
Abstract
Purpose
Dovitinib (TKI258) is an oral multi-tyrosine kinase inhibitor of FGFR, VEGFR, PDGFR β, and c-Kit. Since dovitinib is able to cross the blood–brain barrier and targets brain tumor-relevant pathways, we conducted a phase I trial to demonstrate its safety in recurrent glioblastoma (GBM).
Patients and methods
Patients with first or second GBM recurrence started treatment with the maximal tolerated dose (MTD) previously established in systemic cancer patients (500 mg/d, 5 days on/2 days off). A modified 3 + 3 design in three cohorts (500, 400, 300 mg) was used.
Results
Twelve patients were enrolled. Seventy-two adverse events (AEs) occurred and 16.7 % of AEs were classified as ≥CTC grade 3 toxicity, mainly including hepatotoxicity and hematotoxicity. Only one out of six patients of the 300-mg cohort showed grade 3 toxicity. The PFS-6 rate was 16.7 %, and it was not associated with detection of the FGFR-TACC gene fusion in the tumor.
Conclusion
Dovitinib is safe in patients with recurrent GBM and showed efficacy in only some patients unselected for target expression. The recommended phase II dose of 300 mg would be substantially lower than the recently established MTD in systemic cancer patients. Further personalized trials are recommended.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The malignancy of glioblastoma (GBM) is mediated by its highly infiltrative nature and the tremendous capacity of resistance to therapeutic approaches. Most patients experience progressive or recurrent disease as soon as 7 months after diagnosis, still under the effect of first-line therapy (Chinot et al. 2014; Gilbert et al. 2014). Despite several available salvage therapies, the median survival time after first recurrence remains short (8–12 months; Batchelor et al. 2013; Taal et al. 2014). There is still no universally accepted standard of care for patients with relapsed GBM although many trials have been performed in the last decades. Neurosurgical intervention and/or re-irradiation are applicable for a subset of patients at the time of relapse. Neither cytotoxic chemotherapies nor inhibition of signal cascades with small molecules or antibodies or the combination of both were able to substantially alter the course of the disease. Even though median overall survival rates have shown to increase during the last several years, this may at least to some extent be due to an improved and embracing care of patients by specialized neurooncology centers and palliative services.
The detection of druggable genetic and transcriptional alterations in cancer serves as a rationale for target-specific therapies and has changed treatment strategies in many cancer types such as malignant melanoma, lung cancer or renal-cell carcinoma (Chapman et al. 2011; Shaw et al. 2013; Motzer et al. 2013). For GBM, a multitude of genomic alterations are known to result in deregulated signal pathways suitable for the development of molecular-targeted treatment approaches. To date, however, none of the subsequently derived therapies showed appropriate efficacy rates in randomized trials. Ongoing exploration of targeted therapies nevertheless remains a valid approach to identify potential new treatment strategies for GBM patients, because some pathways or pathway combinations have still not been investigated in GBM.
In this context, the oral multi-tyrosine-kinase inhibitor dovitinib (TKI258) could be considered as a promising compound to treat GBM patients. TKI258 is directed against several brain tumor-relevant pathways and targets, namely fibroblast growth factor receptors (FGFR) 1–3, platelet-derived growth factor receptor β, vascular endothelial growth factor receptors (VEGFR) 1–3, as well as c-Kit, RET, TrkA, CSF 1R, and FLT3 (Renhowe et al. 2009). Some mono-targeted approaches are already under investigation in clinical trials. A simultaneous inhibition of the specified targets of TKI258 could therefore still lead to new and interesting avenues for the treatment of GBM.
Dovitinib has been under investigation in many different solid tumors and demonstrated some activity in advanced and pretreated cancer stages (Escudier et al. 2014; Motzer et al. 2014). Since dovitinib was well tolerated and has shown the capacity to cross the blood–brain barrier, its application in brain tumor patients is reasonable. Here, we report the results of a phase I trial of dovitinib in patients with recurrent glioblastoma.
Patients and methods
General trial specifications
This was a prospective, open-label, non-randomized, mono-center, single-arm phase I trial (EudraCT No. 2012-005460-95; ClinicalTrials.gov: NCT01972750) and approved by the local ethics committee. All patients gave written consent before performing any study-related activity. All trial procedures adhered to the principles of the Declaration of Helsinki and the Guidelines of Good Clinical Practice.
The trial was supported by Novartis Pharma GmbH Germany (internal code: CTKI258ADE02T).
Objectives, endpoints, and statistical analysis
Primary objective was to demonstrate that administration of two cycles (2 × 28 days) of dovitinib is safe in patients with relapsed GBM. Secondary objective was to assess preliminary data on activity and toxicity of the given regimen in patients with relapsed GBM and to establish a recommended dose for a phase II study.
Primary endpoint was safety and tolerability, and was based on the frequency of dose-limiting toxicities (DLTs). Secondary endpoints were best tumor response (CR, PR) according RANO criteria (Wen et al. 2010), overall safety, disease control rate (CR + PR + SD), progression-free survival rate at 6 months (PFS-6), and overall survival (OS) after initiation of therapy.
Analyses are descriptive only; therefore, no statistical rationale had to be explained. The intent-to-treat and safety population included all enrolled subjects who received at least one dose of study medication.
Inclusion/exclusion criteria
Adult female and male subjects (age ≥18 years) with first or second recurrence of histologically confirmed GBM and a Karnofsky performance score (KPS) >60 %, ECOG ≤2, or WHO <2 were enrolled. Further criteria were:
Prior treatment with radiotherapy and temozolomide was required, and a maximum of two prior chemotherapies was permitted. Radiotherapy within 4 weeks prior to the diagnosis of progression was prohibited. Patients were required to have adequate organ function as measured by absolute neutrophil count (ANC) ≥1500/mm3, platelets ≥100,000/mm3, a hemoglobin >9 g/dL, total bilirubin ≤1.5 × upper limit of normal (ULN), aspartate/alanine aminotransferase levels ≤3.0 × ULN, and serum creatinine ≤ 1.5 × ULN. Resurgery before study inclusion was allowed and had to confirm disease progression histologically. They were not enrolled if MRI showed evidence of current/active intratumoral hemorrhage, the corrected QT-time (QTc) was elongated (>450 ms in male and ≥460 ms in female patients), and/or any impaired cardiac function or clinically significant cardiac diseases could be confirmed.
Patients with any clinically significant medical or surgical condition, according to investigators’ discretion, were excluded from participation. A history of myocardial infarction, cerebral stroke, pulmonary embolism, and untreated deep-vein thrombosis within 6 months prior to starting dovitinib was not allowed.
Treatment plan
In patients with systemic cancer, the MTD for dovitinib was already defined (500 mg/d 5 days on/2 days off). To assess the safety and feasibility of this MTD in brain tumor patients, we used a modification of the traditional 3 + 3 design. A minimum of six patients should be entered in the first cohort and followed for two cycles (2 × 28 days) of dovitinib (500 mg/d 5 days on/2 days off). If ≥2/6 patients exhibited DLT during the first two cycles, a new cohort of six patients would be treated with a reduced dose of dovitinib (400 mg/d 5 days on/2 days off). If ≥2/6 patients of this dose-reduced cohort exhibited DLT during the first two cycles, a new cohort of six patients would be treated with a further reduced dose of dovitinib 300 mg/d 5 days on/2 days off. If <2/6 patients of this new cohort showed DLT, dovitinib 300 mg/d 5 days on/2 days off would be the recommended phase II dose.
Study procedures
During the first cycle, a visit was scheduled at day 15 with examination of vital signs, physical and neurological examination, concomitant medication, KPS, CTCAE (Common Terminology Criteria for Adverse Events) evaluation, ECG, laboratory investigation. Regular visits were performed at the beginning of each cycle (every 28 days). Every 8 weeks after start of treatment, standard cerebral Gd-MRI (Gadolinium-enhanced-MRI) was performed within 5 days before or after scheduled visit. At MRI visits, patients had a physical and neurological examination, and evaluations of KPS, Mini-Mental States, and quality of life. At progression or discontinuation, a visit for disruption of treatment was performed. Follow-up visits were intended after withdrawal every 8–12 weeks until the patient’s death or until the patient was lost to follow-up.
Complete blood count, CRP, serum chemistry (including amylase and lipase), urine analysis, coagulation, and pregnancy test were performed at least every 28 days at the beginning of each cycle.
For cardial safety, a 12-lead electrocardiogram and an echocardiography (ECG, left ventricular function assessed by 2-D ECG) were performed at baseline, an additional ECG was obtained during the first cycle and at the end of treatment.
Safety and dose adjustments
Safety assessments consisted of monitoring and recording all adverse events and serious adverse events, the regular monitoring of coagulation, hematology and blood chemistry, ECG, echocardiography and spirometry (at study start), measurement of vital signs, and a physical examination. All laboratory reports were reviewed by the investigator. Adverse events (AE) were recorded between the visit with the first study-related procedure until 30 days after last study-related procedure (including follow-up) and the relationship to dovitinib was evaluated. The CTCAE version 4.03 was the reference for assessing the severity of AEs. In case of intolerability or dose-limiting toxicities, dovitinib was reduced by one dose level. Reduced dovitinib dosages could not be re-escalated. Only two dose reductions were allowed (Suppl Table 1). A dose interruption (regardless of the reason) lasting >21 days resulted in discontinuation of the study treatment.
Response assessment and survival calculation
Imaging response and progression was evaluated using the response criteria defined by the Response Assessment in Neuro-Oncology Working Group (RANO; Wen et al. 2010) based on the first MRI after initiation of study treatment.
PFS was defined from first day of dovitinib treatment to progression or death, and OS from first day of dovitinib treatment to death. Statistical calculation for median PFS and median OS with 95 % confidence interval was made using IBM’s SPSS Statistics software version 22.
Immunohistochemical analysis
Formalin-fixed paraffin-embedded tissue samples were available from 10/12 patients for post hoc biomarker analysis. After neuropathological routine diagnostics to confirm diagnosis of GBM additional analysis for this study comprised immunohistochemistry with monoclonal antibodies directed against the fibroblast growth factor 3 (FGFR3). Two different antibodies were used for potential detection of the FGFR3 wild type and FGFR3-TACC-fusion protein, respectively, based on the epitope specificity with the one directed against a C-terminal epitope (ab137084, dil. 1/50; Abcam, Cambridge, UK) capable of detecting the wild-type protein, and the one directed against a N-terminal epitope (sc-13121, dil. 1/100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) capable of detecting the fusion protein (Di Stefano et al. 2015). Immunohistochemistry was performed on a Ventana Benchmark XT Immunostainer (Roche Ventana, Darmstadt, Germany). Antibody labeling was assessed microscopically and scored visually by an experienced neuropathologist without access to clinical data (blinded scoring procedure). Tissues were classified as positive or negative for the respective epitope. Density of labeled tumor cells were semiquantitatively estimated as negative (no labeling), low (<30 %), intermediate (30–70 %), or high (>70 %). Intensity of staining was distributed in three subgroups as weak positive (1+), moderate positive (2+), or strong positive (3+).
RNA extraction and assessment of FGFR-TACC gene fusion
In order to test for gene fusion of FGFR1/3 and TACC1/3 as a potential molecular mechanism for correlation to treatment response, we performed PCR using patient cDNA for detection of amplicons spanning the FGFR1/3 and TACC1/3 gene regions. Only samples with tumor content >80 % and sufficient RNA quality (A260/A280 >1.7) were used in this study. Tumor content was determined histologically in FFPE tissue samples before RNA extraction to avoid contamination by normal tissue, hemorrhage, or necrosis. Total RNA was extracted from FFPE samples of our patients (including the two patients who were on treatment for at least 6 months) using the AllPrep® DNA/RNA kit (Qiagen, Hilden, Germany) according to manufacturers’ instructions. One microgram of total RNA was reversely transcribed into cDNA using the Invitrogen SuperScript® III first-strand kit (Thermo Fisher Scientific, Life Technologies GmbH, Darmstadt, Germany) and random hexamers. Two microliters of undiluted cDNA was used as template in a PCR using conditions and primers as described by Singh and colleagues (2012): As a positive control, we used the plasmids pcDNA3.1-FGFR3e18-TACC3e11 (‘FGFR3-TACC3-LONG’) and pcDNA3.1-FGFR3e18-TACC3e4 (‘FGFR3-TACC3-SHORT’, kindly provided by Wei Zhang and David Cogdell, MD Anderson Cancer Center). As internal control, we amplified the housekeeping gene GAPDH.
Results
Patient characteristics
Between November 2013 and June 2015, 12 patients with first (N = 6) or second recurrence (N = 6) of GBM were enrolled in this trial. One patient had a hemiparesis due to infarction following primary surgery and was enrolled despite a KPS score of 60 %. Detailed patient characteristics are given in Table 1 and Supplementary Table 2.
Safety and toxicity evaluation
A total of 33 treatment cycles were initiated. We observed 72 adverse events in 11 patients that were classified as related (possibly, probably, or definitely) to study treatment. Six out of 13 serious adverse events were evaluated as possibly caused by dovitinib. No SUSAR (suspected unexpected severe adverse reaction) occurred. The most common adverse events in our trial were fatigue, increased liver enzymes, diarrhea, and general discomfort. The overall rate of ≥CTC grade 3 toxicity was 16.7 % and mainly restricted to increased liver enzymes, hematotoxicity, and one deep-vein thrombosis. Surprisingly, the spectrum and frequency of adverse events in our brain tumor population differed markedly from patients with systemic tumors as described in the current Investigators Brochure (IB). The so far most common adverse events of dovitinib were observed only to a small extent in our patient population: diarrhea 33.3 versus ~68 %, nausea 25 versus ~67 %, vomiting 8.3 versus ~60 %. As expected, fatigue occurred in half of our patients. In contrast to previous trials, a higher rate of hepatotoxicity detected by elevated liver enzymes (ALAT 50 vs. ~22 %; AP 33.3 vs. ~16 %; GGT 33.3 vs. ~9 %) and a higher rate of hematotoxicity detected by decreased neutrophil count (16.7 vs. ~5 %) and decreased platelet count (16.7 vs. ~10 %) were observed. Table 2 summarizes the observed adverse events classified as related to study treatment.
According to the primary objective, we observed 66 AEs during the administration of two cycles of dovitinib including an extra observation period of 30 days. Some of these AEs were evaluated as DLT (Table 3). Since the first two patients of cohort I (500 mg) and two of four patients of the cohort II (400 mg) exhibited a DLT during the first two cycles, another cohort of patients (III) with 300 mg was enrolled. Only one out of six patients of this cohort developed a DLT. Beyond the second treatment cycle, no DLTs were observed. Therefore, treatment with dovitinib in recurrent GBM patients appears to be safe and tolerable when applied in the lower dosage of 300 mg (5 days on/2 days off per week). The recommended dovitinib dose for a phase II brain tumor trial therefore is 300 mg (5 days on/2 days off).
Response and survival
Radiographic response on the first MRI control 8 weeks after study inclusion was assessed in 11/12 patients. One patient was discontinued from therapy due to significant clinical deterioration rejecting further MRIs. None of the patients had complete or partial response. Four patients achieved stable disease, and seven patients had progressive disease on first MRI after study inclusion.
The median PFS after the initiation of dovitinib treatment was 1.8 months (95 % CI 1.7–1.9 months). Two patients were progression-free after 6 months (PFS-6 16.7 %). The median OS since initiation of dovitinib treatment was 9.5 months (95 % CI 2.6–16.4 months).
Biomarker analysis (FGFR3 and FGFR3-TACC3 expression)
Since it is known that only a small subset of glioma patients contains a FGFR-TACC-fusion protein and benefits from a FGFR-directed targeted therapy (Di Stefano et al. 2015; Singh et al. 2012; Tabernero et al. 2015), we analyzed our patients’ cohort for this potential biomarker.
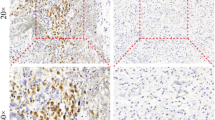
Ten out of ten patients including the two patients who showed a comparably long PFS under TKI258 showed positive immunolabeling results with the C-terminal-directed FGFR3 antibody in the tumor cells, indicating FGFR3 wild type and probably the usual expression levels in GBMs. Distribution of immunopositive tumor cells varied from <30 to >70 % while intensity was weak to strong (Fig. 1a–c). Since FGFR3 is expressed in normal brain tissue, some neurons and glial cells were observed immunopositive as well.

Expression of FGFR3. Immunohistochemical FGFR3 labeling of tumor cells varied from low (a), intermediate (b), to high (c) frequencies. Scale bars, 50 µm. RT-PCR detected no FGFR3-TACC3 fusion protein in the investigated samples (d). Overexpressing plasmids were used as positive controls for PCR, and the housekeeping gene GAPDH was used to check for cDNA integrity
Interestingly, neither the labeling with the N-terminal directed FGFR3 antibody, suitable to detect the FGFR3-TACC-fusion protein, nor PCR analysis for FGFR-TACC gene fusion showed a positive signal in the investigated patients, in particular in the two patients who were on treatment for at least 6 months (for FGFR3-TACC3 see Fig. 1d).
Discussion
This phase I trial showed that dovitinib is a safe and a (clinically) feasible treatment in patients with recurrent GBM. However, the spectrum and frequency of side effects differed between our brain tumor patients and the recently described patients with systemic tumors. Our recommended phase II dose of 300 mg would be substantially lower than the recently established MTD (500 mg) in systemic cancer patients. Moreover, with a PFS-6 of ~17 %, the molecular-targeted compound dovitinib showed only moderate efficacy at best, but in a population not profiled for their putative responsiveness to a molecular-targeted therapy and in a setting of a phase I trial.
The reason for the different toxicity profiles in our cohort remains unclear. One explanation could include the different pretreatment regimes applied in brain tumor patients compared to patients with systemic cancers. Temozolomide is widely administered in patients with GBM. Brain tumor patients were excluded from previously published trials with dovitinib. Regarding the observed adverse events in our trial, one might speculate that temozolomide followed by dovitinib results in a higher risk of hepatotoxicity and hematotoxicity. Importantly, although dovitinib is able to cross the blood–brain barrier, we did not observe increased CNS toxicity in our cohort, e.g., a higher frequency of seizures or alterations in patients’ consciousness.
The overall efficacy of dovitinib was limited and not encouraging in our trial. However, it is important to note that assessing preliminary data on efficacy of the given regimen was not the primary objective of our phase I trial and should therefore not be overrated. Nevertheless, two patients were on treatment for at least 6 months, which indicates clinical response on dovitinib in at least a small subgroup of patients. Interestingly, one of these patients showed clinical response despite the presence of unfavorable prognostically relevant characteristics (KPS 60, unmethylated MGMT promotor; Suppl Table 2).
Benefit for a subgroup only is a frequently observed result of targeted trials (Gilbert et al. 2012; Hutterer et al. 2014; Wen et al. 2014), and all of the applied treatment regimens failed to improve overall survival rates in the whole, unselected patient population (Batchelor et al. 2013; Reardon et al. 2005; Wick et al. 2010; Stupp et al. 2014). Assessing the activity of a targeted approach without prior profiling of the relevant target expression in the respective trial cohort might be the reason for these discouraging results. Therefore, identifying subgroups of patients based on expression/methylation profiles prior to treatment should be encouraged in future brain tumor trials to relate likely responders to the molecular-targeted approach.
A very small subset of glioma patients exhibits a FGFR-TACC-fusion protein (Di Stefano et al. 2015; Singh et al. 2012). In some recently published studies, a clinical response was observed in FGFR3-TACC3-positive glioma treated with an FGFR inhibitor supporting clinical studies of FGFR inhibition in FGFR-TACC-positive patients (Di Stefano et al. 2015; Tabernero et al. 2015). Although one of the major targets of TKI258 is FGFR (esp. FGFR3), we could not detect a FGFR-TACC gene fusion in our two patients with a PFS > than 6 months. One explanation would be that activity of the multi-kinase inhibitor dovitinib was associated with targets other than the FGFR in these patients. A further limitation was that only tissue from the primary and not from the recurrent tumor was available for molecular analysis. A recently published comprehensive analysis of genomic changes between primary and recurrent tumor tissue revealed substantial genetic differences so that stereotactic biopsies of tumor recurrence and molecular re-analysis are strongly encouraged (Kim et al. 2015). But even in case of recurrent tumor biopsy, the expression of targets may vary in the tissue due to intratumoral heterogeneity (Snuderl et al. 2011; Szerlip et al. 2012).
In summary, the results of our phase I trial showed that dovitinib 300 mg 5 days on/2 days off is safe in patients with recurrent GBM and will be the recommended phase II dose. A future phase II trial should be designed in a personalized approach in order to assess more reliably data on the activity of the targeted therapy. The treatment may be particularly suited for those patients who express the main targets of dovitinib in their recurrent GBM tissue.
References
Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, Degroot J, Gattamaneni R, Cher L, Rosenthal M, Payer F, Jürgensmeier JM, Jain RK, Sorensen AG, Xu J, Liu Q, van den Bent M (2013) Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 31:3212–3218. doi:10.1200/JCO.2012.47.2464
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, BRIM-3 Study Group (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. doi:10.1056/NEJMoa1103782
Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T (2014) Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370:709–722. doi:10.1056/NEJMoa1308345
Di Stefano AL, Fucci A, Frattini V, Labussiere M, Mokhtari K, Zoppoli P, Marie Y, Bruno A, Boisselier B, Giry M, Savatovsky J, Touat M, Belaid H, Kamoun A, Idbaih A, Houillier C, Luo FR, Soria JC, Tabernero J, Eoli M, Paterra R, Yip S, Petrecca K, Chan JA, Finocchiaro G, Lasorella A, Sanson M, Iavarone A (2015) Detection, characterization, and inhibition of FGFR-TACC fusions in IDH Wild-type glioma. Clin Cancer Res 21:3307–3317. doi:10.1158/1078-0432.CCR-14-2199
Escudier B, Grünwald V, Ravaud A, Ou YC, Castellano D, Lin CC, Gschwend JE, Harzstark A, Beall S, Pirotta N, Squires M, Shi M, Angevin E (2014) Phase II results of Dovitinib (TKI258) in patients with metastatic renal cell cancer. Clin Cancer Res 20:3012–3022. doi:10.1158/1078-0432.CCR-13-3006
Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M (2012) Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol 106:147–153. doi:10.1007/s11060-011-0650-1
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJ Jr, Mehta MP (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370:699–708. doi:10.1056/NEJMoa1308573
Hutterer M, Nowosielski M, Haybaeck J, Embacher S, Stockhammer F, Gotwald T, Holzner B, Capper D, Preusser M, Marosi C, Oberndorfer S, Moik M, Buchroithner J, Seiz M, Tuettenberg J, Herrlinger U, Wick A, Vajkoczy P, Stockhammer G (2014) A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (SURGE 01-07). Neuro Oncol 16:92–102. doi:10.1093/neuonc/not161
Kim J, Lee IH, Cho HJ, Park CK, Jung YS, Kim Y, Nam SH, Kim BS, Johnson MD, Kong DS, Seol HJ, Lee JI, Joo KM, Yoon Y, Park WY, Lee J, Park PJ, Nam DH (2015) Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 28:318–328. doi:10.1016/j.ccell.2015.07.013
Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, Boleti E, Fife K, Jin J, Jones R, Uemura H, De Giorgi U, Harmenberg U, Wang J, Sternberg CN, Deen K, McCann L, Hackshaw MD, Crescenzo R, Pandite LN, Choueiri TK (2013) Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 369:722–731. doi:10.1056/NEJMoa1303989
Motzer RJ, Porta C, Vogelzang NJ, Sternberg CN, Szczylik C, Zolnierek J, Kollmannsberger C, Rha SY, Bjarnason GA, Melichar B, De Giorgi U, Grünwald V, Davis ID, Lee JL, Esteban E, Urbanowitz G, Cai C, Squires M, Marker M, Shi MM, Escudier B (2014) Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol 15:286–296. doi:10.1016/S1470-2045(14)70030-0
Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, Desjardins A, Sathornsumetee S, Provenzale JM, Herndon JE 2nd, Dowell JM, Badruddoja MA, McLendon RE, Lagattuta TF, Kicielinski KP, Dresemann G, Sampson JH, Friedman AH, Salvado AJ, Friedman HS (2005) Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol 23:9359–9368
Renhowe PA, Pecchi S, Shafer CM, Machajewski TD, Jazan EM, Taylor C, Antonios-McCrea W, McBride CM, Frazier K, Wiesmann M, Lapointe GR, Feucht PH, Warne RL, Heise CC, Menezes D, Aardalen K, Ye H, He M, Le V, Vora J, Jansen JM, Wernette-Hammond ME, Harris AL (2009) Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: a novel class of receptor tyrosine kinase inhibitors. J Med Chem 52:278–292. doi:10.1021/jm800790t
Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, Moro-Sibilot D, Camidge DR, Mok T, Hirsh V, Riely GJ, Iyer S, Tassell V, Polli A, Wilner KD, Jänne PA (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368:2385–2394. doi:10.1056/NEJMoa1214886
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu K, Gao Z, Ceccarelli M, Riccardi R, Brat DJ, Guha A, Aldape K, Golfinos JG, Zagzag D, Mikkelsen T, Finocchiaro G, Lasorella A, Rabadan R, Iavarone A (2012) Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231–1235. doi:10.1126/science.1220834
Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, Louis DN, Iafrate AJ (2011) Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20:810–817. doi:10.1016/j.ccr.2011.11.005
Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, Steinbach JP, Wick W, Tarnawski R, Nam DH, Hau P, Weyerbrock A, Taphoorn MJ, Shen CC, Rao N, Thurzo L, Herrlinger U, Gupta T, Kortmann RD, Adamska K, McBain C, Brandes AA, Tonn JC, Schnell O, Wiegel T, Kim CY, Nabors LB, Reardon DA, van den Bent MJ, Hicking C, Markivskyy A, Picard M, Weller M (2014) Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 15:1100–1108. doi:10.1016/S1470-2045(14)70379-1
Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, Leversha MA, Mikkelsen T, Brennan CW (2012) Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA 109:3041–3046. doi:10.1073/pnas.1114033109
Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH, Boerman D, de Vos FY, Dinjens WN, Enting RH, Taphoorn MJ, van den Berkmortel FW, Jansen RL, Brandsma D, Bromberg JE, van Heuvel I, Vernhout RM, van der Holt B, van den Bent MJ (2014) Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 15:943–953. doi:10.1016/S1470-2045(14)70314-6
Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, Calvo E, Moreno V, Adamo B, Gazzah A, Zhong B, Platero SJ, Smit JW, Stuyckens K, Chatterjee-Kishore M, Rodon J, Peddareddigari V, Luo FR, Soria JC (2015) Phase I Dose-Escalation Study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 33:3401–3408. doi:10.1200/JCO.2014.60.7341
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, Tsien C, Mikkelsen T, Wong ET, Chamberlain MC, Stupp R, Lamborn KR, Vogelbaum MA, van den Bent MJ, Chang SM (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28:1963–1972. doi:10.1200/JCO.2009.26.3541
Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, Robins HI, Lieberman FS, Gilbert MR, Mehta MP, Drappatz J, Groves MD, Santagata S, Ligon AH, Yung WK, Wright JJ, Dancey J, Aldape KD, Prados MD, Ligon KL (2014) Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol 16:567–578. doi:10.1093/neuonc/not247
Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M, Hong S, Musib L, Liepa AM, Thornton DE, Fine HA (2010) Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol 28:1168–1174. doi:10.1200/JCO.2009.23.2595
Acknowledgments
We thank all the patients and their relatives for participation in this trial. The trial was funded by Novartis, the manufacturer of dovitinib.
Funding
The trial was supported by Novartis Pharma GmbH Germany (internal code: CTKI258ADE02T).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Niklas Schäfer and Frederic Mack received honoraria and travel fees from Roche. Ulrich Herrlinger received honoraria from Roche and Mundipharma, has a consulting role to declare for Roche and Mundipharma, and received research funding from Roche and Medac. Martin Glas received honoraria from Roche and Mundipharma, has a consulting role to declare for Novartis, Roche, Mundipharma, received travel fees from Medac, and received research funding from Novartis. All other authors declare that they have no conflict of interest.
Ethical standards
The trial was registered (EudraCT No. 2012-005460-95; ClinicalTrials.gov: NCT01972750) and approved by the local ethical committee. All patients gave written consent before performing of any study-related activity. All trial procedures adhered to the principles of the Declaration of Helsinki and the Guidelines of Good Clinical Practice.
Additional information
Björn Scheffler and Martin Glas have contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Schäfer, N., Gielen, G.H., Kebir, S. et al. Phase I trial of dovitinib (TKI258) in recurrent glioblastoma. J Cancer Res Clin Oncol 142, 1581–1589 (2016). https://doi.org/10.1007/s00432-016-2161-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-016-2161-0