
Abstract
Circulating tumor DNA (ctDNA) has garnered much excitement over the past few years for its potential clinical utility as a surrogate for tumor biopsies in early cancer detection and prognosis. Numerous studies have demonstrated that ctDNA is shed into the circulation and is elevated in disease states such as cancer. Despite the low levels of ctDNA in the “sea” of normal DNA, advances in next generation sequencing (NGS) and digital polymerase chain reaction (PCR) technologies have led to dramatic improvements in variant detection sensitivity and specificity. These technologies allow the quantification of ctDNA, providing both prognostic and predictive information. Here, we review the history of cell-free DNA and different technologies for the detection of ctDNA in cancer and describe the different modalities for using ctDNA in clinical oncology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Our ability to adequately treat and manage disease hinges on an accurate and proper diagnosis. Historically, tissue biopsies have been the gold standard in oncology, providing histopathological information on whether or not a lesion is malignant. In addition to determining whether a lesion may be malignant, tissue biopsies provide a reservoir of genetic information. In a seminal study, Wood et al. utilized Sanger sequencing to unravel the range and frequencies of genetic alterations that make up breast and colorectal tumors, now known as the “cancer genome landscape” [1]. The hope is that this genetic information can reveal what genes might be altered or disrupted that promote cancer, namely “drivers,” and how these alterations can be targeted.
However, despite their utility, there are challenges to acquiring tissue biopsies, such as surgical biopsies, and how they are processed for subsequent analysis. Surgical biopsies are inherently invasive and, depending on the stage of the disease and the amount of tissue isolated, can be limiting in quantity for genomic analysis. Most tumor tissues are subsequently preserved in formalin-fixed, paraffin-embedded (FFPE) blocks for pathological interpretation and staining. However, this process crosslinks and fragments DNA, jeopardizing their structural integrity and introduces challenges for sequencing and interrogating genomic alterations. Lastly, cancer is an inherently heterogeneous disease, where different areas of the same tumor or different metastases can arise from different subclonal populations, namely intra- and intertumoral heterogeneity, respectively. Sampling a single site provides only a single spatial and temporal snapshot and unlikely to reflect the dynamic tumor heterogeneity in patients [2].
In light of this, development of noninvasive techniques such as liquid biopsies as a surrogate for tissue biopsies has garnered increased interest. Compared to surgical biopsies, blood draws are minimally invasive and provide a source of cell-free DNA (cfDNA) for serial sampling to monitor disease. cfDNA is derived from all cells, including both normal and cancers cells, the latter more commonly referred to as ctDNA. Current methods and considerations for the extraction of cfDNA and ctDNA have been reviewed elsewhere [3]. Since the DNA of cancer cells harbors somatic alterations, that is, variants, amplifications, and rearrangements not present in normal cells, cancer DNA presents a unique “fingerprint” that can be used to differentiate cancer from non-cancerous cells. Indeed, studies have demonstrated that the mutational profile generated from analyzing ctDNA generally reflects the same somatic alterations found in patients’ cancers, and can sometimes capture mutations not present in the initial biopsy [4, 5]. Here, we explore the origins of ctDNA, how ctDNA is becoming a surrogate for tissue biopsies, and the various applications for ctDNA in cancer diagnosis, detection, and disease monitoring.
Origin of cfDNA
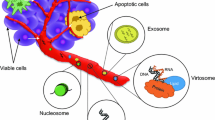
Although circulating DNA is currently a major and relatively new focus of maternal fetal medicine and cancer research, in 1948, Mandel and Metais were the first investigators to publish their findings on cfDNA, referring to free floating DNA that circulates in human blood [6]. At the time, the clinical implications of these findings were largely unappreciated and consequently, the potential utility of these findings unrealized. Moreover, the mechanism of how cfDNA enters the circulation was a mystery. In fact, it is still unclear precisely how cfDNA enters the circulation and whether these circulating nucleic acids have any biological function. Despite knowledge regarding the existence of cfDNA, it would not be until several decades later that Leon et al. reported cancer patients having higher levels of total circulating DNA compared to healthy controls [7]. Specifically, Leon et al. observed greater levels of circulating DNA in the plasma of patients who had advanced cancer compared to individuals with localized disease. This phenomenon was further corroborated in subsequent studies [8,9,10,11]. Unfortunately, due to the lack of specificity in these assays and the wide degree of variability between patients, clinical validity and utility were never proven for the use of total circulating DNA as a prognostic or predictive biomarker for clinical oncology.
While the exact mechanisms by which cfDNA is released into the blood remains unknown, current evidence from several studies suggests that these DNA fragments are derived from necrotic or apoptotic cells that have been engulfed by macrophages [12, 13]. Moreover, compared to healthy controls, the average fragment length of ctDNA found in lung cancer patients for the BRAF V600E mutant allele is shorter than the fragment length of the wild-type allele (132–145 vs. 165 bp, respectively), suggesting a means to discriminate between these populations of cell-free DNA [14]. Other modes of secretion have been proposed whereby DNA fragments are actively released into the circulation [15,16,17,18]. It is possible and likely that the release of cfDNA into the circulation by cells involves multiple processes depending on the disease (e.g., cancer or inflammation) or physiologic state (e.g., pregnancy), as well as the chronicity of the situation (e.g., early stage versus metastatic cancer or first versus third trimester of pregnancy) [19,20,21]. Lastly, cfDNA, and more specifically ctDNA, can be present in a number of bodily fluids including blood, urine, and saliva, though the majority of studies have focused on the use of plasma as described below. Serum has also been used as source of ctDNA and considerations between plasma and serum for mutation detection have been reviewed elsewhere [22].
Detection of ctDNA
While the large majority of circulating cfDNA originates from normal cells, advances in digital PCR and NGS have allowed for the discrimination of ctDNA from wild-type DNA in blood with high specificity and sensitivity. Digital PCR was first described by Vogelstein and Kinzler and is based on the principle of diluting DNA such that single DNA molecules can be separated into individual compartments [23]. The term “digital” refers to the binary aspect of such a dilution strategy since the idea is that each compartment will have 0 or 1 molecule of DNA. Although the concept of digital PCR was thus put forth, it would not be until several years later that new methods of high-throughput digital PCR would enable its use for cancer research, the first platform termed BEAM for Beads, Emulsions, Amplification, and Magnetics [12]. “BEAMing” as it is now called allowed for a semi-automated method of assaying hundreds of thousands of individual digital PCR compartments, in this case water in oil emulsions. Once separated, DNA molecules are PCR amplified on magnetic beads massively and in parallel, and fluorescent probes specific for the mutation or wild-type DNA are then utilized for detection using a flow cytometer. With this technology in place, the application of digital PCR to oncology could now be tested. A series of studies by the Vogelstein group utilized BEAMing to identify APC variants in early and advanced stage colorectal cancer (CRC) patients [12, 24]. BEAMing was capable of identifying ctDNA with high sensitivity and specificity in patients with metastatic colorectal cancer and tracking response to therapies and progression of disease due to its quantitative nature [24]. This seminal work provided the foundation for future studies evaluating digital PCR in clinical oncology. However, BEAMing, now known as “first generation” digital PCR, was not as sensitive for detecting ctDNA in early-stage colorectal cancer patients, prompting for more sensitive techniques to detect low levels of mutational burden.
Advancements in next generation sequencing technologies have greatly assisted with the detection of low frequency mutations in plasma. In one such study, Forshew et al. used tagged-amplicon deep sequencing (namely TAm-Seq) to examine TP53 mutations in ctDNA from 46 advanced ovarian cancer patients, detecting mutations with allele frequencies as low as 2% [25]. While the aforementioned study focused primarily on the metastatic setting, Newman et al. developed a sequencing technique called cancer personalized profiling by deep sequencing or CAPP-Seq to interrogate the levels ctDNA in patients with non-small cell lung cancer (NSCLC) [26]. Using CAPP-Seq, investigators were able to correctly identify 85% of patients with stage II–IV NSCLC and 96% of patients as cancer free. Additionally, in patients with stage I disease, they were able to achieve a sensitivity of 50% and specificity of 96%, marking the first time NGS-based methods were utilized for ultralow detection of ctDNA in the early-stage setting. Concurrent with the advancements of NGS technologies, advancements in second generation digital PCR platforms, namely droplet digital PCR (ddPCR), have also allowed for the interrogation of ctDNA in the early-stage setting [27, 28].
Markers for screening
In oncology, circulating biomarkers have been increasingly helpful in measuring disease burden, which is traditionally assessed by radiographic imaging. This is especially critical when imaging is unable to determine the presence or absence of tumor. Historically, blood-based protein markers such as cancer antigen (CA) 19-9, CA15-3, CA27.29, prostate-specific antigen (PSA), and carcinoembryonic antigen (CEA) have been utilized to monitor patients during treatment [29]. However, not all cancer subtypes have an analogous protein biomarker and protein biomarkers may be elevated under conditions not associated with tumor progression. Additionally, protein biomarkers can persist for weeks, widening their assessment of disease to a window of weeks to months [30,31,32]. While elevated levels of protein markers found in plasma have been associated with disease burden, their largest shortcomings are their limited specificity and sensitivity, exhibiting significantly lower sensitivities when compared to ctDNA in colorectal and breast cancer patients [24, 33].
In contrast, ctDNA is largely able to overcome the shortcomings associated with protein biomarkers and disease assessment. First, ctDNA is specific for cancer cells and, in many cases, reflects the somatic changes found in an individual’s tumor [28]. Second, the short half-life of ctDNA of approximately 2 h in vivo allows precise monitoring of changes in tumor burden or disease progression [24, 34, 35]. Notably, Diehl et al. demonstrated that ctDNA correlates significantly with levels of tumor burden. After surgery, patients who underwent complete resections were observed to have a sharp drop in ctDNA levels, with a 99% median decrease in ctDNA after discharge. Lastly, changes in ctDNA levels can predate changes in imaging or protein markers by up to a few months [24, 36], making it an ideal substrate for monitoring progressive disease.
While changes to levels of detectable ctDNA have largely correlated with tumor burden, studies investigating the prognostic value of cfDNA levels present within patients have provided mixed results. For example, Huang et al. quantified the levels of cfDNA using real-time PCR and hypothesized that the amount of cfDNA present would be able to discriminate between patients with breast cancer and those with benign breast disease [37]. While the plasma DNA concentrations in breast cancer patients was significantly higher compared to patients with benign disease, possibly due to increased turnover of cells, there was no association observed between plasma DNA levels and clinicopathological parameters. However, Garcia et al. found in a prospective study involving 147 breast cancer patients and 35 healthy controls that the presence of plasma tumor DNA at diagnosis, as quantified by loss of heterozygosity (LOH) at six different microsatellite markers and TP53 mutations analyzed over a follow-up period, was consistently linked to shorter overall survival [38]. These findings are refuted by another study comparing circulating tumor cells (CTC) and levels of ctDNA where ctDNA levels provided no prognostic impact on time to progression (TTP) or overall survival but CTC numbers were correlated with overall survival and marginally with TTP [39]. Collectively, the studies highlight that while cancer patients have higher levels of cfDNA compared to healthy controls, the quantification of total cfDNA concentrations alone provide limited diagnostic information.
Concordance between tissue biopsies and ctDNA
While liquid biopsies remain investigational, one of the hurdles regarding utilizing ctDNA has been validating the concordance between mutations found in the plasma with those found in the tumor. Initial studies addressing this concern involved patients with advanced colorectal cancer where the concordance between tumor tissue and ctDNA was 100% [12, 24]. Additionally, our group was one of the first to address concordance between tumor and ctDNA in patients with metastatic breast cancer by analyzing PIK3CA mutations by BEAMing [40]. In a retrospective cohort of 49 tumors and temporally matched samples that were analyzed for PIK3CA mutations, there was 100% concordance between FFPE samples and ctDNA. However, in a prospective cohort of 60 patients, there was 72.5% concordance between BEAMing of PIK3CA mutations in ctDNA and standard sequencing of archival tissue DNA. It was revealed that because the prospective study did not require a contemporaneous tissue and blood sample from each patient, the discordant results were only present in patients’ whose tumor samples were greater than 3 or more years prior to the time of blood draw. This finding would no longer be unexpected considering knowledge of tumor heterogeneity in breast cancer [2]. Similarly, Board et al. assayed for PIK3CA mutations in patients with metastatic breast cancer using a modified allele-specific PCR approach and found a concordance of 95% in 41 cases with matched tumor and plasma samples [41]. However, in 30 localized breast cancers where 14 samples contain a PIK3CA mutation, no PIK3CA mutations were detected in matched plasma. It is quite possible the discrepancy in concordance between these studies is due to difference in the sensitivity between the techniques that were used.
In metastatic cancer patients, ctDNA detection is relatively easier than in early-stage disease. This is likely due to the increased tumor burden in metastatic disease, as well as cancer cell necrosis and apoptosis, leading to a disproportionately higher level of ctDNA in blood. Indeed, one recent study has shown that mutations found in metastatic breast cancer tissues could be detected readily in ctDNA using next generation sequencing (NGS) [4]. We recently published similar results comparing a new metastatic biopsy with blood obtained at the time of study entry in triple-negative metastatic breast cancer patients [42]. These studies and others demonstrate that ctDNA does indeed capture the majority of mutations found in corresponding metastatic tissue biopsies. That said, many groups had already demonstrated that NGS can readily be used for ctDNA detection in metastatic patients using a candidate gene panel or amplicon sequencing approach [43, 44]. Although these studies provided the first proof of principle for using blood as a way to assess a cancer’s mutational profile in a relatively noninvasive, repeatable method, these studies indicated that the current digital PCR and NGS technologies did not yet have reliable sensitivity for detecting cancer at its earliest stages.
Predicting relapse
Though the studies described above collectively began the initial enthusiasm of using ctDNA as a “liquid biopsy,” the clinical utility of ctDNA has not been definitively proven, though ongoing studies are addressing this very issue. Given the relatively low sensitivity of ctDNA detection for early-stage solid tumors in past studies, an unanswered question is whether ctDNA can be used as a genetic biomarker for early-stage disease. Recent advances in digital PCR technologies, namely droplet digital PCR (ddPCR), have increased the throughput and therefore sensitivity of these platforms making this potentially feasible.
In a prospective study conducted by Beaver et al., ddPCR was employed to detect PIK3CA mutations in plasma from patients with early-stage (stage I–III) breast cancer [27]. Primary breast tumors and matched pre- and post-surgery blood samples were collected from 29 patients. DNA was isolated from these tumors and analyzed by both Sanger sequencing and ddPCR for PIK3CA mutations. Sanger sequencing identified a total of 10 PIK3CA mutations which was subsequently verified by ddPCR. However, ddPCR was able to detect an additional five mutations not found by Sanger sequencing, with two mutations present at differing allelic fractions in one tumor. Furthermore, of the 15 mutations that were detected via ddPCR in the tumor samples, 14 were detected in the pre-surgical ctDNA while no mutations were found in PIK3CA wild-type tumors, yielding a sensitivity of 93.3% and specificity of 100%. Interestingly, 10 patients who were positive for PIK3CA mutations in pre-surgery ctDNA by ddPCR had persistent ctDNA post-surgery, with one triple-negative metaplastic breast cancer patients relapsing within 26 months, providing a proof of principle that early-stage detection can predict for relapse. However, the short median follow-up and small sample size of this study prevent definitive conclusions about the prognostic ability of ctDNA in early-stage breast cancer.
More recently, other studies focusing on early-stage breast cancer highlight that serial monitoring of ctDNA may predict for relapse. In a retrospective analysis, Olsson et al. used whole-genome sequencing to identify tumor specific rearrangements and ddPCR to serially monitor 20 patients with primary breast cancer [45]. The presence of tumor-specific rearrangements after surgery was highly accurate for postsurgical discrimination between patients that did or did not recur and ctDNA detection preceded clinical detection in 86% of patients with an average lead time of 11 months. Similarly, Oshiro et al. examined serum ctDNA in early-stage breast cancer and found that patients were stratified into “ctDNA high” versus “ctDNA low” or “ctDNA free” exhibited a shorter recurrence free-survival and overall survival [46]. Most recently, Garcia-Murillas et al. assessed whether analysis of ctDNA in plasma can be used to monitor for minimal residual disease [47]. Using samples collected from prospective studies involving 55 early-stage breast cancer patients receiving neoadjuvant therapy, detection of ctDNA in plasma after completion of curative treatment was associated with metastatic relapse with high accuracy. Their results also found that mutation tracking in serial samples was able to predict for relapse with a median lead time of approximately 8 months before clinical relapse. These studies demonstrate that mutation tracking in early-stage disease may predict for relapse and that subsequent adjuvant therapeutic intervention can be tailored to patients presenting with minimal residual disease.
Monitoring therapeutic response and drug resistance
Beyond the concept of liquid biopsy for evaluating mutational status, assessment of ctDNA to detect response to therapies and drug resistance mutations could be useful for the treatment of metastatic disease. The ability to monitor the emergence of drug resistance affords the possibility of earlier therapeutic intervention and improved clinical outcomes. One of the first examples of targeted therapies directed at specific somatic alterations is the use of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) to treat patients with metastatic non-small cell lung cancer (NSCLC) [48,49,50]. Previously, Taniguchi et al. utilized BEAMing to assay plasma from NSCLC patients to identify potential candidates for EGFR-TKIs [51]. In 32 out of 44 patients, activating mutations were detected in plasma DNA, indicative of clinical benefit with gefitinib. However, of 23 patients who were treated with EGFR-TKIs, they also detected a second site T790M mutation in ctDNA in 10 patients, which has been previously identified to impart gefitinib resistance [52]. Taniguichi et al. demonstrated that ctDNA analysis can be utilized for predicting patients who would respond and determine those who would develop resistance. Subsequently, Oxnard et al. demonstrated that serial monitoring of ctDNA can detect T790M mutations weeks to months before the development of clinical recurrence and patients who could benefit from second and third generation EGFR kinase inhibitors such as rociletinib [53]. In another study, Piotrowska et al. monitored ctDNA from patients with T790M lung cancer mutations undergoing rociletinib treatment. They were able to demonstrate that half of the T790M-positive EGFR-mutant lung cancers treated with rociletinib become T790 wild-type after progression, suggesting that reversion to T790 wild-type is a form of resistance to rociletinib [54]. Lastly, Chabon et al. used a targeted capture panel with the aforementioned CAPP-Seq to study resistance in 43 NSCLC patients with T790M mutations on rociletinib treatment, citing changes in MET copy number as an emerging form of resistance [55]. Together, these studies highlight how ctDNA analysis by digital PCR and NGS is able to undercover novel changes responsible for drug resistance in NSCLC.
In colorectal cancer, molecular profiling of tumor tissues is now commonly performed to assess for clinically relevant genes, such as the presence of KRAS mutations that might predict for lack of response to EGFR-targeted antibody therapy [56]. However, most patients with KRAS wild-type tumors may not respond to EGFR therapies due to oncogenic activation downstream of EGFR proteins [57]. Similar to utilizing ctDNA analysis to monitor the emergence of drug resistance in lung cancer, early studies in metastatic CRC patients utilized BEAMing to identify KRAS mutations in ctDNA that are responsible for resistance to antibody-mediated EGFR-targeted therapy [58, 59]. More recently, Siravegna et al. exploited ctDNA analysis to genotype colorectal tumors and monitor clonal evolution during treatment to EGFR-targeted therapies [60]. They were able to identify somatic alterations in the EGFR pathway in addition to KRAS mutations that were responsible for primary and acquired resistance to EGFR blockade. In addition, they were able to show that upon withdrawal of EGFR-specific antibodies in patients with KRAS mutations, ctDNA levels of KRAS mutations declined, demonstrating the dynamic nature and evolution of CRC cells during drug treatment. Beyond identifying resistance mutations, ctDNA may also be used to monitor responses to therapy. A study investigating ctDNA levels as an early marker of therapeutic response in patients with metastatic colorectal cancer found that decreased ctDNA levels obtained shortly after systemic therapies correlated with computed tomography (CT) responses at 8–10 weeks [61]. This opens the possibility of changing therapies earlier for patients who are predicted not to respond to a new therapy in the metastatic setting, potentially extending the lives of patients with metastatic disease. The ability to distinguish between patients who have responded and those who need further treatment can also help avoid unnecessary treatments in CRC patients who would not benefit, including those with emerging drug resistance mutations.
Recently, multiple studies have shown that liquid biopsies could be used to monitor the emergence of endocrine resistance in breast cancer. Several groups demonstrated that metastatic breast cancer patients who progressed on endocrine therapies developed mutations in the gene encoding estrogen receptor-alpha, ESR1 [62,63,64,65,66]. In a retrospective analysis, our group determined that patients with metastatic breast cancer treated with endocrine therapies were found to contain ESR1 mutations in both their tissue and plasma when blood was collected less than a year after their tissue biopsies [67]. In a prospective cohort where blood and tissue were taken simultaneously, ESR1 mutations were found in the ctDNA of patients who were not positive in their corresponding tissue, highlighting the emergence of resistance clones not present when sampling one tissue biopsy by NGS. Intriguingly, some blood samples contained multiple ESR1 mutations at distinct clonal frequencies, arguing for parallel yet separate clonal populations of resistance. Subsequently, other groups also detected the emergence of ESR1 mutations in patients with metastatic breast cancer [68, 69]. While activating mutations in ESR1 are thought to be associated with metastatic breast cancer, Wang et al. demonstrated that it is possible to detect ESR1 mutations in primary tumors, albeit at low allelic frequencies [70]. Similar to the aforementioned studies, Schiavon et al. found a high concordance between tumor and blood for the detection of ESR1 mutations [71]. However, they found patients with ESR1 mutations have a substantially shorter progression-free survival (PFS) on subsequent aromatase inhibitor-based therapies and that the prevalence of ESR1 mutations is markedly higher if patients were exposed to endocrine therapies in the metastatic setting compared to the adjuvant setting. More recently, ESR1 mutational status was used to predict for resistance or sensitivity to certain combinations of endocrine therapies [72]. Collectively, these studies underscore the opportunities afforded by monitoring breast cancer patients for ESR1 mutations to ascertain which therapies and treatment schedule may be the most effective.
Conclusion
In summary, analysis of ctDNA by digital PCR and NGS technologies holds tremendous promise to noninvasively detect tumor-specific alterations in blood. Due to the high sensitivity of these technologies and short half-life of ctDNA, ctDNA provides a quantitative and qualitative molecular snapshot to monitor tumor burden, response to therapy, track genomic evolution and tumor heterogeneity, identify candidates for targeted therapies, and detect the emergence of drug resistance. While the majority of the work regarding ctDNA has been done in patients with advanced cancer where the levels of ctDNA are relatively high, advancements in these technologies have allowed for detection and monitoring of early-stage disease. The hope is that early detection of cancer can afford opportunities for treatment when cancer is most amenable to cure, while at the same time, sparing these early-stage patients from overtreatment, i.e. measuring the absence of minimal residual disease may define cure and preclude the need for adjuvant therapies. While digital PCR and NGS technologies have opened doors for early detection of cancer mutations, there are still questions that remain to be answered. To date, most studies have been conducted retrospectively with limited patient numbers, or retrospectively analyzed samples from pooled prospective clinical studies. Larger, prospective studies dedicated to directly answering the clinical validity/utility of ctDNA are needed before its use can be incorporated into routine clinical practice. Moreover, it is still unknown how the detection of somatic changes in ctDNA will influence treatment and whether or not this will affect progression-free and overall survival. Until further research is able to prove otherwise, ctDNA is unlikely to replace tissue biopsies and information from both types are likely to complement each other. It is not definitively clear whether all tumor types shed ctDNA in detectable amounts and consequently, liquid biopsies would miss these mutations. Despite the utility of liquid biopsies, to date, detection of ctDNA fails to pinpoint the exact origin of the tumor or subclonal population. Clearly, further work is needed to resolve these issues. However, the ability to detect ctDNA and its promise for carrying clinical oncology into an era of truly personalized medicine is apparent. With further research and validation, detection of ctDNA will bring about a paradigm shift on how we manage and treat all cancer patients.
References
Wood LD, Parsons DW, Jones S et al (2007) The genomic landscapes of human breast and colorectal cancers. Science 318:1108–1113
Gerlinger M, Rowan AJ, Horswell S et al (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366:883–892
Benesova L, Belsanova B, Suchanek S et al (2013) Mutation-based detection and monitoring of cell-free tumor DNA in peripheral blood of cancer patients. Anal Biochem 433:227–234
Rothe F, Laes JF, Lambrechts D et al (2014) Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol 25:1959–1965
Lebofsky R, Decraene C, Bernard V et al (2015) Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol 9:783–790
Mandel P, Metais P (1948) Nucleic acids of human blood plasma. CR Seances Soc Biol Paris 142:241–243
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ (1977) Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37:646–650
Shapiro B, Chakrabarty M, Cohn EM, Leon SA (1983) Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 51:2116–2120
Stroun M, Anker P, Lyautey J, Lederrey C, Maurice PA (1987) Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol 23:707–712
Maebo A (1990) Plasma DNA level as a tumor marker in primary lung cancer. Nihon Kyobu Shikkan Gakkai Zasshi 28:1085–1091
Fournie GJ, Courtin JP, Laval F et al (1995) Plasma DNA as a marker of cancerous cell death. Investigations in patients suffering from lung cancer and in nude mice bearing human tumours. Cancer Lett 91:221–227
Diehl F, Li M, Dressman D et al (2005) Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 102:16368–16373
Choi JJ, Reich CF 3rd, Pisetsky DS (2005) The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology 115:55–62
Underhill HR, Kitzman JO, Hellwig S et al (2016) Fragment length of circulating tumor DNA. PLoS Genet 12:e1006162
Stroun M, Anker P (1972) Nucleic acids spontaneously released by living frog auricles. Biochem J 128:100P–101P
Anker P, Stroun M, Maurice PA (1975) Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res 35:2375–2382
Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P (2001) About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 313:139–142
Rogers JC, Boldt D, Kornfeld S, Skinner A, Valeri CR (1972) Excretion of deoxyribonucleic acid by lymphocytes stimulated with phytohemagglutinin or antigen. Proc Natl Acad Sci U S A 69:1685–1689
Ke WL, Zhao WH, Wang XY (2015) Detection of fetal cell-free DNA in maternal plasma for Down syndrome, Edward syndrome and Patau syndrome of high risk fetus. Int J Clin Exp Med 8:9525–9530
Benachi A, Letourneau A, Kleinfinger P et al (2015) Cell-free DNA analysis in maternal plasma in cases of fetal abnormalities detected on ultrasound examination. Obstet Gynecol 125:1330–1337
Wagner AJ, Mitchell ME, Tomita-Mitchell A (2014) Use of cell-free fetal DNA in maternal plasma for noninvasive prenatal screening. Clin Perinatol 41:957–966
El Messaoudi S, Rolet F, Mouliere F, Thierry AR (2013) Circulating cell free DNA: preanalytical considerations. Clin Chim Acta 424:222–230
Vogelstein B, Kinzler KW (1999) Digital PCR. Proc Natl Acad Sci U S A 96:9236–9241
Diehl F, Schmidt K, Choti MA et al (2008) Circulating mutant DNA to assess tumor dynamics. Nat Med 14:985–990
Forshew T, Murtaza M, Parkinson C et al (2012) Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4:136ra68
Newman AM, Bratman SV, To J et al (2014) An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 20:548–554
Beaver JA, Jelovac D, Balukrishna S et al (2014) Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res 20:2643–2650
Bettegowda C, Sausen M, Leary RJ et al (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6:224ra24
Hayes DF, Zurawski VR Jr, Kufe DW (1986) Comparison of circulating CA15-3 and carcinoembryonic antigen levels in patients with breast cancer. J Clin Oncol 4:1542–1550
Yoshimasu T, Maebeya S, Suzuma T et al (1999) Disappearance curves for tumor markers after resection of intrathoracic malignancies. Int J Biol Markers 14:99–105
Ito K, Hibi K, Ando H et al (2002) Usefulness of analytical CEA doubling time and half-life time for overlooked synchronous metastases in colorectal carcinoma. Jpn J Clin Oncol 32:54–58
Riedinger JM, Wafflart J, Ricolleau G et al (2006) CA 125 half-life and CA 125 nadir during induction chemotherapy are independent predictors of epithelial ovarian cancer outcome: results of a French multicentric study. Ann Oncol 17:1234–1238
Lehner J, Stotzer OJ, Fersching D, Nagel D, Holdenrieder S (2013) Circulating plasma DNA and DNA integrity in breast cancer patients undergoing neoadjuvant chemotherapy. Clin Chim Acta 425:206–211
Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM (1999) Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 64:218–224
Fleischhacker M, Schmidt B (2007, 1775) Circulating nucleic acids (CNAs) and cancer—a survey. Biochim Biophys Acta:181–232
Dawson SJ, Tsui DW, Murtaza M et al (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368:1199–1209
Huang ZH, Li LH, Hua D (2006) Quantitative analysis of plasma circulating DNA at diagnosis and during follow-up of breast cancer patients. Cancer Lett 243:64–70
Garcia JM, Garcia V, Silva J et al (2006) Extracellular tumor DNA in plasma and overall survival in breast cancer patients. Genes Chromosomes Cancer 45:692–701
Madic J, Kiialainen A, Bidard FC et al (2015) Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int J Cancer 136:2158–2165
Higgins MJ, Jelovac D, Barnathan E et al (2012) Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res 18:3462–3469
Board RE, Wardley AM, Dixon JM et al (2010) Detection of PIK3CA mutations in circulating free DNA in patients with breast cancer. Breast Cancer Res Treat 120:461–467
Parsons HA, Beaver JA, Cimino-Mathews A, et al 2017 Individualized Molecular Analyses Guide Efforts (IMAGE): a prospective study of molecular profiling of tissue and blood in metastatic triple negative breast cancer. Clin Cancer Res 23(2):379–386
Murtaza M, Dawson SJ, Tsui DW et al (2013) Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497:108–112
Leary RJ, Kinde I, Diehl F et al (2010) Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2:20ra14
Olsson E, Winter C, George A et al (2015) Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med 7:1034–1047
Oshiro C, Kagara N, Naoi Y et al (2015) PIK3CA mutations in serum DNA are predictive of recurrence in primary breast cancer patients. Breast Cancer Res Treat 150:299–307
Garcia-Murillas I, Schiavon G, Weigelt B et al (2015) Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 7:302ra133
Lynch TJ, Bell DW, Sordella R et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139
Paez JG, Janne PA, Lee JC et al (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500
Rosell R, Moran T, Queralt C et al (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361:958–967
Taniguchi K, Uchida J, Nishino K et al (2011) Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res 17:7808–7815
Pao W, Miller VA, Politi KA et al (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2:e73
Oxnard GR, Paweletz CP, Kuang Y et al (2014) Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res 20:1698–1705
Piotrowska Z, Niederst MJ, Karlovich CA et al (2015) Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov 5:713–722
Chabon JJ, Simmons AD, Lovejoy AF et al (2016) Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 7:11815
Wong NA, Gonzalez D, Salto-Tellez M et al (2014) RAS testing of colorectal carcinoma—a guidance document from the Association of Clinical Pathologists Molecular Pathology and Diagnostics Group. J Clin Pathol 67:751–757
De Roock W, Claes B, Bernasconi D et al (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11:753–762
Diaz LA Jr, Williams RT, Wu J et al (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486:537–540
Misale S, Yaeger R, Hobor S et al (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486:532–536
Siravegna G, Mussolin B, Buscarino M et al (2015) Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 21:827
Tie J, Kinde I, Wang Y et al (2015) Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol 26:1715–1722
Li S, Shen D, Shao J et al (2013) Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 4:1116–1130
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A et al (2013) D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 73:6856–6864
Jeselsohn R, Yelensky R, Buchwalter G et al (2014) Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 20:1757–1767
Toy W, Shen Y, Won H et al (2013) ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45:1439–1445
Robinson DR, Wu YM, Vats P et al (2013) Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45:1446–1451
Chu D, Paoletti C, Gersch C et al (2016) ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin Cancer Res 22:993–999
Sefrioui D, Perdrix A, Sarafan-Vasseur N et al (2015) Short report: monitoring ESR1 mutations by circulating tumor DNA in aromatase inhibitor resistant metastatic breast cancer. Int J Cancer 137:2513–2519
Guttery DS, Page K, Hills A et al (2015) Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin Chem 61:974–982
Wang P, Bahreini A, Gyanchandani R et al (2016) Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res 22:1130–1137
Schiavon G, Hrebien S, Garcia-Murillas I et al (2015) Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 7:313ra182
Fribbens C, O’Leary B, Kilburn L et al (2016) Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 34:2961–2968
Acknowledgments
This work was supported by the Avon Foundation and the Breast Cancer Research Foundation. We would also like to thank and acknowledge the support of NIH P30 CA006973, the Sandy Garcia Charitable Foundation, the Commonwealth Foundation, the Mike and Dianne Canney Foundation, the Marcie Ellen Foundation, and the Helen Golde Trust. None of the funding sources influenced the design, interpretation, or submission of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
B.H.P. is a member of the scientific advisory boards of Horizon Discovery, LTD, and Loxo Oncology, has ownership interest in Loxo Oncology, and has research contracts with Genomic Health, Inc. and Foundation Medicine, Inc. Under separate licensing agreements between Horizon Discovery, LTD, and The Johns Hopkins University, B.H.P. is entitled to a share of royalties received by the University on sales of products. The terms of this arrangement are being managed by the Johns Hopkins University, in accordance with its conflict of interest policies. D.C declares no potential conflicts.
Rights and permissions
About this article

Cite this article
Chu, D., Park, B.H. Liquid biopsy: unlocking the potentials of cell-free DNA. Virchows Arch 471, 147–154 (2017). https://doi.org/10.1007/s00428-017-2137-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-017-2137-8