
Abstract
Various stressors may disrupt the redox homeostasis of an organism by causing oxidative and nitrosative stress that may activate stressor-specific pathways and provoke specific responses. Chronic social isolation (CSIS) represents a mild chronic stress that evokes a variety of neurobehavioral changes in rats similar to those observed in people with psychiatric disorders, including depression. Most rodent studies have focused on the effect of social isolation during weaning or adolescence, while its effect in adult rats has not been extensively examined. In this review, we discuss the current knowledge regarding the involvement of oxidative/nitrosative stress pathways in the prefrontal cortex and hippocampus of adult male rats exposed to CSIS, focusing on hypothalamic-pituitary-adrenocortical (HPA) axis activity, behavior parameters, antioxidative defense systems, stress signaling mediated by nuclear factor-kappa B (NF-κB), and mitochondria-related proapoptotic signaling. Although increased concentrations of corticosterone (CORT) have been shown to induce oxidative and nitrosative stress, we suggest a mechanism underlying the glucocorticoid paradox whereby a state of oxidative/nitrosative stress may exist under basal CORT levels. This review also highlights the differential susceptibility of prefrontal cortex and hippocampus to oxidative stress following CSIS and suggests a possible cellular pathway of stress tolerance that preserves the hippocampus from molecular damage and apoptosis. The differential regulation of the transcriptional factor NF-κB, and the enzymes inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) following CSIS may be one functional difference between the response of the prefrontal cortex and hippocampus, thus identifying potentially relevant targets for antidepressant treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Living organisms in today’s environment are repeatedly exposed to stressors of various origins, which lead to activation of the sympatho-adrenomedullary system and hypothalamic-pituitary-adrenocortical (HPA) axis, resulting in the release of catecholamines and glucocorticoids (GCs) from the adrenal gland (McEwen 2008). The effects of GCs in acute stressful situations can be classified as adaptive (De Kloet et al. 2005; McEwen 2008), while chronic stress, especially chronic psychosocial stress, can be maladaptive. During the acute stress response, physiological processes divert mobilized energy from reserves among various organs, such that the body is again prepared for future events. In contrast, elevated levels of GCs released during chronic stress may lead to an increase in the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that can directly induce mitochondrial dysfunction (Papadopoulos et al. 1997), suppression of hippocampal synaptic plasticity (Joëls et al. 2004), and trigger proapoptotic signaling that results in apoptotic cell death (Cregan et al. 2002).
Various chronic stressors can disrupt redox homeostasis of the organism, causing oxidative/nitrosative stress that leads to the activation of intracellular signaling pathways involved in psychiatric disorders (Maes et al. 2011). Particularly interesting are those stressors with a psychosocial component, as it is known that chronic psychosocial stress in adulthood modulates brain structure and function, resulting in cognitive deficits and an increased risk for psychiatric disorders (De Kloet et al. 2005; Lupien et al. 2009). The most common stressors reported as risk factors for psychiatric disorders, such as depression, are of a social nature in humans (Brown and Prudo 1981; House 2001) and in social animals (Fuchs 2005). For example, the lack of social stimuli associated with social isolation precludes the ability to modulate adaptive responses to new situations (Ishida et al. 2003). In experimental animals, social isolation may be achieved by keeping animals individually housed, with normal auditory and olfactory experiences, but no visual or tactile contact with other animals in the colony (Garzón and Del Río 1981). Since rats naturally live in groups, chronic social isolation (CSIS) is continuous and qualitatively different from other types of chronic stress.
Most changes induced by chronic stress are observed in the hippocampus and prefrontal cortex, brain regions related to the pathophysiology of depression that play a role in mediating the effects of stress on GC regulation (Joëls and Baram 2009; Maes et al. 2011). For example, changes in gray matter volume and reduced neurogenesis in the hippocampus identified in post-mortem studies of patients with depression (Wainwright and Galea 2013) can be induced in adult rats by CSIS (Stranahan et al. 2006). Moreover, stress-induced impairment in prefrontal cortex function and plasticity is thought to be a core pathological feature of several neuropsychiatric disorders (Goto et al. 2010). Stress-induced alterations in HPA axis activity and their relation to oxidative stress may be caused by increased levels of GCs (McIntosh et al. 1998a, b). Given that GCs accomplish their functions via glucocorticoid receptors (GR), dysregulation (hyper- or hypo-activity) of the HPA axis induced by chronic stress may result from altered negative feedback control in the higher centers of the axis, i.e. the hippocampus and prefrontal cortex (Mizoguchi et al. 2003; Filipović et al. 2005). Furthermore, alterations in GCs play a key role in the development of depressive disorders (De Kloet et al. 1998; Holsboer and Ising 2010).
Chronic stress may impair antioxidant defenses, leading to oxidative damage (Liu and Mori 1994), whereby the extent of stress-triggered effects is related to the duration and type of stress (Pacak et al. 1998). The defense mechanisms against oxidative stress include a cascade of antioxidant enzymes such as cytosolic (but not exclusively) copper-zinc superoxide dismutase (CuZnSOD) (Chang et al. 1988) and mitochondrial manganese superoxide dismutase (MnSOD), which catalyzes the dismutation of superoxide anion (O ·−2 ) to oxygen and hydrogen peroxide (H2O2), which is further detoxified by the enzymes catalase (CAT) and glutathione peroxidase (GPx) (Chelikani et al. 2004). CAT (primarily localized in peroxisomes) reduces H2O2 into molecular oxygen and water (Halliwell 2011). GPx is localized in the mitochondria and cytosol and performs a reduction of H2O2 to water and organic hydroperoxides to their corresponding alcohols in the presence of glutathione (GSH) which is oxidized to glutathione disulfide (GSSG) (Dringen 2000). The reduction of GSSG back to GSH is catalyzed by glutathione reductase (GLR) using reduced nicotinamide adenine dinucleotide phosphate (NADPH) (Andreyev et al. 2005; Couto et al. 2013). GSH is a non-enzymatic component of antioxidant defense that plays a central role in maintaining physiological redox status, of which the GSH/GSSG ratio within cells is an indicator of cellular oxidative stress. It has been shown that chronic stress may affect levels of GSH (Madrigal et al. 2001b; Ahmad et al. 2010), and some psychiatric disorders are characterized by GSH depletion (Gawryluk et al. 2011). In addition, the NADPH oxidase (NOX) family, which transfers electrons across biological membranes to catalyze the reduction of molecular oxygen and generate O ·−2 , has also been implicated in psychiatric disorders (Sorce and Krause 2009), and previous studies have shown that NOX2-derived oxidative stress is involved in the development of anxiety-like symptoms following social isolation in rodents (Schiavone et al. 2009).
In addition to ROS, the exposure of organisms to stressors may lead to the overproduction of nitric oxide (NO). Although NO is necessary for the function of the nervous system, including roles in synaptic plasticity, neuromodulation and other physiological roles, if present in high concentrations, it can combine with O ·−2 and form the highly reactive and toxic peroxynitrite (ONOO−) (Patel and Darley-Usmar 1996), which is powerful tyrosine-nitrating agent (Schliess et al. 2002) and may also lead to nitrosylation of antioxidant enzymes (Anand and Stamler 2012). The concentration of NO within biological systems is regulated by the activity of nitric oxide synthase (NOS) isoforms: constitutively expressed neural (nNOS), endothelial (eNOS) and inducible (iNOS) forms. The NOS enzymes are widely distributed within the mammalian brain, and NOS-positive neurons are located in the hippocampus and cerebral cortex (Olivenza et al. 2000). Although nNOS is a constitutive enzyme responsible for producing the NO that can act as a neurotransmitter, its expression is also influenced by certain stressors and may be involved in depressive-like behavior in rodents (McLeod et al. 2001). In contrast, persistent activation of iNOS, mainly regulated at the transcription level, is associated with pathological inflammatory processes (Brown 2007) and may also be responsible for stress-induced depression (Haroon et al. 2012).
One factor that may link oxidative stress and brain damage is the redox-sensitive transcription factor nuclear factor-kappa B (NF-κB) (Jin et al. 2008). NF-κB is localized in the cytoplasm as an inactive form through its interaction with the inhibitory protein I-kappa B (IκB) (Muriach et al. 2010). It can be activated by ROS (Li and Karin 1999), resulting in the proteolytic degradation of IkB with concomitant nuclear translocation of the p50 and p65 heterodimer of NF-κB, which then acts on NF-κB target genes (Senftleben et al. 2001). Stress activates NF-κB in brain cells as early as 4 h after the onset of stress in rats (Madrigal et al. 2006), stimulating the expression of a variety of genes responsible for cell injury or cell protection. Genes which in their promoters contain NF-κB binding sites that contribute to the activation of oxidative/nitrosative and inflammatory mediators are iNOS (Xie et al. 1994), nNOS (Hall et al. 1994; Li et al. 2007), and cyclooxygenase-2 (COX-2) (Plummer et al. 1999; Maes et al. 2007b), while genes with NF-κB binding sites that protect cells include CuZnSOD (Meyer et al. 1993; Kim et al. 1994), MnSOD (Xu et al. 1999), and B cell lymphoma (Bcl) family genes such as Bcl-2 and Bcl-xL (Tamatani et al. 1999; Chen et al. 1999). Moreover, ROS/RNS and GSH levels may be critical determinants of NF-κB activation (Mihm et al. 1995).
In addition, to adapt to environmental changes and survive injury, cells synthesize heat shock proteins (HSPs). While HSP70 is involved in cellular repair and protective mechanisms (Georgopoulos and Welch 1993; Morimoto and Tissieres 1994), the degree of HSP70i inducible form depends on the type and duration of exposure to stressors (Kiang 2004). The expression of HSP70i blocks NF-κB activation and NF-κB-dependent gene expression (Malhotra and Wong 2002). Moreover, HSP70i induction protects neurons from apoptosis (Arieli et al. 2003) and also suppresses microglial activation (Heneka et al. 2000), which upon chronic stress exposure represents a significant source of ROS (Tynan et al. 2010; Hinwood et al. 2012). In addition to HSP70i, the small HSP27 exerts its antiapoptotic effect at the level of the mitochondria via a series of signal transduction events, such as the phosphorylation and inactivation of Bad (Bcl-xL/Bcl-2 associated death promoter) (Datta et al. 1997), the inhibition of Bax-mediated mitochondrial membrane injury (Havasi et al. 2008), and the inhibition of caspases and cytochrome c release (Garrido et al. 1999; Charette et al. 2000). Furthermore, the heme oxygenase (HO) system, consisting of constitutive HO-2 and the inducible isoform HO-1(HSP32), also has a protective role against ROS damage in rat brain (Scapagnini et al. 2002; Muñoz-Sánchez and Chánez-Cárdenas 2014). These isozymes catalyze the NADPH- and cytochrome P450 reductase-dependent degradation of heme to carbon monoxide, ferrous iron and biliverdin, which in the presence of biliverdin reductase is reduced to the antioxidant bilirubin (Maines et al. 1996) that counteracts NO and RNS activity (Mancuso et al. 2006). HO-2 is the predominant HO isoenzyme in the adult rodent brain and is regulated entirely by adrenal GCs (Raju et al. 1997), while the transcription factors AP-1, NF-κB, and mitogen-activated protein kinases have been implicated in HO-1 regulation (Ryter et al. 2006).
Compromised HPA axis functioning in socially isolated adult male rats
Regulation of the HPA system, which may be responsible for individual differences in susceptibility to stress, is highly effected by social isolation. Relatively few studies have investigated the effect of social isolation in adult rats. The effects of this psychosocial stress on corticosterone (CORT) levels in adult male rats have been inconsistent among studies (Table 1). Increased CORT levels have been reported (Ferland and Schrader 2011), whereas other groups observed no changes (Scaccianoce et al. 2006; Filipović and Pajović 2009) or reduced CORT levels (Miachon et al. 1993). This inconsistency may arise due to differences in the nature and/or length of the social isolation, as well as in the age of animals at its onset (Serra et al. 2007). The duration of isolation between studies varies from 2 to 13 weeks. Miachon et al. (1993) showed that 13 weeks of social isolation produced a significant increase in catecholamine turnover in the hippocampus, cortex and cerebellum, accompanied by increased adrenocorticotropic hormone (ACTH) and decreased basal plasma CORT levels. Filipović and Pajović (2009) showed that 3 weeks of social isolation resulted in CORT levels similar to basal values in adult male Wistar rats. However, decreased responsiveness of the HPA axis of socially isolated rats in response to novel acute immobilization or cold stressors relative to those acute stressors alone indicates compromised HPA axis activity, as reflected by a lesser increase in CORT levels (Filipović and Pajović 2009). As GR shuttling between the cytoplasm and nucleus is essential for proper HPA axis activity, an unchanged CORT response during CSIS resulted from reduced nuclear translocation of cytosolic GR and increased cytosolic retention in the hippocampus and prefrontal cortex, suggesting diminished GR-negative feedback control (Mizoguchi et al. 2003; Dronjak et al. 2004; Filipović et al. 2005) (Fig. 1). The retention of cytosolic GR and disabling of its translocation to the nucleus, where it functions as a transcription factor, may lead the body into a state in which there is no cessation of the transmission of stress signals and thus induce an allostatic load. Moreover, a decreased secretion of corticotrophin-releasing hormone (CRH) following long-term isolation (Sánchez et al. 1998) may also result in unaltered CORT levels. Serra et al. (2005) showed that an intraperitoneal injection of dexamethasone (a synthetic GC) caused a decrease in CORT levels, but this decrease was significantly lower in isolated relative to control rats, suggesting that social isolation impairs the glucocorticoid negative feedback regulation of CORT secretion. This study also demonstrated that reduced efficacy of this regulatory system may be a consequence of an isolation-induced decrease in GR in the pituitary, the hypothalamus and the hippocampus.

Effect of chronic social isolation (CSIS) on hypothalamic-pituitary-adrenocortical (HPA) axis functioning. The physiological response to stress involves the activation of HPA axis. Paraventricular nuclei of the hypothalamus secrete corticotropin releasing hormone (CRH), which stimulates the pituitary gland to secrete adrenocorticotropic hormone (ACTH) which acts on adrenal gland stimulating the secretion of corticosterone (CORT). In turn, CORT acts back on the hypothalamus, pituitary glands, prefrontal cortex and hippocampus limiting the activity of the HPAaxis (a). Compromised HPA axis functioning of adult male socially isolated rats may be a consequence of incomplete nuclear translocation of cytosolic glucocorticoid receptor (GR) and its cytosolic retention in the hippocampus and prefrontal cortex. This suggests that dysregulation of the HPA axis induced by stress results from a partial disruption of GR negative feedback control in the higher centers of the rat brain (b). GRE glucocorticoid response element
Chronic social isolation provokes depressive- and anxiety-like behaviors in adult male rats
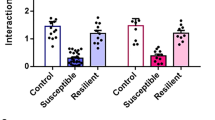
The concept of isolation “stress” in rats is derived from studies in the early 1960s that reported social isolates as abnormally reactive to handling, anxiogenic, and overly emotional (Wiberg and Grice 1963; Hatch et al. 1965), which led to the term “isolation-induced stress syndrome” (Holson et al. 1991). CSIS is a variety of chronic mild stress that represent a more natural stressor in rodents, as it has been shown to evoke a variety of neurobehavioral changes in rats similar to those changes observed in humans with psychiatric disorders, including depression (Heim and Nemeroff 2001; Heinrich and Gullone 2006). Although it is apparent that depressive symptoms such as suicidal tendencies and recurrent thoughts of death cannot be modeled in rats, it is possible to study specific behavioral domains in relation to psychiatric endophenotypes such as anxiety, anhedonia, sleep disturbances, and hormonal dysregulation (Gould and Gottesman 2006). Deprivation of social interaction in rats causes aggressiveness, (Serra et al. 2005; Sandi and Haller 2015), cognitive impairments as evidenced by spatial memory deficits, impaired maze learning (Einon 1980), and hyper-reactivity to novel environments (Lapiz et al. 2003; Zlatković et al. 2014b). A lack of social interaction in adult rats results in a reduced number of specific subpopulations of hippocampal neurons, such as parvalbumin-positive neurons (Harte et al. 2007; Filipović et al. 2013). Enhanced anxiety-like behavior in socially isolated rats has been observed using open field and elevated plus maze testing (Hall 1998; Weiss et al. 2004). For example, 3 weeks of social isolation in adult male Wistar rats resulted in a reduction in the percentage of open arm entries and a general decrease in locomotion (total number of arm entries) in the elevated plus maze, indicative of anxiogenic behavior (Djordjevic et al. 2015), and after only 2 weeks of social isolation, adult rats spent less time in the light compartment of the light–dark box, again indicating anxiety-like behavior (Carrier and Kabbaj 2012). Furthermore, Spasojevic et al. (2007) showed that 3 weeks of CSIS in adult male rats led to anxiety behavior, a reduced duration of grooming, more defecations and urinations, increased reluctance to step down to a test platform, and an increased number of vertical rears. A recent study reported that 6 weeks of social isolation resulted in spatial memory deficits in middle-aged rats, as indicated by a significantly increased latency to find a hidden platform in the Morris water maze test relative to controls (Ren et al. 2015). Moreover, 3 weeks of social isolation in adult male Wistar rats led to depressive-like behaviors, including increased immobility and less time swimming and climbing in the forced swim test, indicating despair behavior, and reduced sucrose preference, indicative of an impaired sensitivity to reward and anhedonia (Zlatković et al. 2014b) (Fig. 2). These results are in agreement with findings from Brenes and Fornaguera (2009), who also reported despair behavior in isolated rats in the forced swim test, and Carrier and Kabbaj (2012), who demonstrated that 3 weeks of CSIS induced depressive-like symptoms, such as anhedonia, in adult male rats. A notable effect of CSIS was observed in the marble burying test, in which socially isolated rats displayed anxiety-like behaviors and neophobia, assessed by an increase in burying behavior, an active effort of rodents to hide unfamiliar objects (Fig. 2). Although this test is primarily used to test potential antidepressant treatments (Borsini et al. 2002), increased burying behavior in chronically stressed animals may indicate a heightened anxiety (Farley et al. 2010). CSIS-induced behavioral despair, anhedonia and anxiety have been identified as a correlate of depression (Sandi and Richter-Levin 2009).

Social isolation in adult male Wistar rats for 3 weeks causes anxiety-like behavior, including an increase in the number of marbles buried, despair behavior (increased immobility time in forced swim test) and increased depressive-like behavior (reduced sucrose preference)
Compromised brain SOD activity in socially isolated adult male rats
In addition to compromised HPA axis activity, CSIS causes brain oxidative stress and leads to dysregulation of antioxidative enzymes that may contribute to psychiatric disorders (Colaianna et al. 2013). A previous study has shown that increased CORT levels during chronic stress decreased the activity of antioxidative enzymes in rat brain, indicating a direct effect of CORT on the induction of oxidative stress (Zafir and Banu 2009). High levels of GCs may increase glutamate release and calcium mobilization in neurons leading to the calcium-dependent activation of NOS, and the subsequent production of toxic NO levels and mitochondrial dysfunction. Moreover, GCs may induce neuronal oxidative stress directly through enhanced mitochondrial respiration and oxidative phosphorylation (Spiers et al. 2015). In addition, the NOX2 enzyme is considered a major source of ROS in the central nervous system that may be responsible for CSIS-induced oxidative stress (Schiavone et al. 2009). In fact, the earliest neuropathological alterations in socially isolated rats were increased expression of NOX2 and signs of oxidative stress in the prefrontal cortex. The NOX2-derived oxidative stress led to increased glutamate levels and a reduced the number of parvalbumin-positive inhibitory neurons. The application of the antioxidant/NOX inhibitor apocynin during 7 weeks of CSIS prevented development of the signs of oxidative stress, such as oxidized nucleic acid 8-hydroxy-2′-deoxyguanosine, redox-sensitive transcription factor c-fos, and hypoxia-inducible factor-1alpha, which have been found to be increased in the prefrontal cortex and nucleus accumbens of socially isolated rats. Also, in these brain regions, apocynin treatment prevented a CSIS-induced decrease in parvalbumin immunoreactivity, as well as behavioral changes associated with CSIS, such as increased spontaneous locomotor activity in the open field test and a decreased discrimination index in the novel object recognition test (Schiavone et al. 2009). It was also revealed that the application of apocynin for 3 weeks fully reversed CSIS-induced behavioral alterations when applied after 4 weeks (from week 4 to week 7 of CSIS), but only partially when administered after 7 weeks of post-weaning isolation (from week 7 to week 10 of CSIS) (Schiavone et al. 2012). An excessive increase in ROS production can inhibit antioxidative activity of CAT by oxidizing the heme group in its active site (Spiers et al. 2015), where high levels of H2O2 may inactivate CuZnSOD activity by oxidizing its thiol groups (Halliwell and Gutteridge 1989).
Additionally, it is known that CuZnSOD expression is regulated by GCs (Kim et al. 1994) via the GR, which acts as a hormone dependent transcriptional factor (McKay and Cidlowski 2000; Gass et al. 2001) that traffics continuously between the cytoplasm and nucleus when liganded to GCs, thus mediating the final effects of GCs (Madan and DeFranco 1993). Accordingly, although CuZnSOD activity is primary protective, a lack of coupling with respective peroxidase activity of CAT and GPx (McIntosh et al. 1998a) may result in an SOD-driven accumulation of toxic H2O2, which further negatively modulates GR function (Okamoto et al. 1999; Zhou et al. 2011), causing a decrease in glucocorticoid-inducible gene expression (Makino et al. 1996) and consequent CuZnSOD expression. Moreover, GCs also regulate the expression of HO-2, as the promoter region of the gene encoding HO-2 contains a glucocorticoid response element (Muñoz-Sánchez and Chánez-Cárdenas 2014). Although there are no published data pertaining to the effect of social isolation on this enzyme, Chen et al. (2005) demonstrated that a chronic restraint stress-induced increase in plasma CORT levels decreased HO-2 protein levels in hippocampal neurons, likely a consequence of incomplete nuclear translocation of cytosolic GR. Furthermore, venlafaxine, an antidepressant, and quetiapine, an atypical antipsychotic, effectively prevented the decrease in HO-2 protein in hippocampal neurons of stressed rats (Chen et al. 2005).
Adult male Wistar rats that exhibited CORT levels similar to basal values following 3 weeks of social isolation did not show changes in cytosolic CuZnSOD and mitochondrial MnSOD protein levels in the hippocampus (Filipović et al. 2009). Interestingly, an acute stressor (2 h of immobilization) that caused a significant increase in serum CORT levels and CuZnSOD mRNA in the hippocampus failed to activate the transcription of CuZnSOD gene when applied in combination with CSIS, despite the fact that CORT levels were increased compared to CSIS alone (Filipović and Pajović 2009).This lack of upregulation of CuZnSOD protein expression partly resulted from a compromised HPA axis, i.e. impaired nucleo-cytoplasmic GR shuttling (Dronjak et al. 2004; Filipović et al. 2005), which likely prevented activation of the SOD promoter and caused a lack of significant upregulation of SOD, as well as ROS defense inefficiency (Fig. 3). Concurrently, the total SOD activity in the hippocampal cytosolic fraction of socially isolated rats was unchanged (Zlatković et al. 2014b). Moreover, there are different findings related to SOD activity following CSIS stress in rat brain. Social isolation for 8 weeks decreased the activities of CAT, GPx, SOD, and the total antioxidant capacity, but increased levels of H2O2, in the prefrontal cortex and hippocampus of rats (Shao et al. 2015). In addition, CuZnSOD expression is under the control of NF-κB (Meyer et al. 1993; Kim et al. 1994), which can be activated by H2O2 (Bowie and O’Neill 2000). H2O2 may trigger a positive feed-forward cycle with NF-κB, causing the accumulation of toxic H2O2 and thus diminishing CuZnSOD activity. Möller et al. (2011) reported that post-weaning social isolation for 8 weeks increased SOD activity, increased the GSH/GSSG ratio, and increased lipid peroxidation in striatal and frontal cortical tissue while application of the antipsychotic clozapine reversed the behavioral changes and cortico-striatal redox disturbances associated with social isolation. Isolation stress in the prepubertal period led to increased SOD and complex IV activities in the prefrontal cortex of male rats, effects still observed in adulthood (Krolow et al. 2012). These alterations in brain oxidative stress parameters are paralleled by deficits in prepulse inhibition and social and self-directed interactive behaviors (Schiavone et al. 2013).

Possible mechanism that explains the lack of upregulation of copper-zinc superoxide dismutase (CuZnSOD) protein expression in the hippocampus of socially isolated adult male Wistar rats for 3 weeks. Protein expression of the CuZnSOD is under glucocorticoid receptor (GR) regulation (left). Incomplete nuclear translocation of cytosolic GR in socially isolated rats exposed to an acute stressor and its cytosolic retention is partially unable to activate the CuZnSOD promoter, leading to a lack of significant upregulation of CuZnSOD and ROS defense inefficiency (right). GRE glucocorticoid response element
In addition, CSIS acting either directly or indirectly may shift the antioxidant/prooxidant balance toward a more prooxidant state, with more oxidative stress produced in mitochondria in the prefrontal cortex (Filipović et al. 2011). The fact that CSIS resulted in no change in serum CORT levels relative to controls suggests a mechanism underlying the glucocorticoid paradox whereby a state of oxidative stress may also exist under CORT levels similar to basal values. Furthermore, overexpression of nNOS and iNOS with a concomitant increase in NO in the prefrontal cortex of socially isolated rats (Filipović et al. 2013) caused nitrosative stress during chronic stress (Leza et al. 1998; Olivenza et al. 2000). Concurrently, a significant decrease in mitochondrial MnSOD activity was found, suggesting that its detoxifying capacity was compromised by nitrosative stress (Filipović et al. 2011). Decreased mitochondrial MnSOD activity could be due to high levels of NO and ONOO− which have been shown to inhibit MnSOD activity, typically via nitration of the tyrosine residue at the enzyme active site (Lawler and Song 2002; Stojanović et al. 2005), resulting in dityrosine formation that may lead to the amplification of oxidative stress by allowing the accumulation of O ·−2 and subsequently trigger apoptosis (Radi et al. 2002). A corresponding decrease in the MnSOD activity may be regulated at the posttranslational level by lysine acetylation (Tao et al. 2010; Ozden et al. 2011), independent of regulation of its protein synthesis (Hopper et al. 2006). Nonetheless, mitochondrial MnSOD activity may be regulated via mitochondrial-localized p53 by its physical interaction with p53,which inhibits its activity (Candas and Li 2014), in accordance with data from Filipović et al. (2011). Accordingly, compromised mitochondrial MnSOD activity may lead to increased oxidant production within mitochondria, causing nitration of other mitochondrial proteins (Cruthirds et al. 2003). A significant decrease in mitochondrial MnSOD protein levels and reciprocal increase in the cytosolic fraction of the prefrontal cortex of socially isolated rats exposed to novel acute immobilization or cold stress has been reported (Filipović et al. 2009). Given that MnSOD is encoded in the nuclear chromatin, synthesized as a precursor in the cytoplasm, and transported to mitochondria via the mitochondria targeting sequence may assembling into an active enzyme with the incorporation of a manganese ion in the mitochondrial matrix, increased cytosolic MnSOD protein levels may be derived from its translocation from mitochondria and/or the inappropriate transport of newly synthesized MnSOD into mitochondria (changes in the mitochondrial targeting domain) (Cruthirds et al. 2003). Regardless of the mechanisms, the appearance of MnSOD protein in the cytosolic fraction clearly indicates a loss of mitochondrial membrane integrity (Jin et al. 2005). At the same time, the presence of cytochrome c protein in the cytosol fraction of the prefrontal cortex following 3 weeks of social isolation in adult male rats indicates a loss of mitochondrial membrane integrity, a hallmark of mitochondrial dysfunction (Jin et al. 2005), and further ROS production by inhibition of the mitochondrial respiratory chain (Cai and Jones 1998). Moreover, ROS generated under chronic stress has been shown to contribute to the release of cytochrome c into the cytosol following opening of the permeability transition pore (Petrosillo et al. 2001).
These data indicate that following 3 weeks of social isolation in adult male Wistar rats, the prefrontal cortex is a target of the maladaptive response to stress (Cerqueira et al. 2007). In contrast, no change in the hippocampal protein levels of MnSOD following CSIS indicates preserved integrity of the mitochondrial membrane, and greater resistance to oxidative stress compared to the prefrontal cortex.
Compromised brain glutathione antioxidant defenses in socially isolated adult male rats
An important component of the non-enzymatic antioxidant system is GSH. GSH is the major redox buffer (Giustarini et al. 2004) and an essential cofactor for a number of enzymes, playing a role in protecting cells from oxidative stress and xenobiotics, as well as maintaining the thiol redox state (Dringen 2000; Aoyama et al. 2008). It also functions as a storage and transport form of cysteine (Janáky et al. 2000) and can serve as neuromodulator/neurotransmitter (Janáky et al. 1999). Changes in GSH and the enzyme activity of GPx and GLR may indicate a deficit in antioxidative defense. Three weeks of social isolation in adult male rats caused a significant decrease of GSH content in the prefrontal cortex (Zlatković et al. 2014b) (Fig. 4). This GSH depletion may be a consequence of its oxidation during the detoxification of H2O2 and/or lipid peroxides (Gupta et al. 2005), its participation in the maintenance of non-GSH sulfhydryl proteins in a reduced state, its increased consumption via increased glutathione S-transferase activity (Tew and Ronai 1999) or increased GPx activity that uses GSH for the catalytic reduction of H2O2. Increased GPx protein expression and its activity in CSIS rats is likely the result of an elevated production of lipid peroxides (Zlatković et al. 2014b). Intensified GSH consumption during CSIS and unchanged protein expression and activity of GLR likely diminishes GSH recycling due to the inability of GLR to compensate for increased GSH consumption by GPx. Moreover, unchanged GLR may indirectly cause an increase in H2O2 production, which may activate NF-κB signaling (Kobayashi et al. 2008). Hence, increased activity of GPx associated with unchanged activity of GLR may shift redox balance GSH/GSSG towards a more prooxidant state. Moreover, an impaired peroxidase-reductase system resulting in decreased GSH content may lead to the accumulation of peroxidizable products related to the initiation of proapoptotic signaling in the rat prefrontal cortex (Filipović et al. 2011). However, Möller et al. (2011) reported that 8 weeks of post-weaning social isolation increased GSH/GSSG ratio in the rat frontal cortex, and elevated levels of malondialdehyde, a lipid peroxidation product, suggesting CSIS-induced oxidative cell damage. These inconsistencies may be attributable to species differences, the age at which the animals were isolated (adult versus post-weaning), and/or the duration of the social isolation (3 versus 8 weeks). In both cases, the presence of oxidative stress was found, but stress-induced changes in the antioxidative defense system were likely age- and time-dependent. Treatment with N-acetyl cysteine, the GSH precursor and antioxidant, during the last 2 weeks of the 8-week long social isolation effectively reversed the bio-behavioural effects of CSIS (Möller et al. 2013).

Possible mechanisms that lead to increased oxidative stress in the hippocampus and prefrontal cortex of adult male rats exposed to chronic social isolation (CSIS) stress. In the prefrontal cortex, CSIS causes decreased glutathione (GSH) and manganese superoxide dismutase (MnSOD), increased glutathione peroxidase (GPx), and unchanged glutathione reductase (GLR) causing oxidative stress. Hence, a CSIS-induced shift in the prooxidant-antioxidant balance toward prooxidant state activates nuclear transcription factor-kappa B (NF-κB), which stimulates the expression of a variety of genes that contribute to activation of oxidative/nitrosative and inflammatory mediators. In contrast, an increase of inducible heat shock protein 70 (HSP70i) in the rat hippocampus indicates a protective effect by attenuation of the nuclear translocation of NF-κB, suggesting cellular pathways of stress tolerance that preserve the hippocampus from molecular damage
Interestingly, in contrast to the prefrontal cortex, 3 weeks of social isolation in adult male rats resulted in a decrease in GSH content and reduced protein level and activity of GPx and GLR in the hippocampus (Todorović et al. 2014) (Fig. 4). These results indicate that CSIS compromised the GSH-dependent defense system, promoting a prooxidative state in the hippocampus. Moreover, compromised GSH content has been associated with stress-induced behavioral depression and cognitive impairments (Dean et al. 2009). Given that GSSG is converted back to GSH by GLR using NADPH as a reducing power, decreased hippocampal GLR activity in socially isolated rats may result from an NADPH deficiency (Singh et al. 2008) or increased H2O2 concentration (Gutierrez-Correa and Stoppani 1997). In addition, compromised GPx activity has also been associated with stress-induced behavioral depression in animal models (Eren et al. 2007). Nonetheless, a decrease in GLR activity may result in further deterioration during a state of oxidative damage, compromising GSH restoration. These results suggest that the GPx/GLR cycle in the prefrontal cortex and hippocampus is compromised following CSIS stress (Todorović et al. 2014).
CSIS-induced nitrosative stress by NF-κB activation and iNOS protein expression in the prefrontal cortex of adult male rats
In biological tissue, NO is generated by specific NOS isoforms and plays a role in synaptic plasticity, neuromodulation and other physiological functions. However, the overproduction of NO, as a result of nNOS and iNOS overexpression (Ridnour et al. 2004), is caused by prolonged activation of the glutamate receptor during stress (Musazzi et al. 2011), together with an increased ROS formation due to NOX activation and mitochondrial respiration. In addition, the regionally selective activation of microglia by chronic stress in rats (Tynan et al. 2010) results in the release of high concentrations of NO, promoting nitrosative stress (Cassina et al. 2002). Increases in nNOS and iNOS protein expression in the prefrontal cortex of adult male rats exposed to 3 weeks of social isolation has been shown to cause nitrosative stress (Zlatković and Filipović 2012). One explanation for nitrosative stress following CSIS may be the activation of NF-κB (Maes et al. 2007a, b) (Fig. 5). As NO can upregulate NF-κB (Connelly et al. 2001), the induction of both NOS protein expression isoforms in CSIS likely causes persistent NO production that may mediate NF-κB activation. Accordingly, activated NF-κB in the nucleus may interact with kappa B elements in the NOS2 5′ flanking region, triggering iNOS gene transcription (Davis et al. 2005). This has been confirmed with the use of pyrrolidinedithiocarbamate, an inhibitor of NF-κB activation, which decreased the activity and expression of iNOS in stressed animals (Madrigal et al. 2001a). Given that NO functions as a proapoptotic molecule, primarily activating the mitochondrial apoptotic pathway (Pacher et al. 2007), increased iNOS levels associated with increased NO following CSIS may be related to the activation of proapoptotic signaling in the prefrontal cortex of adult male rats (Filipović et al. 2011; Zlatković and Filipović 2012). Considering that CSIS increases the expression of both NOS isoforms in the prefrontal cortex of adult male rats, whereby oxidative stress and glutamate can increase NF-κB activation (Pizzi et al. 2005), a positive feedback loop between glutamate, NF-κB and NOS changes in the prefrontal cortex may be involved in the behavioral consequences of stress.

Schematic representation of iNOS-mediated release of NO and its downstream effects in the cytoplasm of the prefrontal cortex of socially isolated adult male Wistar rats. In an environment of oxidative stress, the activation of nuclear factor-kappa B (NF-κB) leads to increased production of the inducible isoform of nitric oxide synthase (iNOS), neural isoform of nitric oxide synthase (nNOS), and proinflammatory mediator cyclooxygenase-2 (COX-2). The increase in COX-2 expression results in an increase of prostaglandins (PGE) and inflammatory cytokines, causing inflammation. High concentrations of NO produced by increased iNOS can then interact with the superoxide anion radical (O ·−2 ), resulting in the formation of the neurotoxic radical peroxynitrite (ONOO−). Increased NO can also covalently bond to protein thiol groups, causing S-nitrosylation of proteins such as S-nitrosoglutathione. Increased ONOO− is capable of causing oxidation, hydroxylation and nitration, as well as macromolecule damage together with hydroxyl radicals (OH−). Superoxide is degraded by manganese superoxide dismutase (MnSOD) and then by catalase and glutathione peroxidase (GPx) with concomitant oxidation of GSH to GSSG. Glutathione reductase (GLR) catalyzes the reduction of GSSG back to GSH using reduced nicotinamide adenine dinucleotide phosphate (NADPH). In the prefrontal cortex of socially isolated adult male rats, decreased activity of MnSOD may result in increased O ·−2 levels and reinforce oxidative stress
Moreover, a stress-induced shift of redox balance toward a prooxidant state may activate NF-κB, which translocates into the nucleus and induces the transcription of cyclooxygenase-2 (COX-2), an inflammatory marker. Our group reported that 3 weeks of social isolation increased COX-2 protein expression in the prefrontal cortex of adult male rats (Zlatković et al. 2014b). Increased COX-2 and iNOS protein expression are caused by an upregulated production of NF-κB (Maes et al. 2007b). The activation of COX-2 may cause the release of additional free radicals and inflammatory cytokines (Arimoto and Bing 2003), as well as the biosynthesis of prostaglandins, that further contribute to the cellular prooxidant state. Furthermore, prostaglandin itself may also cause cell damage by inducing glutamate release from astrocytes or apoptosis (Vesce et al. 2007). Once expressed, iNOS and COX-2 may generate large amounts of ROS that mediate the oxidation of cellular components (Madrigal et al. 2003) and are involved in the activation of proapoptotic signaling in the prefrontal cortex. The activation of NF-κB can also upregulate HO-1 expression (Muñoz-Sánchez and Chánez-Cárdenas 2014), which may either confer cytoprotection by converting the prooxidant heme and hemoproteins to the antioxidants biliverdin and bilirubin, or conversely, produce carbon monoxide and ferrous iron, which may reinforce oxidative stress (Song et al. 2012). Prabakaran et al. (2004) demonstrated that HO-1 was upregulated in the prefrontal cortex of patients suffering from schizophrenia. In coculture paradigms, overexpression of glial HO-1 enhanced the vulnerability of nearby neuronal constituents to oxidative insult (Song et al. 2007). In addition, mitochondria have been identified as a selective target for the protective effects of HSP70 against oxidative injury (Calabrese et al. 2000). The lack of initiation of HSP response during 3 weeks of social isolation may be a factor of the mitochondria-related proapoptotic cascade and apoptosis in the prefrontal cortex of adult rats (Filipović et al. 2011).
In contrast to the prefrontal cortex, 3 weeks of social isolation results in no activation of NF-κB and unaltered COX-2 protein expression in the hippocampus of adult male Wistar rats (Zlatković et al. 2014b). Protective responses triggered in the hippocampus may be mediated by increased HSP70i protein expression (Zlatković et al. 2014a), which likely keeps NF-κB in an inactive state and partially prevents greater damage, such as that observed in the prefrontal cortex (Fig. 4). Previous data have shown that the overproduction of NO and a depleted redox GSH status are critical factors in the induction of cytoprotective HSP70 (Hao et al. 1999; Calabrese et al. 2000). HSP70i upregulation likely stabilizes the cytoplasmic NF-κB/IκB complex (Malhotra and Wong 2002) and prevents NF-κB translocation into the nucleus (Zheng et al. 2008), resulting in decreased iNOS protein expression (Heneka et al. 2000). A lack of activated NF-κB levels following CSIS have been demonstrated to be due to unaltered CORT levels or a feedback loop of the HSP70i pathway in the chronic stress state that stabilizes NF-κB. Although nNOS protein expression has been shown to be upregulated following 3 weeks of social isolation, increased HSP70i showed anti-apoptotic effects as evidenced by the absence of cleaved caspase-3 and apoptosis (Kiang 2004), findings also demonstrated in the hippocampus (Filipović et al. 2011). Nevertheless, despite a lack of neurotoxicity, an observed increase in hippocampal NO levels that most likely originates from nNOS may still be involved in depressive-like behavior in socially isolated rats (Zhou et al. 2007).
Mitochondria-related proapoptotic signaling in the prefrontal cortex but not in hippocampus of socially isolated adult male rats
Chronic stress may induce apoptosis via genomic and non-genomic actions of elevated GCs, as well as affecting mitochondrial functions (Zhang et al. 2006). Oxidative stress is a known initiator of apoptotic signaling, whereby ROS generated from mitochondria may cause p53-mediated apoptotic signaling independently of its transcriptional activity (Mihara et al. 2003). p53 is a tumor suppressor protein and transcription activator that modulates the expression of numerous target genes that control apoptosis (Morselli et al. 2008). After activation, p53 may rapidly translocate from the cytoplasm to mitochondria (detectable at 30 min–1 h) (Moll et al. 2005), where ROS plays a signaling role in the mitochondrial migration of p53 (Nithipongvanitch et al. 2007). Its translocation causes permeabilization of the outer mitochondrial membrane by forming an inhibitory complex with protective Bcl-2 family proteins, resulting in mitochondrial cytochrome c release to the cytoplasm and caspase activation, triggering apoptotic cell death (Mihara et al. 2003; Chipuk et al. 2004). Bcl-2 family proteins, whose members may be antiapoptotic (Bcl-2) or proapoptotic (Bcl-2-associated X protein, Bax), regulate mitochondrial membrane permeability during apoptosis (Shimizu et al. 1999). Moreover, an increase in the prosurvival molecule Bcl-2 in neurons and inhibition of p53 translocation has been linked to overexpression of HO-1 (Panahian et al. 1999). Specifically, the interaction between Bcl-2, p53 and HO-1 may involve the heme-regulating motifs of HO-2 (McCoubrey et al. 1997). Bax is a soluble protein present predominantly in the cytosol that, during the induction of apoptosis, shifts to mitochondrial membranes, causing the release of cytochrome c preceding caspase activation (Kroemer and Reed 2000), while Bcl-2 is present in mitochondria and functions as a repressor of apoptosis (Reed et al. 1998). The ratio of Bcl-2/Bax in mitochondria determines the cellular response to cell death signals transmitted by mitochondria (Desagher and Martinou 2000). While overexpression of Bcl-2 (a higher Bcl-2/Bax ratio) protects cells from apoptosis, the translocation of Bax to the mitochondria induces cytochrome c release that can trigger apoptosis (Hsu et al. 1997). Moreover, sustained NO overproduction via iNOS can induce apoptosis via mitochondrial Bax translocation (Ghatan et al. 2000).
Three weeks of CSIS in adult male Wistar rats caused an increase in protein levels of cytosolic cytochrome c and cleaved caspase-3 activation, leading to apoptotic cell death in the prefrontal cortex (Filipović et al. 2011) (Fig. 6, left part). These results suggest that CSIS compromised mitochondrial membrane integrity and caused a loss of mitochondrial function (Cruthirds et al. 2003). Moreover, proapoptotic signaling initiated by CSIS in the adult male rat prefrontal cortex enhanced the proapoptotic response to subsequent acute immobilization or cold stressors by sustained NO overproduction, accompanied by the translocation of cytosolic p53 and proapoptotic Bax protein to mitochondria. Also, mitochondrial membrane antiapoptotic Bcl-2 protein translocation to cytoplasm, mitochondrial cytochrome c release into the cytoplasm, and caspase-3 activation occurred, causing apoptosis (Filipović et al. 2011). Interestingly, the effects of CSIS following exposure to a subsequent acute stressor were not mediated by the regulation of Bax as a proapoptotic factor, but rather by increased cytosolic antiapoptotic Bcl-2 protein, resulting from its translocation from mitochondria in the prefrontal cortex (Cao et al. 2001). Given that the Bax/Bcl-2 ratio was unchanged in the hippocampus and upregulated in the prefrontal cortex, it is likely that CSIS exerts opposing actions on Bax and Bcl-2 in a tissue-specific manner (prefrontal cortex versus hippocampus), indicating that the prefrontal cortex is a key target of the maladaptive response to stress (Cerqueira et al. 2007). In addition, HSP70i induction in the hippocampus of socially isolated rats protects neurons from apoptosis (Belay and Brown 2003; Arieli et al. 2003) through its ability to inhibit NF-κB activation (Malhotra and Wong 2002) (Fig. 6, right part), increase the Bcl-2 stability during oxidative stress (Jiang et al. 2009), inhibit translocation of Bax into mitochondria, and suppress mitochondrial cytochrome c release (Didelot et al. 2006). Furthermore, HSP70i interferes with the formation of the apoptosome (Beere et al. 2000; Saleh et al. 2000) by preventing apoptosomal caspase activation. Nonetheless, direct interaction of HSP27 with one or more components of the permeability transition pore on the mitochondrial outer-membrane prevents the release of cytochrome c (Stetler et al. 2008). HSP70i may also inhibit apoptosome formation and/or the recruitment of caspase-9 to the complex by binding to cytochrome c or Apaf-1 (Bratton and Salvesen 2010). A recent study showed that oxidative stress increased protein levels of HSP27 only after 24 h in cultured rat hippocampal neurons (Bartelt-Kirbach and Golenhofen 2014). Unfortunately, at this time point, there are no data concerning the role of HSP27 in CSIS-induced alterations, but this issue should be addressed in future studies.

Schematic representation of p53-mediated mitochondrial proapoptotic signaling in response to 3 weeks of chronic social isolation (CSIS) stress and subsequent acute stressors (2 h of immobilization or cold) in brain regions of adult male Wistar rats. In the prefrontal cortex (left part), CSIS induces activation of nuclear factor-kappa B (NF-κB) that translocates to the nucleus followed by protein expression of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) that leads to an overproduction of nitric oxide (NO), causing oxidative/nitrosative state. Moreover, CSIS in the prefrontal cortex reinforced the proapoptotic response to aforementioned subsequent acute stressors via p53 mitochondrial translocation, which is followed by translocation of cytosolic Bcl-2-associated X protein (Bax) to mitochondria and B-celllymphoma-2 (Bcl-2) from the mitochondrial membrane to the cytoplasm, mitochondrial cytochrome c release into the cytoplasm, and caspase-3 activation. In contrast, the upregulation of inducible heat shock protein 70 (HSP70i) inhibits NF-κB activation that may provide cellular protection against CSIS-induced oxidative/nitrosative stress in the rat hippocampus (right part), as well as apoptosome formation, preventing caspase-3 activation and apoptosis
Beyond the prefrontal cortex and hippocampus
Although this review has focused on the effect of CSIS on oxidative and nitrosative stress pathways in the prefrontal cortex and hippocampus, stress-induced alterations in other brain regions likely also serve to impair adaptive stress responses. For example, the amygdala also plays an important role in social behaviors (Sandi et al. 2008). Given that the prefrontal cortex has robust projections to the amygdala (Del Arco and Mora 2009), initiating a glucocorticoid cascade through the HPA axis (Jankord and Herman 2008), dysregulation of amygdalar function may also be associated with anxiety and mood disorders. For example, increases in anxiety-like behavior are associated with a loss of GABAergic interneurons in the basolateral amygdala (Truitt et al. 2009) and reduced inhibitory synaptic transmission (Chen et al. 2013). In fact, hyperactivity of the amygdala has been observed in patients with social anxiety disorder (Phan et al. 2006). The entorhinal cortex, which has extensive reciprocal connections with the hippocampus and amygdala (Pitkänen et al. 2000), has also been demonstrated to play a role in psychosocial stress (Blanchard et al. 1991; Lucassen et al. 2001) and the regulation of HPA axis activity (Umegaki et al. 2006; Zhu et al. 2008), though its specific function during social isolation has only been addressed in a few studies. For example, in mice, 4 weeks of social isolation significantly lowered serotonin (5-HT1A) postsynaptic receptor densities in the frontal and entorhinal cortex, as well as in limbic regions (Schiller et al. 2003). Interestingly, the amygdala and entorhinal cortex are necessary for the processing of complex constructs such as emotional learning (Sah et al. 2003; Green and McCormick 2013) and spatial cognition (Burgess 2008; Kunz et al. 2015), respectively, which are altered following exposure to chronic stress (Avital et al. 2006; Green and McCormick 2013) and may be relevant to CSIS. Thus, further studies characterizing the role of pathways mediating CSIS in these and other brain regions will complement the findings discussed here in the prefrontal cortex and hippocampus.
Conclusion
Exposure of an organism to chronic psychosocial stress may lead to activation of the HPA axis and increased release of GCs, causing oxidative stress that is implicated in several mental disorders, including depression and anxiety. However, adult male Wistar rats exposed to CSIS for 3 weeks showed unchanged serum CORT levels that may illustrate the mechanism underlying the glucocorticoid paradox, in which a state of oxidative stress might also exist under CORT levels similar to basal values. Moreover, a prooxidant state may, at least in part, result from the sustained overproduction of NO and increased iNOS protein expression. Furthermore, CSIS-induced oxidative and nitrosative stress in the rat prefrontal cortex was mediated by NF-κB activation accompanied by an increased iNOS protein expression which compromised antioxidative enzyme activity. Mitochondrial proapoptotic signaling initiated by CSIS in the prefrontal cortex was reinforced by subsequent acute stressor via p53 mitochondrial translocation, Bax and Bcl-2 proteins redistribution between mitochondrial and cytoplasmic compartments, mitochondrial cytochrome c release into the cytoplasm, and the activation of caspase-3, causing apoptosis (Fig. 7). In contrast, the upregulation of HSP70i protected rat hippocampus from CSIS-induced neurotoxicity. The differential regulation of NF-κB, iNOS and COX-2 following CSIS may be one functional difference between the prefrontal cortex and hippocampus, as well as an indicator of differential sensitivity of these rat brain structures to oxidative stress. The observed different thresholds for stress susceptibility of the prefrontal cortex and hippocampus may also be due to alternative signaling pathways operating in these brain regions (Mizoguchi et al. 2003). All aforementioned conditions may compromise the adaptive stress responses in the hippocampus and prefrontal cortex of adult male rats that are closely related to behavioral depressive- and anxiety-like symptoms. Hence, oxidative and nitrosative mechanisms may be potential targets for therapeutic strategies and the development of drugs for the treatment of stress-induced depressive/anxious states.

Maladaptive stress response in the prefrontal cortex of chronically isolated rats. Chronic social isolation stress compromises hypothalamic-pituitary-adrenocortical (HPA) axis functioning and causes oxidative and nitrosative stress, likely triggered by nuclear factor-kappa B (NF-κB) activation and concomitant inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) upregulation, which results in increased NO and prostaglandin (PGE) production. In addition, NADPH oxidase (NOX)-derived reactive oxygen species (ROS), together with a compromised antioxidative defense contribute to a cellular prooxidant state. Superoxide anion radical (O ·−2 ) may either react with NO and form radical peroxynitrite (ONOO−), which negatively affects the activity of manganese superoxide dismutase (MnSOD), or may be processed by copper-zinc superoxide dismutase (CuZnSOD) or MnSOD and be converted to hydrogen peroxide (H2O2). A social isolation-induced decrease in catalase (CAT) activity contributes to H2O2 accumulation. A prooxidant environment causes depletion of glutathione GSH, a major redox buffer, which together with an impared peroxidase-reductase system exacerbates the difficulty in responding to prooxidative insult and causes accumulation of peroxidizable products promoting apoptotic signaling. Increased levels of NO promote mitochondrial Bcl-2-associated X protein (Bax) and p53 translocation; ROS and p53 cause mitochondrial membrane permeabilization which leads to cytochrome c release in the cytoplasm, apoptosome formation, and caspase-3 activation. Finally, all described changes may be manifested as changes in behavior, such as depressive- and anxiety-like behavior and behavioral despair. Numbers indicate proposed inducible heat shock protein 70 (HSP70i)-mediated protection in the hippocampus: 1inhibition of NF-κB activation and NF-κB-dependent gene expression; 2inhibition of Bax translocation into mitochondria, cytochrome c release in the cytoplasm, and prevention of apoptosomal caspase activation
References
Ahmad A, Rasheed N, Banu N, Palit G (2010) Alterations in monoamine levels and oxidative systems in frontal cortex, striatum, and hippocampus of the rat brain during chronic unpredictable stress. Stress 13:355–364. doi:10.3109/10253891003667862
Anand P, Stamler JS (2012) Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med (Berl) 90:233–244. doi:10.1007/s00109-012-0878-z
Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70:200–214
Aoyama K, Watabe M, Nakaki T (2008) Regulation of neuronal glutathione synthesis. J Pharmacol Sci 108:227–238
Arieli Y, Eynan M, Gancz H et al (2003) Heat acclimation prolongs the time to central nervous system oxygen toxicity in the rat. Possible involvement of HSP72. Brain Res 962:15–20
Arimoto T, Bing G (2003) Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dis 12:35–45
Avital A, Ram E, Maayan R et al (2006) Effects of early-life stress on behavior and neurosteroid levels in the rat hypothalamus and entorhinal cortex. Brain Res Bull 68:419–424. doi:10.1016/j.brainresbull.2005.09.015
Bartelt-Kirbach B, Golenhofen N (2014) Reaction of small heat-shock proteins to different kinds of cellular stress in cultured rat hippocampal neurons. Cell Stress Chaperones 19:145–153. doi:10.1007/s12192-013-0452-9
Beere HM, Wolf BB, Cain K et al (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2:469–475. doi:10.1038/35019501
Belay HT, Brown IR (2003) Spatial analysis of cell death and Hsp70 induction in brain, thymus, and bone marrow of the hyperthermic rat. Cell Stress Chaperones 8:395–404
Blanchard DC, Cholvanich P, Blanchard RJ et al (1991) Serotonin, but not dopamine, metabolites are increased in selected brain regions of subordinate male rats in a colony environment. Brain Res 568:61–66
Borsini F, Podhorna J, Marazziti D (2002) Do animal models of anxiety predict anxiolytic-like effects of antidepressants? Psychopharmacology 163:121–141. doi:10.1007/s00213-002-1155-6
Bowie A, O’Neill LA (2000) Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol 59:13–23
Bratton SB, Salvesen GS (2010) Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci 123:3209–3214. doi:10.1242/jcs.073643
Brenes JC, Fornaguera J (2009) The effect of chronic fluoxetine on social isolation-induced changes on sucrose consumption, immobility behavior, and on serotonin and dopamine function in hippocampus and ventral striatum. Behav Brain Res 198:199–205. doi:10.1016/j.bbr.2008.10.036
Brown GC (2007) Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans 35:1119–1121. doi:10.1042/BST0351119
Brown GW, Prudo R (1981) Psychiatric disorder in a rural and an urban population: 1. Aetiology of depression. Psychol Med 11:581–599
Burgess N (2008) Spatial cognition and the brain. Ann N Y Acad Sci 1124:77–97. doi:10.1196/annals.1440.002
Cai J, Jones DP (1998) Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 273:11401–11404
Calabrese V, Copani A, Testa D et al (2000) Nitric oxide synthase induction in astroglial cell cultures: effect on heat shock protein 70 synthesis and oxidant/antioxidant balance. J Neurosci Res 60:613–622
Candas D, Li JJ (2014) MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid Redox Signal 20:1599–1617. doi:10.1089/ars.2013.5305
Cao G, Minami M, Pei W et al (2001) Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab 21:321–333. doi:10.1097/00004647-200104000-00001
Carrier N, Kabbaj M (2012) Testosterone and imipramine have antidepressant effects in socially isolated male but not female rats. Horm Behav 61:678–685. doi:10.1016/j.yhbeh.2012.03.001
Cassina P, Peluffo H, Pehar M et al (2002) Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J Neurosci Res 67:21–29
Cerqueira JJ, Mailliet F, Almeida OFX et al (2007) The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci 27:2781–2787. doi:10.1523/JNEUROSCI.4372-06.2007
Chang LY, Slot JW, Geuze HJ, Crapo JD (1988) Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J Cell Biol 107:2169–2179
Charette SJ, Lavoie JN, Lambert H, Landry J (2000) Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol 20:7602–7612
Chelikani P, Fita I, Loewen PC (2004) Diversity of structures and properties among catalases. Cell Mol Life Sci 61:192–208. doi:10.1007/s00018-003-3206-5
Chen F, Demers LM, Vallyathan V et al (1999) Involvement of 5′-flanking kappaB-like sites within bcl-x gene in silica-induced Bcl-x expression. J Biol Chem 274:35591–35595
Chen Z, Xu H, Haimano S et al (2005) Quetiapine and venlafaxine synergically regulate heme oxygenase-2 protein expression in the hippocampus of stressed rats. Neurosci Lett 389:173–177. doi:10.1016/j.neulet.2005.07.040
Chen J, Song Y, Yang J et al (2013) The contribution of TNF-α in the amygdala to anxiety in mice with persistent inflammatory pain. Neurosci Lett 541:275–280. doi:10.1016/j.neulet.2013.02.005
Chipuk JE, Kuwana T, Bouchier-Hayes L et al (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014. doi:10.1126/science.1092734
Colaianna M, Schiavone S, Zotti M et al (2013) Neuroendocrine profile in a rat model of psychosocial stress: relation to oxidative stress. Antioxid Redox Signal 18:1385–1399. doi:10.1089/ars.2012.4569
Connelly L, Palacios-Callender M, Ameixa C et al (2001) Biphasic regulation of NF-kappa B activity underlies the pro- and anti-inflammatory actions of nitric oxide. J Immunol 166:3873–3881
Couto N, Malys N, Gaskell SJ, Barber J (2013) Partition and turnover of glutathione reductase from Saccharomyces cerevisiae: a proteomic approach. J Proteome Res 12:2885–2894. doi:10.1021/pr4001948
Cregan SP, Fortin A, MacLaurin JG et al (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158:507–517. doi:10.1083/jcb.200202130
Cruthirds DL, Novak L, Akhi KM et al (2003) Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Arch Biochem Biophys 412:27–33
Datta SR, Dudek H, Tao X et al (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Davis RL, Sanchez AC, Lindley DJ et al (2005) Effects of mechanistically distinct NF-kappaB inhibitors on glial inducible nitric-oxide synthase expression. Nitric Oxide 12:200–209. doi:10.1016/j.niox.2005.04.005
De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M (1998) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19:269–301. doi:10.1210/edrv.19.3.0331
De Kloet ER, Joëls M, Holsboer F (2005) Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6:463–475. doi:10.1038/nrn1683
Dean O, Bush AI, Berk M et al (2009) Glutathione depletion in the brain disrupts short-term spatial memory in the Y-maze in rats and mice. Behav Brain Res 198:258–262. doi:10.1016/j.bbr.2008.11.017
Del Arco A, Mora F (2009) Neurotransmitters and prefrontal cortex-limbic system interactions: implications for plasticity and psychiatric disorders. J Neural Transm 116:941–952. doi:10.1007/s00702-009-0243-8
Desagher S, Martinou JC (2000) Mitochondria as the central control point of apoptosis. Trends Cell Biol 10:369–377
Didelot C, Schmitt E, Brunet M et al (2006) Heat shock proteins: endogenous modulators of apoptotic cell death. Handb Exp Pharmacol 172:171–198
Djordjevic J, Djordjevic A, Adzic M et al (2015) Alterations in the Nrf2-Keap1 signaling pathway and its downstream target genes in rat brain under stress. Brain Res 1602:20–31. doi:10.1016/j.brainres.2015.01.010
Dringen R (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol 62:649–671
Dronjak S, Gavrilović L, Filipović D, Radojcić MB (2004) Immobilization and cold stress affect sympatho-adrenomedullary system and pituitary-adrenocortical axis of rats exposed to long-term isolation and crowding. Physiol Behav 81:409–415. doi:10.1016/j.physbeh.2004.01.011
Einon D (1980) Spatial memory and response strategies in rats: age, sex and rearing differences in performance. Q J Exp Psychol 32:473–489. doi:10.1080/14640748008401840
Eren İ, Nazıroğlu M, Demirdaş A et al (2007) Venlafaxine modulates depression-induced oxidative stress in brain and medulla of rat. Neurochem Res 32:497–505. doi:10.1007/s11064-006-9258-9
Farley S, Apazoglou K, Witkin JM et al (2010) Antidepressant-like effects of an AMPA receptor potentiator under a chronic mild stress paradigm. Int J Neuropsychopharmacol 13:1207–1218. doi:10.1017/S1461145709991076
Ferland CL, Schrader LA (2011) Cage mate separation in pair-housed male rats evokes an acute stress corticosterone response. Neurosci Lett 489:154–158. doi:10.1016/j.neulet.2010.12.006
Filipović D, Pajović SB (2009) Differential regulation of CuZnSOD expression in rat brain by acute and/or chronic stress. Cell Mol Neurobiol 29:673–681. doi:10.1007/s10571-009-9375-5
Filipović D, Gavrilović L, Dronjak S, Radojcić MB (2005) Brain glucocorticoid receptor and heat shock protein 70 levels in rats exposed to acute, chronic or combined stress. Neuropsychobiology 51:107–114. doi:10.1159/000084168
Filipović D, Zlatković J, Pajović SB (2009) The effect of acute or/and chronic stress on the MnSOD protein expression in rat prefrontal cortex and hippocampus. Gen Physiol Biophys 28 Spec No:53–61
Filipović D, Zlatković J, Inta D et al (2011) Chronic isolation stress predisposes the frontal cortex but not the hippocampus to the potentially detrimental release of cytochrome c from mitochondria and the activation of caspase-3. J Neurosci Res 89:1461–1470. doi:10.1002/jnr.22687
Filipović D, Zlatković J, Gass P, Inta D (2013) The differential effects of acute vs. chronic stress and their combination on hippocampal parvalbumin and inducible heat shock protein 70 expression. Neuroscience 236:47–54. doi:10.1016/j.neuroscience.2013.01.033
Fuchs E (2005) Social stress in tree shrews as an animal model of depression: an example of a behavioral model of a CNS disorder. CNS Spectr 10:182–190
Garrido C, Bruey JM, Fromentin A et al (1999) HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J 13:2061–2070
Garzón J, Del Río J (1981) Hyperactivity induced in rats by long-term isolation: further studies on a new animal model for the detection of antidepressants. Eur J Pharmacol 74:287–294
Gass P, Reichardt HM, Strekalova T et al (2001) Mice with targeted mutations of glucocorticoid and mineralocorticoid receptors: models for depression and anxiety? Physiol Behav 73:811–825
Gawryluk JW, Wang J-F, Andreazza AC et al (2011) Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol 14:123–130. doi:10.1017/S1461145710000805
Georgopoulos C, Welch WJ (1993) Role of the major heat shock proteins as molecular chaperones. Annu Rev Cell Biol 9:601–634. doi:10.1146/annurev.cb.09.110193.003125
Ghatan S, Larner S, Kinoshita Y et al (2000) p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol 150:335–347
Giustarini D, Rossi R, Milzani A et al (2004) S-glutathionylation: from redox regulation of protein functions to human diseases. J Cell Mol Med 8:201–212. doi:10.1111/j.1582-4934.2004.tb00275.x
Goto Y, Yang CR, Otani S (2010) Functional and dysfunctional synaptic plasticity in prefrontal cortex: roles in psychiatric disorders. Biol Psychiatry 67:199–207. doi:10.1016/j.biopsych.2009.08.026
Gould TD, Gottesman II (2006) Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav 5:113–119. doi:10.1111/j.1601-183X.2005.00186.x
Green MR, McCormick CM (2013) Effects of stressors in adolescence on learning and memory in rodent models. Horm Behav 64:364–379. doi:10.1016/j.yhbeh.2012.09.012
Gupta S, Pandey R, Katyal R et al (2005) Lipid peroxide levels and antioxidant status in alcoholic liver disease. Indian J Clin Biochem 20:67–71. doi:10.1007/BF02893045
Gutierrez-Correa J, Stoppani AO (1997) Inactivation of yeast glutathione reductase by Fenton systems: effect of metal chelators, catecholamines and thiol compounds. Free Radic Res 27:543–555
Hall FS (1998) Social deprivation of neonatal, adolescent, and adult rats has distinct neurochemical and behavioral consequences. Crit Rev Neurobiol 12:129–162
Hall AV, Antoniou H, Wang Y et al (1994) Structural organization of the human neuronal nitric oxide synthase gene (NOS1). J Biol Chem 269:33082–33090
Halliwell B (2011) Free radicals and antioxidants—quo vadis? Trends Pharmacol Sci 32:125–130. doi:10.1016/j.tips.2010.12.002
Halliwell B, Gutteridge JMC (1989) Free radicals in biology and medicine, 2nd edn. Oxford University Press, Oxford
Hao W, Myhre AP, Palmer JP (1999) Nitric oxide mediates IL-1beta stimulation of heat shock protein but not IL-1beta inhibition of glutamic acid decarboxylase. Autoimmunity 29:93–101
Haroon E, Raison CL, Miller AH (2012) Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37:137–162. doi:10.1038/npp.2011.205
Harte MK, Powell SB, Swerdlow NR et al (2007) Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm 114:893–898. doi:10.1007/s00702-007-0627-6
Hatch AM, Wiberg GS, Zawidzka Z et al (1965) Isolation syndrome in the rat. Toxicol Appl Pharmacol 7:737–745
Havasi A, Li Z, Wang Z et al (2008) Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem 283:12305–12313. doi:10.1074/jbc.M801291200
Heim C, Nemeroff CB (2001) The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry 49:1023–1039
Heinrich LM, Gullone E (2006) The clinical significance of loneliness: a literature review. Clin Psychol Rev 26:695–718. doi:10.1016/j.cpr.2006.04.002
Heneka MT, Sharp A, Klockgether T et al (2000) The heat shock response inhibits NF-kappaB activation, nitric oxide synthase type 2 expression, and macrophage/microglial activation in brain. J Cereb Blood Flow Metab 20:800–811. doi:10.1097/00004647-200005000-00006
Hinwood M, Morandini J, Day TA, Walker FR (2012) Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex 22:1442–1454. doi:10.1093/cercor/bhr229
Holsboer F, Ising M (2010) Stress hormone regulation: biological role and translation into therapy. Annu Rev Psychol 61:81–109. doi:10.1146/annurev.psych.093008.100321
Holson RR, Scallet AC, Ali SF, Turner BB (1991) “Isolation stress” revisited: isolation-rearing effects depend on animal care methods. Physiol Behav 49:1107–1118
Hopper RK, Carroll S, Aponte AM et al (2006) Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry 45:2524–2536. doi:10.1021/bi052475e
House JS (2001) Social isolation kills, but how and why? Psychosom Med 63:273–274. doi:10.1097/00006842-200103000-00011
Hsu YT, Wolter KG, Youle RJ (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 94:3668–3672
Ishida H, Mitsui K, Nukaya H et al (2003) Study of active substances involved in skin dysfunction induced by crowding stress. I. Effect of crowding and isolation on some physiological variables, skin function and skin blood perfusion in hairless mice. Biol Pharm Bull 26:170–181
Janáky R, Ogita K, Pasqualotto BA et al (1999) Glutathione and signal transduction in the mammalian CNS. J Neurochem 73:889–902
Janáky R, Shaw CA, Varga V et al (2000) Specific glutathione binding sites in pig cerebral cortical synaptic membranes. Neuroscience 95:617–624
Jankord R, Herman JP (2008) Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci 1148:64–73. doi:10.1196/annals.1410.012
Jiang B, Wang K, Liang P et al (2009) ATP-binding domain of heat shock protein 70 is essential for its effects on the inhibition of the release of the second mitochondria-derived activator of caspase and apoptosis in C2C12 cells. FEBS J 276:2615–2624. doi:10.1111/j.1742-4658.2009.06989.x
Jin Z-Q, Zhou H-Z, Cecchini G et al (2005) MnSOD in mouse heart: acute responses to ischemic preconditioning and ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 288:H2986–H2994. doi:10.1152/ajpheart.01144.2004
Jin W, Wang H, Yan W et al (2008) Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediat Inflamm 2008:725174. doi:10.1155/2008/725174
Joëls M, Baram TZ (2009) The neuro-symphony of stress. Nat Rev Neurosci 10:459–466. doi:10.1038/nrn2632
Joëls M, Karst H, Alfarez D et al (2004) Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress 7:221–231. doi:10.1080/10253890500070005
Kiang JG (2004) Inducible heat shock protein 70 kD and inducible nitric oxide synthase in hemorrhage/resuscitation-induced injury. Cell Res 14:450–459. doi:10.1038/sj.cr.7290247
Kim HT, Kim YH, Nam JW et al (1994) Study of 5′-flanking region of human Cu/Zn superoxide dismutase. Biochem Biophys Res Commun 201:1526–1533
Kobayashi Y, Nishikawa M, Hyoudou K et al (2008) Hydrogen peroxide-mediated nuclear factor kappaB activation in both liver and tumor cells during initial stages of hepatic metastasis. Cancer Sci 99:1546–1552. doi:10.1111/j.1349-7006.2008.00856.x
Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6:513–519. doi:10.1038/74994
Krolow R, Noschang C, Weis SN et al (2012) Isolation stress during the prepubertal period in rats induces long-lasting neurochemical changes in the prefrontal cortex. Neurochem Res 37:1063–1073. doi:10.1007/s11064-012-0709-1
Kunz L, Schröder TN, Lee H et al (2015) Reduced grid-cell-like representations in adults at genetic risk for Alzheimer’s disease. Science 350:430–433. doi:10.1126/science.aac8128
Lapiz MDS, Fulford A, Muchimapura S et al (2003) Influence of postweaning social isolation in the rat on brain development, conditioned behavior, and neurotransmission. Neurosci Behav Physiol 33:13–29
Lawler JM, Song W (2002) Specificity of antioxidant enzyme inhibition in skeletal muscle to reactive nitrogen species donors. Biochem Biophys Res Commun 294:1093–1100. doi:10.1016/S0006-291X(02)00602-2
Leza JC, Salas E, Sawicki G et al (1998) The effects of stress on homeostasis in JCR-LA-cp rats: the role of nitric oxide. J Pharmacol Exp Ther 286:1397–1403
Li N, Karin M (1999) Is NF-kappaB the sensor of oxidative stress? FASEB J 13:1137–1143
Li Y, Li G, Li C, Zhao Y (2007) Identification of nuclear factor-kappaB responsive element within the neuronal nitric oxide synthase exon 1f-specific promoter. Acta Biochim Biophys Sin (Shanghai) 39:247–254
Liu J, Mori A (1994) Involvement of reactive oxygen species in emotional stress: a hypothesis based on the immobilization stress-induced oxidative damage and antioxidant defense changes in rat brain, and the effect of antioxidant treatment with reduced glutathione. Int J Stress Manag 1:249–263. doi:10.1007/BF01857992
Lucassen PJ, Vollmann-Honsdorf GK, Gleisberg M et al (2001) Chronic psychosocial stress differentially affects apoptosis in hippocampal subregions and cortex of the adult tree shrew. Eur J Neurosci 14:161–166
Lupien SJ, McEwen BS, Gunnar MR, Heim C (2009) Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 10:434–445. doi:10.1038/nrn2639
Madan AP, DeFranco DB (1993) Bidirectional transport of glucocorticoid receptors across the nuclear envelope. Proc Natl Acad Sci USA 90:3588–3592
Madrigal JL, Moro MA, Lizasoain I et al (2001a) Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J Neurochem 76:532–538
Madrigal JL, Olivenza R, Moro MA et al (2001b) Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24:420–429. doi:10.1016/S0893-133X(00)00208-6
Madrigal JLM, García-Bueno B, Moro MA et al (2003) Relationship between cyclooxygenase-2 and nitric oxide synthase-2 in rat cortex after stress. Eur J Neurosci 18:1701–1705
Madrigal JLM, García-Bueno B, Caso JR et al (2006) Stress-induced oxidative changes in brain. CNS Neurol Disord Drug Targets 5:561–568
Maes M, Mihaylova I, Bosmans E (2007a) Not in the mind of neurasthenic lazybones but in the cell nucleus: patients with chronic fatigue syndrome have increased production of nuclear factor kappa beta. Neuro Endocrinol Lett 28:456–462
Maes M, Mihaylova I, Kubera M, Bosmans E (2007b) Not in the mind but in the cell: increased production of cyclo-oxygenase-2 and inducible NO synthase in chronic fatigue syndrome. Neuro Endocrinol Lett 28:463–469
Maes M, Galecki P, Chang YS, Berk M (2011) A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog Neuropsychopharmacol Biol Psychiatry 35:676–692. doi:10.1016/j.pnpbp.2010.05.004
Maines MD, Eke BC, Zhao X (1996) Corticosterone promotes increased heme oxygenase-2 protein and transcript expression in the newborn rat brain. Brain Res 722:83–94
Makino Y, Okamoto K, Yoshikawa N et al (1996) Thioredoxin: a redox-regulating cellular cofactor for glucocorticoid hormone action. Cross talk between endocrine control of stress response and cellular antioxidant defense system. J Clin Invest 98:2469–2477. doi:10.1172/JCI119065
Malhotra V, Wong HR (2002) Interactions between the heat shock response and the nuclear factor-kappaB signaling pathway. Crit Care Med 30:S89–S95
Mancuso C, Pani G, Calabrese V (2006) Bilirubin: an endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep 11:207–213. doi:10.1179/135100006X154978
McCoubrey WK, Huang TJ, Maines MD (1997) Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J Biol Chem 272:12568–12574
McEwen BS (2008) Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol 583:174–185. doi:10.1016/j.ejphar.2007.11.071
McIntosh LJ, Cortopassi KM, Sapolsky RM (1998a) Glucocorticoids may alter antioxidant enzyme capacity in the brain: kainic acid studies. Brain Res 791:215–222
McIntosh LJ, Hong KE, Sapolsky RM (1998b) Glucocorticoids may alter antioxidant enzyme capacity in the brain: baseline studies. Brain Res 791:209–214
McKay LI, Cidlowski JA (2000) CBP (CREB binding protein) integrates NF-kappaB (nuclear factor-kappaB) and glucocorticoid receptor physical interactions and antagonism. Mol Endocrinol 14:1222–1234. doi:10.1210/mend.14.8.0506
McLeod TM, López-Figueroa AL, López-Figueroa MO (2001) Nitric oxide, stress, and depression. Psychopharmacol Bull 35:24–41
Meyer M, Schreck R, Baeuerle PA (1993) H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J 12:2005–2015
Miachon S, Rochet T, Mathian B et al (1993) Long-term isolation of Wistar rats alters brain monoamine turnover, blood corticosterone, and ACTH. Brain Res Bull 32:611–614
Mihara M, Erster S, Zaika A et al (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11:577–590
Mihm S, Galter D, Dröge W (1995) Modulation of transcription factor NF kappa B activity by intracellular glutathione levels and by variations of the extracellular cysteine supply. FASEB J 9:246–252
Mizoguchi K, Ishige A, Aburada M, Tabira T (2003) Chronic stress attenuates glucocorticoid negative feedback: involvement of the prefrontal cortex and hippocampus. Neuroscience 119:887–897
Moll UM, Wolff S, Speidel D, Deppert W (2005) Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 17:631–636. doi:10.1016/j.ceb.2005.09.007
Möller M, Du Preez JL, Emsley R, Harvey BH (2011) Isolation rearing-induced deficits in sensorimotor gating and social interaction in rats are related to cortico-striatal oxidative stress, and reversed by sub-chronic clozapine administration. Eur Neuropsychopharmacol 21:471–483. doi:10.1016/j.euroneuro.2010.09.006
Möller M, Du Preez JL, Viljoen FP et al (2013) Social isolation rearing induces mitochondrial, immunological, neurochemical and behavioural deficits in rats, and is reversed by clozapine or N-acetyl cysteine. Brain Behav Immun 30:156–167. doi:10.1016/j.bbi.2012.12.011
Morimoto RI, Tissieres AGC (1994) Molecular chaperone functions of hsp70 and hsp60 in protein folding. In: Frydman JHF (ed) The biology of the heat shock proteins and molecular chaperones. Cold Spring Harbor Laboratory Press, New York, pp 251–283
Morselli E, Galluzzi L, Kroemer G (2008) Mechanisms of p53-mediated mitochondrial membrane permeabilization. Cell Res 18:708–710. doi:10.1038/cr.2008.77
Muñoz-Sánchez J, Chánez-Cárdenas ME (2014) A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev 2014:604981. doi:10.1155/2014/604981
Muriach M, López-Pedrajas R, Barcia JM et al (2010) Cocaine causes memory and learning impairments in rats: involvement of nuclear factor kappa B and oxidative stress, and prevention by topiramate. J Neurochem 114:675–684. doi:10.1111/j.1471-4159.2010.06794.x
Musazzi L, Racagni G, Popoli M (2011) Stress, glucocorticoids and glutamate release: effects of antidepressant drugs. Neurochem Int 59:138–149. doi:10.1016/j.neuint.2011.05.002
Nithipongvanitch R, Ittarat W, Cole MP et al (2007) Mitochondrial and nuclear p53 localization in cardiomyocytes: redox modulation by doxorubicin (Adriamycin)? Antioxid Redox Signal 9:1001–1008. doi:10.1089/ars.2007.1632
Okamoto K, Tanaka H, Ogawa H et al (1999) Redox-dependent regulation of nuclear import of the glucocorticoid receptor. J Biol Chem 274:10363–10371
Olivenza R, Moro MA, Lizasoain I et al (2000) Chronic stress induces the expression of inducible nitric oxide synthase in rat brain cortex. J Neurochem 74:785–791
Ozden O, Park S-H, Kim H-S et al (2011) Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging (Albany NY) 3:102–107
Pacak K, Palkovits M, Yadid G et al (1998) Heterogeneous neurochemical responses to different stressors: a test of Selye’s doctrine of nonspecificity. Am J Physiol 275:R1247–R1255
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424. doi:10.1152/physrev.00029.2006
Panahian N, Yoshiura M, Maines MD (1999) Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem 72:1187–1203
Papadopoulos MC, Koumenis IL, Dugan LL, Giffard RG (1997) Vulnerability to glucose deprivation injury correlates with glutathione levels in astrocytes. Brain Res 748:151–156
Patel RP, Darley-Usmar VM (1996) Using peroxynitrite as oxidant with low-density lipoprotein. Methods Enzymol 269:375–384
Petrosillo G, Ruggiero FM, Pistolese M, Paradies G (2001) Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett 509:435–438
Phan KL, Fitzgerald DA, Nathan PJ, Tancer ME (2006) Association between amygdala hyperactivity to harsh faces and severity of social anxiety in generalized social phobia. Biol Psychiatry 59:424–429. doi:10.1016/j.biopsych.2005.08.012
Pitkänen A, Pikkarainen M, Nurminen N, Ylinen A (2000) Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann N Y Acad Sci 911:369–391
Pizzi M, Sarnico I, Boroni F et al (2005) Inhibition of IkappaBalpha phosphorylation prevents glutamate-induced NF-kappaB activation and neuronal cell death. Acta Neurochir Suppl 93:59–63
Plummer SM, Holloway KA, Manson MM et al (1999) Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene 18:6013–6020. doi:10.1038/sj.onc.1202980
Prabakaran S, Swatton JE, Ryan MM et al (2004) Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry 9(684–97):643. doi:10.1038/sj.mp.4001511
Radi R, Cassina A, Hodara R et al (2002) Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33:1451–1464
Raju VS, McCoubrey WK, Maines MD (1997) Regulation of heme oxygenase-2 by glucocorticoids in neonatal rat brain: characterization of a functional glucocorticoid response element. Biochim Biophys Acta 1351:89–104
Reed JC, Jurgensmeier JM, Matsuyama S (1998) Bcl-2 family proteins and mitochondria. Biochim Biophys Acta 1366:127–137
Ren Q-G, Gong W-G, Wang Y-J et al (2015) Citalopram attenuates tau hyperphosphorylation and spatial memory deficit induced by social isolation rearing in middle-aged rats. J Mol Neurosci 56:145–153. doi:10.1007/s12031-014-0475-4
Ridnour LA, Thomas DD, Mancardi D et al (2004) The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem 385:1–10. doi:10.1515/BC.2004.001
Ryter SW, Alam J, Choi AMK (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86:583–650. doi:10.1152/physrev.00011.2005
Sah P, Faber ESL, Lopez De Armentia M, Power J (2003) The amygdaloid complex: anatomy and physiology. Physiol Rev 83:803–834. doi:10.1152/physrev.00002.2003
Saleh A, Srinivasula SM, Balkir L et al (2000) Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2:476–483. doi:10.1038/35019510
Sánchez MM, Aguado F, Sánchez-Toscano F, Saphier D (1998) Neuroendocrine and immunocytochemical demonstrations of decreased hypothalamo-pituitary-adrenal axis responsiveness to restraint stress after long-term social isolation. Endocrinology 139:579–587. doi:10.1210/endo.139.2.5720
Sandi C, Haller J (2015) Stress and the social brain: behavioural effects and neurobiological mechanisms. Nat Rev Neurosci 16:290–304. doi:10.1038/nrn3918
Sandi C, Richter-Levin G (2009) From high anxiety trait to depression: a neurocognitive hypothesis. Trends Neurosci 32:312–320. doi:10.1016/j.tins.2009.02.004
Sandi C, Cordero MI, Ugolini A et al (2008) Chronic stress-induced alterations in amygdala responsiveness and behavior–modulation by trait anxiety and corticotropin-releasing factor systems. Eur J Neurosci 28:1836–1848. doi:10.1111/j.1460-9568.2008.06451.x
Scaccianoce S, Del Bianco P, Paolone G et al (2006) Social isolation selectively reduces hippocampal brain-derived neurotrophic factor without altering plasma corticosterone. Behav Brain Res 168:323–325. doi:10.1016/j.bbr.2005.04.024
Scapagnini G, D’Agata V, Calabrese V et al (2002) Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res 954:51–59
Schiavone S, Sorce S, Dubois-Dauphin M et al (2009) Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry 66:384–392. doi:10.1016/j.biopsych.2009.04.033
Schiavone S, Jaquet V, Sorce S et al (2012) NADPH oxidase elevations in pyramidal neurons drive psychosocial stress-induced neuropathology. Transl Psychiatry 2:e111. doi:10.1038/tp.2012.36
Schiavone S, Jaquet V, Trabace L, Krause K-H (2013) Severe life stress and oxidative stress in the brain: from animal models to human pathology. Antioxid Redox Signal 18:1475–1490. doi:10.1089/ars.2012.4720
Schiller L, Jähkel M, Kretzschmar M et al (2003) Autoradiographic analyses of 5-HT1A and 5-HT2A receptors after social isolation in mice. Brain Res 980:169–178
Schliess F, Görg B, Fischer R et al (2002) Ammonia induces MK-801-sensitive nitration and phosphorylation of protein tyrosine residues in rat astrocytes. FASEB J 16:739–741. doi:10.1096/fj.01-0862fje
Senftleben U, Cao Y, Xiao G et al (2001) Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293:1495–1499. doi:10.1126/science.1062677
Serra M, Pisu MG, Floris I, Biggio G (2005) Social isolation-induced changes in the hypothalamic-pituitary-adrenal axis in the rat. Stress 8:259–264. doi:10.1080/10253890500495244
Serra M, Sanna E, Mostallino MC, Biggio G (2007) Social isolation stress and neuroactive steroids. Eur Neuropsychopharmacol 17:1–11. doi:10.1016/j.euroneuro.2006.03.004
Shao Y, Yan G, Xuan Y et al (2015) Chronic social isolation decreases glutamate and glutamine levels and induces oxidative stress in the rat hippocampus. Behav Brain Res 282:201–208. doi:10.1016/j.bbr.2015.01.005
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487. doi:10.1038/20959
Singh R, Lemire J, Mailloux RJ, Appanna VD (2008) A novel strategy involved in [corrected] anti-oxidative defense: the conversion of NADH into NADPH by a metabolic network. PLoS One 3:e2682. doi:10.1371/journal.pone.0002682
Song L, Song W, Schipper HM (2007) Astroglia overexpressing heme oxygenase-1 predispose co-cultured PC12 cells to oxidative injury. J Neurosci Res 85:2186–2195. doi:10.1002/jnr.21367
Song W, Zukor H, Lin S-H et al (2012) Schizophrenia-like features in transgenic mice overexpressing human HO-1 in the astrocytic compartment. J Neurosci 32:10841–10853. doi:10.1523/JNEUROSCI.6469-11.2012
Sorce S, Krause K-H (2009) NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 11:2481–2504. doi:10.1089/ARS.2009.2578
Stojanović S, Stanić D, Nikolić M, Raicević S, Spasić M, Niketić V (2005) Manganese superoxide dismutase (MnSOD) catalyzes NO-dependent tyrosine residue nitration. J Serb Chem Soc 70:601–608
Spasojevic N, Gavrilovic L, Varagic VV, Dronjak S (2007) Effects of chronic diazepam treatments on behavior on individually housed rats. Arch Biol Sci 59:113–117. doi:10.2298/ABS0702113S
Spiers JG, Chen H-JC, Sernia C, Lavidis NA (2015) Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front Neurosci 8:456. doi:10.3389/fnins.2014.00456
Stetler RA, Cao G, Gao Y et al (2008) Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci 28:13038–13055. doi:10.1523/JNEUROSCI.4407-08.2008
Stranahan AM, Khalil D, Gould E (2006) Social isolation delays the positive effects of running on adult neurogenesis. Nat Neurosci 9:526–533. doi:10.1038/nn1668
Tamatani M, Che YH, Matsuzaki H et al (1999) Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem 274:8531–8538
Tao R, Coleman MC, Pennington JD et al (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40:893–904. doi:10.1016/j.molcel.2010.12.013
Tew KD, Ronai Z (1999) GST function in drug and stress response. Drug Resist Updat 2:143–147. doi:10.1054/drup.1999.0086
Todorović N, Bošković M, Filipović D (2014) Fluoxetine failed to prevent isolation-induced changes of glutathione-dependent defense in rat hippocampus. Physical Chemistry 2014, 12th International conference on fundamental and applied aspects of Physical Chemistry, vol 3, pp 1121–1124
Truitt WA, Johnson PL, Dietrich AD et al (2009) Anxiety-like behavior is modulated by a discrete subpopulation of interneurons in the basolateral amygdala. Neuroscience 160:284–294. doi:10.1016/j.neuroscience.2009.01.083
Tynan RJ, Naicker S, Hinwood M et al (2010) Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 24:1058–1068. doi:10.1016/j.bbi.2010.02.001
Umegaki H, Yamamoto A, Suzuki Y, Iguchi A (2006) Stimulation of the hippocampal glutamate receptor systems induces stress-like responses. Neuro Endocrinol Lett 27:339–343
Vesce S, Rossi D, Brambilla L, Volterra A (2007) Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int Rev Neurobiol 82:57–71. doi:10.1016/S0074-7742(07)82003-4
Wainwright SR, Galea LAM (2013) The neural plasticity theory of depression: assessing the roles of adult neurogenesis and PSA-NCAM within the hippocampus. Neural Plast 2013:805497. doi:10.1155/2013/805497
Weiss IC, Pryce CR, Jongen-Rêlo AL et al (2004) Effect of social isolation on stress-related behavioural and neuroendocrine state in the rat. Behav Brain Res 152:279–295. doi:10.1016/j.bbr.2003.10.015
Wiberg GS, Grice HC (1963) Long-term isolation stress in rats. Science 142:507
Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269:4705–4708
Xu Y, Kiningham KK, Devalaraja MN et al (1999) An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA Cell Biol 18:709–722. doi:10.1089/104454999314999
Zafir A, Banu N (2009) Induction of oxidative stress by restraint stress and corticosterone treatments in rats. Indian J Biochem Biophys 46:53–58
Zhang L, Zhou R, Li X et al (2006) Stress-induced change of mitochondria membrane potential regulated by genomic and non-genomic GR signaling: a possible mechanism for hippocampus atrophy in PTSD. Med Hypotheses 66:1205–1208. doi:10.1016/j.mehy.2005.11.041
Zheng Z, Kim JY, Ma H et al (2008) Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab 28:53–63. doi:10.1038/sj.jcbfm.9600502
Zhou Q-G, Hu Y, Hua Y et al (2007) Neuronal nitric oxide synthase contributes to chronic stress-induced depression by suppressing hippocampal neurogenesis. J Neurochem 103:1843–1854. doi:10.1111/j.1471-4159.2007.04914.x
Zhou Q-G, Zhu L-J, Chen C et al (2011) Hippocampal neuronal nitric oxide synthase mediates the stress-related depressive behaviors of glucocorticoids by downregulating glucocorticoid receptor. J Neurosci 31:7579–7590. doi:10.1523/JNEUROSCI.0004-11.2011
Zhu W, Zhang R, Hu C, Umegaki H (2008) Effect of the entorhinal cortex on diurnal ACTH and corticosterone release in rats. Neuro Endocrinol Lett 29:159–162
Zlatković J, Filipović D (2012) Bax and B-cell-lymphoma 2 mediate proapoptotic signaling following chronic isolation stress in rat brain. Neuroscience 223:238–245. doi:10.1016/j.neuroscience.2012.08.005
Zlatković J, Bernardi RE, Filipović D (2014a) Protective effect of Hsp70i against chronic social isolation stress in the rat hippocampus. J Neural Transm 121:3–14. doi:10.1007/s00702-013-1066-1
Zlatković J, Todorović N, Bošković M et al (2014b) Different susceptibility of prefrontal cortex and hippocampus to oxidative stress following chronic social isolation stress. Mol Cell Biochem 393:43–57. doi:10.1007/s11010-014-2045-z
Acknowledgments
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia, grants 173044 and 173023, and grants from the Deutsche Forschungsgemeinschaft (SFB636-TP3) and German Ministry of Education and Research (BMBF, 01GQ1003B) awarded to P.G.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Filipović, D., Todorović, N., Bernardi, R.E. et al. Oxidative and nitrosative stress pathways in the brain of socially isolated adult male rats demonstrating depressive- and anxiety-like symptoms. Brain Struct Funct 222, 1–20 (2017). https://doi.org/10.1007/s00429-016-1218-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-016-1218-9