer. Nucl Med Biol 78–79:23–30
Li J, Shayan G, Avery L, Jie HB, Gildener-Leapman N, Schmitt N, Lu BF, Kane LP, Ferris RL (2016) Tumor-infiltrating Tim-3(+) T cells proliferate avidly except when PD-1 is co-expressed: evidence for intracellular cross talk. Oncoimmunology 5:e1200778
Lino DOC, Freitas IA, Meneses GC, Martins AMC, Daher EF, Rocha JHC, Silva Junior GB (2019) Interleukin-6 and adhesion molecules VCAM-1 and ICAM-1 as biomarkers of post-acute myocardial infarction heart failure. Braz J Med Biol Res 52:e8658
Liu JF, Wu L, Yang LL, Deng WW, Mao L, Wu H, Zhang WF, Sun ZJ (2018) Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res 37:44
Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, Chen H (2018) The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer 9:176–189
Lu X, Yang L, Yao D, Wu X, Li J, Liu X, Deng L, Huang C, Wang Y, Li D, Liu J (2017) Tumor antigen-specific CD8(+) T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol 313:43–51
Luster TA, He J, Huang X, Maiti SN, Schroit AJ, de Groot PG, Thorpe PE (2006) Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti-tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J Biol Chem 281:29863–29871
Martinez-Flores JA, Serrano M, Perez D, Lora D, Paz-Artal E, Morales JM, Serrano A (2015) Detection of circulating immune complexes of human IgA and beta 2 glycoprotein I in patients with antiphospholipid syndrome symptomatology. J Immunol Methods 422:51–58
Masuda A, Yoshida M, Shiomi H, Morita Y, Kutsumi H, Inokuchi H, Mizuno S, Nakamura A, Takai T, Blumberg RS, Azuma T (2009) Role of Fc receptors as a therapeutic target. Inflamm Allergy Drug Targets 8:80–86
Mokdad AA, Zhu H, Beg MS, Arriaga Y, Dowell JE, Singal AG, Yopp AC (2019) Efficacy and safety of bavituximab in combination with sorafenib in advanced hepatocellular carcinoma: a single-arm, open-label, phase II clinical trial. Target Oncol 14:541–550
Murphy KA, Erickson JR, Johnson CS, Seiler CE, Bedi J, Hu P, Pluhar GE, Epstein AL, Ohlfest JR (2014) CD8+ T cell-independent tumor regression induced by Fc-OX40L and therapeutic vaccination in a mouse model of glioma. J Immunol 192:224–233
Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K (2009) Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113:3821–3830
Naqvi AR, Fordham JB, Nares S (2016) MicroRNA target Fc receptors to regulate Ab-dependent Ag uptake in primary macrophages and dendritic cells. Innate Immun 22:510–521
Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ (2011) Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res 71:3540–3551
Ocana-Guzman R, Torre-Bouscoulet L, Sada-Ovalle I (2016) TIM-3 regulates distinct functions in macrophages. Front Immunol 7:229
Okimoto RA, Bivona TG (2015) AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer (auckl) 6:27–34
Park M, Kang KW (2019) Phosphatidylserine receptor-targeting therapies for the treatment of cancer. Arch Pharm Res 42:617–628
Park SY, Kim SY, Jung MY, Bae DJ, Kim IS (2008) Epidermal growth factor-like domain repeat of stabilin-2 recognizes phosphatidylserine during cell corpse clearance. Mol Cell Biol 28:5288–5298
Park SY, Kim SY, Kang KB, Kim IS (2010) Adaptor protein GULP is involved in stabilin-1-mediated phagocytosis. Biochem Biophys Res Commun 398:467–472
Park SY, Bae DJ, Kim MJ, Piao ML, Kim IS (2012) Extracellular low pH modulates phosphatidylserine-dependent phagocytosis in macrophages by increasing stabilin-1 expression. J Biol Chem 287:11261–11271
Park SY, Yun Y, Lim JS, Kim MJ, Kim SY, Kim JE, Kim IS (2016) Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat Commun 7:10871
Park SY, Kim IS (2019) Stabilin receptors: role as phosphatidylserine receptors. Biomolecules 9:387
Pistillo MP, Carosio R, Banelli B, Morabito A, Mastracci L, Ferro P, Varesano S, Vene R, Poggi A, Roncella S (2020) IFN-gamma upregulates membranous and soluble PD-L1 in mesothelioma cells: potential implications for the clinical response to PD-1/PD-L1 blockade. Cell Mol Immunol 17:410–411
Quan H, Kim Y, Park HC, Yang HC (2018) Effects of phosphatidylserine-containing supported lipid bilayers on the polarization of macrophages. J Biomed Mater Res A 106:2625–2633
Ran S, Gao B, Duffy S, Watkins L, Rote N, Thorpe PE (1998) Infarction of solid Hodgkin’s tumors in mice by antibody-directed targeting of tissue factor to tumor vasculature. Cancer Res 58:4646–4653
Ran S, Downes A, Thorpe PE (2002) Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res 62:6132–6140
Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE (2005) Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res 11:1551–1562
Romero D (2016) Immunotherapy: PD-1 says goodbye, TIM-3 says hello. Nat Rev Clin Oncol 13:202–203
Sabatos-Peyton CA, Nevin J, Brock A, Venable JD, Tan DJ, Kassam N, Xu F, Taraszka J, Wesemann L, Pertel T et al (2018) Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 7:e1385690
Samuel MS, Lundgren-May T, Ernst M (2007) Identification of putative targets of DNA (cytosine-5) methylation-mediated transcriptional silencing using a novel conditionally active form of DNA methyltransferase 3a. Growth Factors 25:426–436
Serinkan BF, Gambelli F, Potapovich AI, Babu H, Di Giuseppe M, Ortiz LA, Fabisiak JP, Kagan VE (2005) Apoptotic cells quench reactive oxygen and nitrogen species and modulate TNF-alpha/TGF-beta1 balance in activated macrophages: involvement of phosphatidylserine-dependent and -independent pathways. Cell Death Differ 12:1141–1144
Sharma B, Kanwar SS (2018) Phosphatidylserine: a cancer cell targeting biomarker. Semin Cancer Biol 52:17–25
Shurin MR, Potapovich AI, Tyurina YY, Tourkova IL, Shurin GV, Kagan VE (2009) Recognition of live phosphatidylserine-labeled tumor cells by dendritic cells: a novel approach to immunotherapy of skin cancer. Cancer Res 69:2487–2496
Stasi I, Cappuzzo F (2014) Profile of bavituximab and its potential in the treatment of non-small-cell lung cancer. Lung Cancer (auckl) 5:43–50
Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, Huang L, Vijh S, Johnson S, Bonvini E, Koenig S (2007) Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res 67:8882–8890
Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, Leighty RM, Roers A, Karp CL, Muller W, Trinchieri G (2013) Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J Clin Invest 123:4859–4874
Sun C, Zhang L, Zhang W, Liu Y, Chen B, Zhao S, Li W, Wang L, Ye L, Jia K et al (2020) Expression of PD-1 and PD-L1 on tumor-infiltrating lymphocytes predicts prognosis in patients with small-cell lung cancer. Onco Targets Ther 13:6475–6483
Tang SQ, Tang LL, Mao YP, Li WF, Chen L, Zhang Y, Guo Y, Liu Q, Sun Y, Xu C, Ma J (2020) The pattern of time to onset and resolution of immune-related adverse events caused by immune checkpoint inhibitors in cancer: a pooled analysis of 23 clinical trials and 8,436 patients. Cancer Res Treat. https://doi.org/10.4143/crt.2020.790
Tormoen GW, Blair TC, Bambina S, Kramer G, Baird J, Rahmani R, Holland JM, McCarty OJT, Baine MJ, Verma V et al (2020) Targeting MerTK enhances adaptive immune responses after radiation therapy. Int J Radiat Oncol Biol Phys 108:93–103
Twarda-Clapa A, Labuzek B, Krzemien D, Musielak B, Grudnik P, Dubin G, Holak TA (2018) Crystal structure of the FAS1 domain of the hyaluronic acid receptor stabilin-2. Acta Crystallogr D Struct Biol 74:695–701
van der Meer JH, van der Poll T, van’t Veer C (2014) TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood 123:2460–2469
Vaupel P, Multhoff G (1887) Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol 2017:8
Wang T, Chu Z, Lin H, Jiang J, Zhou X, Liang X (2014) Galectin-3 contributes to cisplatin-induced myeloid derived suppressor cells (MDSCs) recruitment in Lewis lung cancer-bearing mice. Mol Biol Rep 41:4069–4076
Wang JL, Yu T, Sun TT, Feng Y, Xiong H, Fang JY (2020) PD-L1 overexpression on tumor-infiltrating lymphocytes related to better prognosis of colorectal cancer. Clin Lab 66. https://doi.org/10.7754/Clin.Lab.2020.200325
Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S, Li T, Chen F, Yang Y (2018) Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer 17:20
Zhang B, Wu Q, Zhou YL, Guo X, Ge J, Fu J (2018) Immune-related adverse events from combination immunotherapy in cancer patients: a comprehensive meta-analysis of randomized controlled trials. Int Immunopharmacol 63:292–298
Zhou J, Jiang Y, Zhang H, Chen L, Luo P, Li L, Zhao J, Lv F, Zou D, Zhang Y, Jing Z (2019) Clinicopathological implications of TIM3(+) tumor-infiltrating lymphocytes and the miR-455-5p/Galectin-9 axis in skull base chordoma patients. Cancer Immunol Immunother 68:1157–1169
Zhou X, Yao Z, Yang H, Liang N, Zhang X, Zhang F (2020) Are immune-related adverse events associated with the efficacy of immune checkpoint inhibitors in patients with cancer? A systematic review and meta-analysis. BMC Med 18:87
Zohar DN, Shoenfeld Y (2018) Antibody targeting of phosphatidylserine for detection and immunotherapy of cancer. Immunotargets Ther 7:51–53
Acknowledgements
Thanks to Li Zhang for helping me with the writing process.
Funding
This work was supported by grants from the Training Project of Key Talents of Youth Medicine in Jiangsu province, China (No.QNRC2016330) and High-level talent “six one projects” top talent scientific research project of Jiangsu Province (No.LGY2019034).
Author information
Authors and Affiliations
Contributions
JZ and ZD drafted the manuscript in detail. JZ and CY researched the literatures and drew figures. ZD and CY counted and plotted the diagram and table. DT and DW critically revised the article for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, J., Dai, Z., Yan, C. et al. Blocking antibody-mediated phosphatidylserine enhances cancer immunotherapy. J Cancer Res Clin Oncol 147, 3639–3651 (2021). https://doi.org/10.1007/s00432-021-03792-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03792-3