
Abstract
Acetaldehyde and acetic acid/acetate, the active metabolites of alcohol (ethanol, EtOH), generate actions of their own ranging from behavioral, physiological, to pathological/cancerogenic effects. EtOH and acetaldehyde have been studied to some depth, whereas the effects of acetic acid have been less well explored. In this study, we investigated the effect of acetic acid on big conductance calcium-activated potassium (BK) channels present in GH3 rat pituitary tumor cells in more detail. In whole cell voltage clamp recordings, extracellular application of acetic acid increased total outward currents in a dose-dependent manner. This effect was prevented after the application of the specific BK channel blocker paxilline. Acetic acid action was pH-dependent—in whole cell current and single BK channel recordings, open probability (Po) was significantly increased by extracellular pH reduction and decreased by neutral or base pH. Acetic acid hyperpolarized the membrane potential, whereas acidic physiological solution had a depolarizing effect. Moreover, acetic acid reduced calcium (Ca2+) oscillations and exocytosis of growth hormone contained secretory granules from GH3 cells. These effects were partially prevented by BK inhibitors—tetraethylammonium or paxillin. In conclusion, our experiments indicate that acetic acid activates BK channels in GH3 cells which eventually contribute to acetic acid-induced membrane hyperpolarization, cessation of Ca2+ oscillations, and decrease of growth hormone release.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabolization of alcohol (ethanol, EtOH) involves in a first step enzymes such as alcohol dehydrogenase, cytochrome P450 (CYP450 2E1), or catalase to produce acetaldehyde. In a second step, acetaldehyde is quickly converted via aldehyde dehydrogenase to more harmless acetic acid (AA) which in a third step is finally oxidized to carbon dioxide and water [18]. EtOH and its metabolites all appear to have effects of their own ranging from well-known delirious to addictive actions, toxic, neurodegenerative, or cancerogenic properties [11, 20, 23, 33, 40]. In the brain, acetate is primarily produced by metabolizing sugar, acetylcholine, and ethanol [51].
EtOH and acetaldehyde have been studied to some depth on the behavioral, addictive, and cellular level, whereas the effects of AA have been less well explored [9, 37]. Compared with ethanol and acetaldehyde which in low doses facilitated locomotor activity in rodents, acetate produced a decrease in motor activity [11, 37]. Although precise mechanisms of the depressant action of acetate in the brain are not known, adenosine was increased after acetate injection [23; 37]. However, inhibition of spontaneous firing of rat cerebral cortical neurons by ionophoretic injection of acetate was not prevented by an adenosine antagonist [38].
Large conductance calcium (Ca2+)-activated potassium (K+) channels, referred as BK or Maxi K+ channels, are prominent targets of EtOH [4, 13, 15, 20, 22, 24, 28, 47]. BK channels are found in a great variety of excitable and non-excitable cells in a huge variety of organisms. The channels are located in cellular plasma membranes but also in membranes of intracellular organelles such as mitochondria, nucleus, endoplasmic reticulum, or the Golgi apparatus. The channels exhibit a particularly high conductivity and under physiological conditions are usually synergistically activated by both—intracellular Ca2+ and by membrane voltage [10, 27, 29]. BK channels play an important role in regulation of electrical excitability of cells such as generation of action potentials, transmitter/hormone secretion, or negative feedback regulation of Ca2+ channels [3, 21, 29].
AA and acetate also appear to be modulators of BK channels [17]. In guinea pig detrusor smooth muscle cells and of recombinant human CHO cells, AA acts as a powerful opener of BK channels [18]. The study was carried out on whole cell preparations which give way to the possibility that AA may not directly act on BK channels but via some cellular signaling mode. We became interested in studying AA since it is one of the three metabolites of EtOH and we have investigated EtOH as well as the combined action of EtOH and acetaldehyde on BK channels previously [19, 24]. It therefore appeared consequent to investigate the effects of AA on BK channels in more detail. We studied AA effects on the single channel level, membrane potential, Ca2+ oscillations, and secretory granule exocytosis reflecting growth hormone release in rat pituitary cells.
Materials and methods
Cell culture
GH3 pituitary tumor cells (German Collection of Microorganisms and Cell Cultures) were cultured in MEM (minimal essential medium, Sigma-Aldrich) supplemented with 7% fetal calf serum and 3% horse serum at 37 °C, 95% humidity, 5% CO2 and fed two times a week. For experiments, cells were grown on poly-D-lysine-coated (0.01% poly-D-lysine for 15 min) coverslips and used for electrophysiological recordings 3 to 4 days after seeding. Culture media were from Sigma (Vienna, Austria) and sera from Invitrogen (Vienna, Austria); all other chemicals were from Sigma.
Solutions
The standard experimental bath solution contained in mM: 145 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, pH 7.2. The pipette solution contained in mM: 145 KCl, 1 MgCl2, and 10 HEPES, pH 7.2 and 5 EGTA, 3.63 CaCl2 (which results in 0.5 μM free Ca2+ as calculated with the Webmaxc extended calculator (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/webmaxc/webmaxcE.htm)). All experiments were carried out at room temperature between 20 and 22 °C. Osmolarities of solutions were controlled with a Micro-Osmometer (Type OM 806, Löser, Germany) and adjusted to 310 mOsm. Within an experimental setting, the difference in osmolarities did not exceed 5 mOsm.
Acetic acid (AA) (CH3COOH) and sodium acetate (CH3COONa) in solution give acetate (CH3COO−) and H+ or Na+, respectively. AA was used in concentrations of 0.005% (0.835 mM), 0.01% (1.67 mM), and 0.02% (3.34 mM), which decreased the pH of solutions to 6.97, 6.78, and 5.7, respectively. Sodium acetate (0.02%) did not change the pH of the extracellular solution. To reveal the impact of low pH on BK channels, we used AA solution (0.02%) with the pH corrected by NaOH to 7.2 (AA corrected) and bath solution with pH = 5.7 achieved by addition of HCl. We also used 0.02% AA with pH = 4.5 and pH = 8 to analyze its effects on single BK channels activity. Tetraethylammonium (TEA, 1 mM) or paxilline (1 μM) were used as inhibitors of BK channels.
Electrophysiology and data analysis
Electrophysiological experiments were performed as described previously [19, 42, 43]. In brief, patch pipettes were fabricated from borosilicate glass (Science Products GmbH, Germany) with resistances of 3–5 MΩ in single channel experiments. In whole cell experiments, the electrode resistances were 2–4 MΩ with an access resistance not exceeding 10 MΩ. An agar bridge was used as reference electrode to prevent any offset from changing solutions. Recordings in the whole cell mode or from excised outside-out patches were made with an Axopatch-200B amplifier connected to a Digidata 1440A interface, using pClamp 10.7 software. Sampling frequency for single channel recordings was 10 kHz. Data were filtered with a low-pass 4-pole Bessel filter set at 1 kHz, which results in a 10–90% rise time of 350 μs and analyzed with Clampfit software (Axon Instruments/Molecular Devices, USA). Channel open probability was expressed as Popen = NPo/n where NPo = [(to)/(to + tc)], Po = open probability for one channel, to = sum of open times, tc = sum of closed times, N = actual number of channels in the patch, and n = maximum number of individual channels observed in the patch. All equations used were standard equations from Clampfit.
Experimental solutions were applied via a gravity-driven, electronically switched perfusion system (ALA Scientific Instruments, USA). For rapid solution exchange (about 300 ms), membrane patches were held in a stream of the experimental solution from a second pipette.
Fluorescence experiments and analysis
FM1-43 is an amphiphilic dye that partitions from water into membranes where it is trapped [5]. The dye is more strongly fluorescent in membranes. Therefore, when added to the medium, an increase of fluorescence indicates exocytosis of secretory granules [7, 26, 32]. Cells were incubated with 4 μM FM1-43 (Synaptogreen C4, Sigma-Aldrich, USA) for 5 min to measure basal exocytosis and for 5 min during KCl (100 mM) application to evoke massive exocytosis of secretory granules. In high K+ containing solution, NaCl was iso-osmotically substituted by KCl. AA containing solution with uncorrected or corrected to pH 7.2 was applied for 3 min before cells were stained with FM1-43.
For analysis of Ca2+ oscillations, the cell membrane permeable calcium indicator Fluo-4 AM (Sigma-Aldrich, USA) was used. Cells were stained with 1 μm Fluo-4 AM for 40 min at 37 °C. Cells were then placed for 10 min in bath solution to ensure that de-esterification of AM esters was completed. Ca2+ oscillations were recorded every 30 s for 3 min in control and for 5 min during application of AA. At the end of each experiment, 100 mM KCl containing solution was applied for measurement of maximum fluorescence.
Fluorescence imaging of stained cells was carried out by using an AxioScope A1 microscope (Carl Zeiss, Germany) equipped with a water immersion objective PlanNeofluar 63×/0.9NA (Carl Zeiss, Germany), an excitation filter (BP 450–490 nm), a beam splitter (FT510), and an emission filter (LP 555 nm). The fluorescence staining was recorded with a high-speed camera AxioCam MRm (Carl Zeiss, Germany). The image acquisition time was 500 ms per frame. The fluorescence intensity was analyzed with ImageJ software (NIH, USA). The peak fluorescence of FM 1-43 staining was expressed as relative change in fluorescence (F-F0)/F0 for each frame of each measurement, where F is the peak fluorescence of stained cell membrane before (basal) or after stimulation of exocytosis by application of 100 mM K+ containing solution and F0 is the background fluorescence.
The mean amplitude, frequency, and duration of Ca2+ oscillations were detected by the built-in function “findpeaks” in the package MATLAB (MathWorks, USA). The fluorescence intensity of calcium signaling was expressed as relative change in fluorescence (F-F0)/F0 for each frame of each measurement, where F is the peak fluorescence of stained cells and F0 is the background fluorescence close to a given cell. Quantitative analysis of fluorescence images was performed using OriginPro 8.5 software (OriginLab, USA).
Statistics
Normality of the sample data was evaluated with the Shapiro-Wilk test and for equal variances using F-test Origin Pro software (OriginLab Corp, USA). Statistical significance between means was calculated using non-parametric Wilcoxon signed rank test for independent samples, Wilcoxon signed rank test for paired samples, and Kruskal-Wallis ANOVA test for group samples in Origin Pro 2018 (OriginLab Corp, USA). Experiments were repeated at least five times; the mean and the SEM (standard error of the mean) were calculated. Differences were considered as statistically significant at p < 0.05.
Results
Acetic acid increases outward K+ currents by activation of BK channels
Whole cell ion currents were elicited in response to a series of voltage pulses from − 80 mV holding potential to + 140 mV in steps of 20 mV. Application of acetic acid (AA, 0.02%, uncorrected with pH = 5.7) significantly increased the amplitude of outward currents at voltages from + 20 mV up to + 140 mV (n = 7; Fig. 1a, b). The effect of AA was dose-dependent increasing currents for all concentrations used (0.005%, 0.01%, 0.02%, V = + 20/+ 40 mV; Fig. 1c). To examine the specificity of AA on BK channels, we used paxilline, a specific inhibitor of BK channels at concentration of 1 μM. Application of paxilline inhibited outward currents and subsequent administration of AA did not show any further effect (n = 6; Fig. 1b).

Effects of acetic acid on outward K+ currents of pituitary GH3 cells. a Original traces of outward currents in control and after application of acetic acid (AA) 0.02% uncorrected. b Current-voltage (I-V) relationship of K+ outward current densities (picoAmpere (pA) vs. picoFarad (pF)—pA/pF). The plot was taken from whole cell recordings of maximum total membrane currents in response to a series of depolarizing voltage pulses from a holding potential of − 80 mV to + 140 mV in 20 mV steps in control (control, open circles), after application of AA 0.02% with uncorrected pH (AA uncorr, pH = 5.7, n = 7, filled circles), after application of the specific BK channel blocker paxilline (pax, 1 μM, open triangles) and after acetic acid application in the presence of paxilline (pax + AA 0.02% uncorr., n = 6, filled triangles). c Dose dependence of the current amplitude at + 20 mV and + 40 mV in response to various concentrations of acetic acid (0.005%, n = 10; 0.01%, n = 5; 0.02%, n = 10) compared with control (100%, stippled line). *p < 0.05 compared with control
The effect of AA was pH-dependent. The pH was adjusted by adding either NaOH or HCl to the bath solutions. In whole cell recordings at + 40 mV steps, current amplitudes in AA (0.02%, uncorrected, pH 5.7) increased by 273%, at corrected pH 7.2 by 136% (n = 9), and with sodium acetate (0.02%, pH 7.2), by 64%, compared with 100% in control, stippled line in Fig. 2d (Fig. 2a–d). Sodium acetate (0.02%) activated outward currents similar to AA but to a lesser extend (n = 5; Fig. 2b, c). On the other hand, an acidic bath solution of pH 5.7 (no AA added) significantly decreased outward currents by 33% compared with control (100%, n = 9; Fig. 2c, d).

Effects of acetic acid, sodium acetate, and low pH bath solution on outward K+ currents. a I-V plot of K+ currents in response to a series of depolarizing voltage pulses from a holding potential of − 80 mV to + 140 mV in 20 mV steps before (control, open circles) and after application of acetic acid 0.02% with corrected pH = 7.2 (AA corr, n = 10). b I-V plot of K+ currents before (control, open circles) and after application of sodium acetate (SA 0.02%, n = 11). c I-V plot of K+ currents in bath solution (control, open circles, pH = 7.2) and in low pH solution (pH = 5.7, n = 9). d Average current amplitudes (%) at voltage step + 40 mV in response to application of 0.02% acetic acid uncorrected (AA uncorr), 0.02% acetic acid with corrected pH = 7.2 (AA corr), sodium acetate 0.02% (SA), and low pH 5.7 bath solution; control (100%) is indicated as horizontal stippled line. *p < 0.05 compared with control, #p < 0.05 compared with AA uncorrected
Acetic acid increases single BK channel activity in a pH-dependent manner
We further investigated the effects AA on the activity of single BK channels in outside-out patches at a holding potential of + 30 mV. The open probability (Po) of BK channels did not significantly change after application of low AA concentration (0.005%, Po 9% compared with control 100%, n = 4). At a concentration of 0.01% AA, BK channel Po increased by 43% (from 0.044 ± 0.015 to 0.061 ± 0.019, n = 6, p < 0.05) after 1 min of application. Po further increased during 2 min by 51% of control (0.064 ± 0.020, p < 0.05), and after 3 min, Po was 34% of control (0.058 ± 0.019). AA at a concentration of 0.02% (pH 5.7, uncorrected) significantly increased Po after 1 min of application by 68% (Fig. 3a, b), after 2 min by 60% and after 3 min of application by 55% (n = 14, p < 0.05) (Tables 1 and 2).

Effects of acetic acid on BK single channel open probability (Po). a Original traces of channel activity during control and after application of acetic acid 0.02% uncorrected pH (AA uncorr). Recordings were taken at + 30 mV and 0.5 μM free Ca2+ in the pipette solution. b Po of BK channels during 3-min application of acetic acid 0.02% (AA uncorr, horizontal bar). c Percentage (%) of BK single channel open probability (Po) during application of 0.02% acetic acid uncorrected (AA uncorr., white column, n = 6); AA 0.02% with corrected pH = 7.2 (AA corr., dashed column, n = 5,); sodium acetate (SA) 0.02% (gray column, n = 5), and bath solution with low pH 5.7 (gray dashed column, n = 6) compared with control (stipples line 100%). d Effect of 0.02% acetic acid at different pH bath solutions: pH 5.7 (uncorrected), pH = 8.0, and pH = 4.5 (stipples line 100%), *p < 0.05 compared with control
Similar effects were observed by application of AA with pH corrected to 7.2 (36% increase, n = 5, p < 0.05) and 0.02% sodium acetate (25% increase, n = 5, p < 0.05) which increased Po only during the first minute of application and these effects were significantly lower compared with AA 0.02%, (pH 5.7, uncorrected) (Fig. 3c; Table 2). In all experimental approaches, the amplitude and the mean channel open time (dwell time) of currents did not change (Table 1).
To investigate the pH effect on AA activated currents in more detail, we compared the effects of AA in a variety of pH solutions. Surprisingly, at pH = 4.5, a marked increase of BK channel Po was observed which was significantly higher compared with the effect of AA 0.02% at pH 5.7 (uncorrected; Fig. 3d). Po increased by 299 % (n = 8, p < 0.05) after 1 min, by 270% after 2 min, and by 228% after 3 min of application (Table 2). At pH = 8, 0.02% AA, Po only slightly increased during the first minute by 9% (n = 6, p > 0.05), with a further decrease by 38% and 50% of control during 2 and 3 min (Fig. 3d; Table 1). Lowering the pH of AA free solution to 5.7 decreased Po by 33% compared with 100% control at pH 7.2 (n = 6, p < 0.05) (Fig. 3c; Table 1). Channel amplitudes and dwell times were not significantly altered (Table 1).
Acetic acid hyperpolarizes GH3 cells
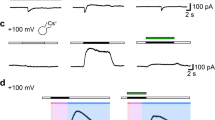
Application of AA (0.02%, uncorrected, pH = 5.7) induced hyperpolarization of the membrane potential from − 42.83 ± 1.19 to − 71.34 ± 1.84 mV (n = 16, p < 0.05) with cessation of spontaneous action potentials (Fig. 4a, top, b). AA (0.02%) with corrected pH to 7.2 or sodium acetate had basically similar, but smaller effects. AA with pH corrected to 7.2 induced hyperpolarization from − 41.38 ± 0.87 mV to − 50.76 ± 1.46 mV (n = 13, p < 0.05) and sodium acetate (0.02%) from − 41.2 ± 0.57 mV to − 51.87 ± 2.5 mV (n = 4, p < 0.05) (Fig. 4b). Low pH solution (pH 5.7, no AA added) depolarized the membrane potential from − 42.25 ± 2.42 mV to − 23.47 ± 2.01 mV (n = 4, p < 0.05) (Fig. 4b). The effects of all substances were completely washable. Inhibition of BK channels by TEA (1 mM) or paxilline (1 μM) induced slight depolarization of membrane: − 44.26 ± 1.13 mV to − 42.76 ± 1.41 in case of TEA (n = 25, p < 0.05) and from − 43.39 ± 1.06 mV to − 40.68 ± 1.05 mV in case of paxilline (n = 20, p < 0.05). Subsequent application of AA uncorrected induced an initial hyperpolarization during the first 30 s of the application to − 63.45 ± 2.84 mV in the presence of TEA (n = 15) (Fig. 4a, bottom) and to − 59.97 ± 4.36 mV in case of the BK channel inhibitor paxilline (n = 6). After 1 min, the membrane potential recovered to − 54.55 ± 3.59 mV in case of TEA (Fig. 4a, bottom) and to − 49.58 ± 3.15 mV (n = 6) in case of paxilline which was significantly lower than the effect of AA (0.02%, uncorrected) alone (p < 0.05). In case of AA with corrected pH = 7.2, the changes of membrane potential after inhibition of BK channels were not significant (− 2.37 ± 1.72 mV in case of TEA, n = 6 and − 2.51 ± 1.23 mV in case of paxilline, n = 7, p > 0.05) (Fig. 4b).

Effects of acetic acid (0.02%) on membrane potential and exocytosis of secretory granules. a Original trace of membrane potential recordings after application of AA with uncorrected (AA uncorr) in control (a, top) and after preliminary incubation with the BK channels inhibitor tetraethylammonium (TEA,1 mM) (a, bottom). b Membrane potential changes (ΔV) after application of AA uncorr (AA uncorr, n = 16) in control and in the presence of TEA (1 mM, n = 15); AA corrected pH = 7.2 (AA corr, n = 13) in control and AA corr + TEA (n = 6); sodium acetate (SA, n = 4) and low pH 5.7 bath solution (n = 4). *p < 0.05 compared with control; # p < 0.05 compared with AA uncorr and & p < 0.05 compared to AA corr. c Membrane fluorescence (ΔF/F0) due to basal and KCl (100 mM) evoked exocytosis in control, and after preliminary incubation of cells with AA uncorr and AA corr. Inserts show fluorescence images of GH3 cells incubated in FM 1-43 (4 μM) for 5 min before (basal) and after stimulation with high KCl containing bath solution in control (1) and after incubation in AA uncorr (2), and AA corr (3). Scale bar 5 μm. c Membrane fluorescence due to basal and KCl evoked exocytosis after incubation in AA uncorr, TEA + AA uncorr, AA corr, and TEA + AA corr relatively to control conditions expressed as 100%. *p < 0.05 compared with control values
Acetic acid decreases exocytosis of secretory granules and Ca2+-oscillations
For analysis of the effect of AA on exocytosis of secretory granules, the fluorescent dye FM 1-43 was used, which labels membranes undergoing exocytosis [16, 32]. In pituitary somatotrophs, diffuse plasma membrane fluorescence in the presence of FM dye is proportional to the amount of vesicular membrane added during exocytosis [7, 26]. AA was added 3 min before dye application to allow for a maximum AA effect. In control conditions, the basal membrane fluorescence was 0.38 ± 0.01 ΔF/F0 (n = 12, 63 cells) reflecting spontaneous exocytosis (Fig. 4c). Incubation of cells in a solution containing 0.02%, AA with uncorrected pH reduced the intensity of basal fluorescence to 0.27 ± 0.01 ΔF/F0 (n = 10, 78 cells; p < 0.05) In the presence of 0.02% AA with corrected pH to 7.2, the fluorescence was reduced to 0.32 ± 0.01 ΔF/F0 (n = 9, 63 cells; p < 0.05) (Fig. 4c). Depolarization of cells by extracellular high KCl containing solution (100 mM) to induce Ca2+ influx into cells increased membrane fluorescence up to 0.59 ± 0.03 ΔF/F0 in control (n = 11, 73 cells) (Fig. 4b). KCl-evoked fluorescence was significantly lower in cells incubated in 0.02% AA uncorrected (0.47 ± 0.03 ΔF/F0, n = 8, 35 cells, p < 0.05, p < 0.05) and in cells treated with AA corrected pH to 7.2 (00.51 ± 0.03 ΔF/F0, n = 9, 40 cells, p < 0.05; Fig. 4c). Preliminary incubation of cells in TEA (1 mM) prevented the decrease of membrane fluorescence, induced by AA with uncorrected and corrected pH (Fig. 4d).
For recording of [Ca2+]i oscillations, GH3 cells were loaded with Fluo-4 AM. Application of 0.02% AA (uncorrected) induced a rapid decrease of the oscillations (Fig. 5a, b). The amplitude of oscillations was almost 25% less than under control conditions at 1 min (from 2.06 ± 0.19 to 0.58 ± 0.15 relative units (r.u.), n = 34, p < 0.05) and further decreased to 15% of control by 3–5 min (Fig. 5a, b). The frequency of oscillations significantly decreased from 4.21 ± 0.50 to 1.30 ± 0.27 pulses per 30 s within the first minute (p < 0.05) and after 2 min in 75% of cells Ca2+ oscillations had completely vanished. AA with pH corrected to 7.2 showed similar effects, but they were less pronounced (Fig. 5a, b). In this case, Ca2+ oscillations did not come to a halt after 5 min of AA application, whereas the amplitude of oscillations decreased from 1.85 ± 0.14 r.u. under control conditions to 1.67 ± 0.14 r.u. by 1 min and to 1.23 ± 0.12 r.u. by 3 min in the presence of AA (n = 64, p < 0.05) (Fig. 5a, b). The frequency of oscillations decreased from 7.32 ± 0.41 to 4.23 ± 0.34 pulses per 30 s after 1 min (n = 64, p < 0.05) and to 4.41 ± 0.38 pulses per 30 s after 3 min (p < 0.05).

Effects of acetic acid (0.02%) on calcium oscillations. a (top) Representative traces of Ca2+ oscillations during a 5-min application of AA uncorrected (AA uncorr) in control and after incubation with the BK channel inhibitor TEA (1 mM). a (bottom) Representative traces of Ca2+ oscillations during a 5-min application of AA corrected (AA corr) in control and after incubation in TEA (1 mM). High KCl (100 mM) containing solution was added at the end of traces causing strong depolarization and Ca2+ influx. b Average amplitudes of Ca2+ oscillations during a 5-min application of AA uncorrected pH = 5.7 (open circles) and pH corrected to 7.2 (black circles). *p < 0.05 compared with control values. c Amplitude of Ca2+ oscillations during 5-min application of AA uncorr in control (open circles) and after incubation of TEA (black circles) compared with control values (100%). *p < 0.05 compared with control, #p < 0.05 compared with AA uncorr. d Amplitude of Ca2+ oscillations during 5-min application of AA corr in control (open circles) and after the incubation in TEA (black circles) compared with control values (100%). *p < 0.05 compared with control, #p < 0.05 compared with AA corr
To reveal the role of BK channels in the inhibitory effects of AA, cells were first incubated in TEA (1 mM). Application of AA uncorrected in the presence of TEA decreased the amplitude of Ca2+ oscillations by 53% by 1 min (from 2.09 ± 0.17 to 0.87 ± 0.10 r.u., n = 72, p < 0.05); however, Ca2+ oscillations did not stop as compared with the effects of AA in control conditions (Fig. 5a, c). The frequency of Ca2+ oscillations (TEA + AA uncorr) decreased after the first minute from 4.64 ± 0.27 to 2.67 ± 0.28 pulses per 30 s (n = 72, p < 0.05) and recovered by 3 min to control values (4.02 ± 0.28, p > 0.05). AA with pH corrected to 7.2 did not significantly change the amplitude of Ca2+ oscillations (n = 86, p > 0.05 (Fig. 5a, d). The frequency of oscillation decreased in this condition after the first minute from 4.41 ± 0.23 to 3.25 ± 0.25 pulses per 30 s (n = 86, p < 0.05) and recovered to 4.02 ± 0.28 pulses per 30 s (p > 0.05) after 3 min. These data indicate that inhibition of BK channels partially prevented the inhibitory effects of AA on Ca2+ oscillations.
Discussion
The metabolic products of EtOH receive increasing attention in the context of EtOH action. In addition to EtOH, acetaldehyde and AA/acetate appear to contribute effects of their own to the pharmacological impact of alcohol. In addition to posttranslational modifications at target sites, EtOH action may be different in various cell types or organs caused by their metabolic capacity. In this report, we focused on the effects of acetic acid (AA) on BK channels present in GH3 cells.
The primary source of exogenous acetate in our body is bacterial metabolism of indigestible carbohydrates resulting in production of short-chain fatty acids acetate, propionate, and butyrate in a 3:1:1 stoichiometry [31]. Independent of the bacterial metabolism of dietary fiber alcohol consumption is another considerable source of AA. After metabolism of ethanol, about 70% of the formed acetate is released into the bloodstream and transported through the body where it rather easily accesses all organs. Acetate after passing the blood-brain barrier via carrier-mediated processes may affect glial cells/neurons in the brain [14]. Another source of acetate in the brain is the metabolism of ethanol via the catalase system. Finally, acetate is metabolized to CO2 and H2O or if it is not metabolized may be used in the formation of acetyl-coA, ketone bodies, fatty acids, and steroids [12, 37]. In the brain, acetate is metabolized in glia cells—not in neurons [46]. While plasma concentrations of acetate in humans under normal conditions range between 50 and 200 μM [39, 41, 45], alcohol consumption can increase plasma acetate to 0.5 mM which is more evident during chronic alcohol consumption [34]. After EtOH application in rodents, blood acetate concentrations were reported to rise up to 1.5 mM [9]. Endogenously, acetate can be also generated during deacetylation of proteins such as histones [48] and can be further used in the production of acetyl-CoA. Recent studies suggest a pathway of de novo acetate production from pyruvate, when the metabolic supply exceeds requirement resulted in the accumulation and excretion of intermediate products [6, 30].
AA is a weak acid and in water dissociates to acetate anions and protons, which induces acidification of the solution. In our experiments, both AA and sodium acetate increased BK channel activity; however, AA appeared more efficient. The only difference between AA and sodium acetate is the pH change caused by addition of AA to the perfusate. We, therefore, suspected that H+ influenced the efficacy of AA at the channels. Indeed, AA solution with pH corrected to 7.2 was less effective—similar to sodium acetate which is not acidic. The potentiating effect appears not to be caused by the pH-sensitive site at the channels since H+ inhibits BK channels by interfering with its external voltage sensor [50]. Indeed, acidification of extracellular solution (without AA added) had the expected inhibitory effect at BK channels.
AA at lower pH (pKa = 4.7) contains a larger non-ionized fraction compared with basic pH where acetate ions predominate in solution. Therefore, we consider that the molecular form of AA is responsible for its effects on BK channels. Indeed, AA uncorrected at pH 5.7 increased K+ channel activity about 1.5 times. Acidification of AA solution to pH 4.5 further increased Po 3-fold compared with control. At neutral pH, the stimulating effect of AA was only about 30% and alkalization of AA containing solution to pH 8 did not change BK channel activity. This suggests that the acidic environment plays a role in the efficacy of the acetate effect on BK channels. Our findings may explain results obtained by of Ghatta et al. (2007) where AA (0.01–0.05%) increased whole-cell BK currents in recombinant CHO cells and sodium acetate failed to do so which may be explained by pH dependence of AA effects on BK channel activity. Similar to our results in their work, acidification of the external solution did not activate BK currents [17]—in fact acidification decreased BK activity.
Acetate has both potentiating and antagonizing effects on cellular activity. General anesthesia was found to be potentiated whereas locomotor activity was reduced by acetate which was three times more effective compared with EtOH [9, 11, 37]. In a mouse model of pyruvate dehydrogenase (PDH) deficiency, i.p. acetate injections were shown to decrease epileptiform activity due to recovery of inhibitory neurone firing [25]. In hippocampal dentate granule neurons, acetate hyperpolarizes the membrane and decreases spike frequency adaptation [12]. One of the cellular mechanisms of inhibitory effects of acetate could be activation of BK channels which results in a reduction of excitability. In fact, in our experiments, AA induced a fast and substantial hyperpolarization and cessation of spontaneous action potentials in GH3 cells, which was followed by decrease of the amplitude and frequency of spontaneous Ca2+ oscillations. Inhibition of BK channels completely prevented the inhibitory effects of AA corrected; however, AA uncorrected still produced some hyperpolarization although less pronounced compared with control. Therefore, the participation of other channels contributing to the resting membrane potential in GH3 cells appears feasible [44]. It is also possible that AA impairs Ca2+ oscillations by inhibition of Ca2+ release from intracellular stores [49].
GH3 cells produce prolactin and growth hormones packed into secretory vesicles which traffic towards and finally fuse with the plasma membrane to release hormones by the exocytosis process [8]. In previous studies, it was shown that acetate inhibits growth hormone hypersecretion in humans; however, cellular mechanisms of acetate effects were not investigated [36]. In our studies, we used the membrane-bound fluorescent dye FM1-43 to analyze spontaneous and KCl-evoked exocytosis of secretory granules [26]. The reduction of the fluorescence signal during basal and KCl stimulated conditions by AA suggests that less vesicles fuse to the plasma membrane and the discharge of their content, such as growth hormone into the extracellular space, is reduced. Hence, we interpret the reduction in fluorescence as reduction of secretory granule exocytosis. A decrease of Ca2+ oscillation due to membrane hyperpolarization will also decrease basal exocytosis. Massive exocytosis triggered by KCl-evoked depolarization depends on activation of voltage-gated Ca2+ channels and AA-mediated BK channel activation decreases the duration of Ca2+ influx. Indeed, inhibition of BK channels prevented the inhibitory effects of AA on basal and KCl stimulated exocytosis of secretory granules.
Impaired growth hormone release and decreased level of the insulin-like growth factors (IGF) during pregnancy due to alcohol intoxication results in growth retardation and delayed development of the offspring [2]. Our results indicate that AA may impact ethanol effects on growth hormone release which plays a role not only on growth and development but is also involved in regulation of cognitive function and neuroprotection in brain pathologies [1, 35].
Conclusion
Alcohol/ethanol and its major metabolic products, acetaldehyde, and acetic acid/acetate have effects of their own on Ca2+-activated K+ (BK) channels. The changes in cellular excitability and intracellular signaling caused by acetic acid/acetate may contribute to the action of ethanol. In tissues, such as the nervous system or the gut where pH changes occur physiologically in the microvicinity of cells or during ischemic events, AA/acetate could potentiate the activity of BK channels. Since BK channels are known to constitute a prominent target of alcohol which is creating a multitude of behavioral and medical symptoms, alcohol metabolites have to be also taken into account for further assessment of the diversified and complicated mechanism of alcohol action.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Abbreviations
- BK:
-
big-conductance calcium-activated potassium channels
- AA:
-
acetic acid
- EtOH:
-
ethanol
- Po:
-
open probability
References
Åberg ND, Brywe KG, Isgaard J (2006) Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. ScientificWorldJournal. 6:53–80. https://doi.org/10.1100/tsw.2006.22
Andreu-Fernández V, Bastons-Compta A, Navarro-Tapia E, Sailer S, Garcia-Algar O (2019) Serum concentrations of IGF-I/IGF-II as biomarkers of alcohol damage during foetal development and diagnostic markers of foetal alcohol syndrome. Sci Rep 9:1–10. https://doi.org/10.1038/s41598-018-38041-0
Berkefeld H, Fakler B, Schulte U (2010) Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev 90:1437–1459. https://doi.org/10.1152/physrev.00049.2009
Bettinger JC, Davies AG (2014) The role of the BK channel in ethanol response behaviors: evidence from model organism and human studies. Front Physiol 5:346. https://doi.org/10.3389/fphys.2014.00346
Betz WJ, Mao F, Smith CB (1996) Imaging exocytosis and endocytosis. Curr Opin Neurobiol 6:365–371. https://doi.org/10.1016/S0959-4388(96)80121-8
Bose S, Ramesh V, Locasale JW (2019) Acetate metabolism in physiology, cancer, and beyond, trends. Cell Biol 29:695–703. https://doi.org/10.1016/j.tcb.2019.05.005
Brumback AC, Lieber JL, Angleson JK, Betz WJ (2004) Using FM1-43 to study neuropeptide granule dynamics and exocytosis. Methods. 33:287–294. https://doi.org/10.1016/j.ymeth.2004.01.002
Burgoyne RD, Morgan A (2003) Secretory granule exocytosis. Physiol Rev 83:581–632. https://doi.org/10.1152/physrev.00031.2002
Carmichael FJ, Israel Y, Crawford M, Minhas K, Saldivia V, Sandrin S, Campisi P, Orrego H (1991) Central nervous system effects of acetate: contribution to the central effects of ethanol. J Pharmacol Exp Ther 259:403–408
Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, Milesi V, Neely A, Alvarez O, Ferreira G, Gonzalez C, Latorre R (2013) A BK (Slo1) channel journey from molecule to physiology. Channels. 7:442–458. https://doi.org/10.4161/chan.26242
Correa M, Arizzi MN, Betz A, Mingote S, Salamone JD (2003) Open field locomotor effects in rats after intraventricular injections of ethanol and the ethanol metabolites acetaldehyde and acetate. Brain Res Bull 62:197–202. https://doi.org/10.1016/j.brainresbull.2003.09.013
Cullen N, Carten PL (1992) Electrophysiological actions of acetate, a metabolite of ethanol, on hippocampal dentate granule neurons: interactions with adenosine. Brain Res 588:49–57. https://doi.org/10.1016/0006-8993(92)91343-D
Davies AG, Pierce-Shimomura JT, Kim H, VanHoven MK, Thiele TR, Bonci A, Bargmann CI, McIntire SL (2003) A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans. Cell 115:655–666. https://doi.org/10.1016/S0092-8674(03)00979-6
Deelchand DK, Shestov AA, Koski DM, Uğurbil K, Henry P-G (2009) Acetate transport and utilization in the rat brain. J Neurochem 109:46–54. https://doi.org/10.1111/j.1471-4159.2009.05895.x
Dopico AM, Bukiya AN, Kuntamallappanavar G, Liu J (2016) Modulation of BK channels by ethanol. In: Int Rev Neurobiol. Academic Press Inc., pp 239-279. https://doi.org/10.1016/bs.irn.2016.03.019
Gaifullina AS, Yakovlev AV, Mustafina AN, Weiger TM, Hermann A, Sitdikova GF (2016) Homocysteine augments BK channel activity and decreases exocytosis of secretory granules in rat GH3 cells. FEBS Lett 590:3375–3384. https://doi.org/10.1002/1873-3468.12381
Ghatta S, Lozinskaya I, Lin Z, Gordon E, Willette RN, Brooks DP, Xu X (2007) Acetic acid opens large-conductance Ca2+-activated K+ channels in guinea pig detrusor smooth muscle cells. Eur J Pharmacol 563:203–208. https://doi.org/10.1016/j.ejphar.2007.02.037
Ghazali R, Patel VB (2016) Alcohol metabolism: general aspects. In: Molecular Aspects of Alcohol and Nutrition. A Vol. Mol. Nutr. Ser. Elsevier Inc., pp 17-21. https://doi.org/10.1016/B978-0-12-800773-0.00002-1
Handlechner AG, Hermann A, Fuchs R, Weiger TM (2013) Acetaldehyde-ethanol interactions on calcium-activated potassium (BK) channels in pituitary tumor (GH3) cells. Front Behav Neurosci 7:58. https://doi.org/10.3389/fnbeh.2013.00058
Harris RA, Trudell JR, Mihic SJ (2008) Ethanol’s molecular targets. Sci Signal 1:re7. https://doi.org/10.1126/scisignal.128re7
Hermann A, Sitdikova GF, Weiger TM (2012) BK Channels-focus on polyamines, ethanol/acetaldehyde and hydrogen sulfide (H2S). In: Kaneez FS (ed) Patch Clamp Technique. InTech, pp 109-142
Hermann A, Sitdikova GF, Weiger TM (2015) Oxidative stress and maxi calcium-activated potassium (BK) channels. Biomolecules. 5(3):1870–1911. https://doi.org/10.3390/biom5031870
Israel Y, Orrego H, Carmichael FJ (1994) Acetate-mediated effects of ethanol. Alcohol Clin Exp Res 18:144–148. https://doi.org/10.1111/j.1530-0277.1994.tb00894.x
Jakab M, Weiger TM, Hermann A (1997) Ethanol activates maxi Ca2+-activated K+ channels of clonal pituitary (GH3) cells. J Membr Biol.157(3):237–45. https://doi.org/10.1007/pl00005895
Jakkamsetti V, Marin-Valencia I, Ma Q, Good LB, Terrill T, Rajasekaran K, Pichumani K, Khemtong C, Hooshyar MA, Sundarrajan C, Patel MS, Bachoo RM, Malloy CR, Pascual JM (2019) Brain metabolism modulates neuronal excitability in a mouse model of pyruvate dehydrogenase deficiency. Sci Transl Med 11:1–30. https://doi.org/10.1126/scitranslmed.aan0457
Kilic G, Angleson JK, Cochilla AJ, Nussinovitch I (2001) Sustained stimulation of exocytosis triggers continuous membrane retrieval in rat pituitary somatotrophs. J Physiol 532:771–783. https://doi.org/10.1111/j.1469-7793.2001.0771e.x
Krabbendam IE, Honrath B, Culmsee C, Dolga AM (2018) Mitochondrial Ca2+-activated K+ channels and their role in cell life and death pathways. Cell Calcium 69:101–111. https://doi.org/10.1016/j.ceca.2017.07.005
Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, Alvarez O (2017) Molecular determinants of BK channel functional diversity and functioning. Physiol Rev 97:39–87. https://doi.org/10.1152/physrev.00001.2016
Lee US, Cui J (2010) BK channel activation: structural and functional insights. Trends Neurosci 33:415–423. https://doi.org/10.1016/j.tins.2010.06.004
Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, Liu J, Lund PJ, Lopes M, Garcia BA, Wellen KE, Kirsch DG, Locasale JW (2018) Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175:502–513.e13. https://doi.org/10.1016/j.cell.2018.08.040
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12:661–672. https://doi.org/10.1038/nrmicro3344
Mustafina AN, Yakovlev AV, Gaifullina AS, Weiger TM, Hermann A, Sitdikova GF (2015) Hydrogen sulfide induces hyperpolarization and decreases the exocytosis of secretory granules of rat GH3 pituitary tumor cells. Biochem Biophys Res Commun 465:825–831. https://doi.org/10.1016/j.bbrc.2015.08.095
Nieminen M, Salaspuro M (2018) Local acetaldehyde – an essential role in alcohol-related upper gastrointestinal tract carcinogenesis. Cancers (Basel) 10:11. https://doi.org/10.3390/cancers10010011
Nuutinen H, Lindros K, Hekali P, Salaspuro M (1985) Elevated blood acetate as indicator of fast ethanol elimination in chronic alcoholics. Alcohol. 2:623–626. https://doi.org/10.1016/0741-8329(85)90090-4
Nyberg F, Hallberg M (2013) Growth hormone and cognitive function. Nat Rev Endocrinol 9:357–365. https://doi.org/10.1038/nrendo.2013.78
Orskov H, Hansen AP, Hansen HE, Alberti KGMM, Noy GA, Nosadini R (1982) Acetate: inhibitor of growth hormone hypersecretion in diabetic and non-diabetic uraemic subjects. Acta Endocrinol 99:551–558. https://doi.org/10.1530/acta.0.0990551
Pardo M, Betz AJ, San MN, López-Cruz L, Salamone JD, Correa M (2013) Acetate as an active metabolite of ethanol: studies of locomotion, loss of righting reflex, and anxiety in rodents. Front Behav Neurosci 7:81. https://doi.org/10.3389/fnbeh.2013.00081
Phillis JW, O’Regan MH, Perkins LM (1992) Actions of ethanol and acetate on rat cortical neurons: ethanol/adenosine interactions. Alcohol. 9:541–546. https://doi.org/10.1016/0741-8329(92)90094-Q
Pomare EW, Branch WJ, Cummings JH (1985) Carbohydrate fermentation in the human colon and its relation to acetate concentrations in venous blood. J Clin Invest 75:1448–1454. https://doi.org/10.1172/JCI111847
Quertemont E, Didone V (2006) Role of acetaldehyde in mediating the pharmacological and behavioral effects of alcohol. Alcohol Res Health 29:258–265
Richards RH, Dowling JA, Vreman HJ, Feldman C, Weiner MW (1976) Acetate levels in human plasma. Proc Clin Dial Transplant Forum 6:73–79 https://europepmc.org/article/med/1029892 (accessed September 9, 2020)
Sitdikova GF, Weiger TM, Hermann A (2010) Hydrogen sulfide increases calcium-activated potassium (BK) channel activity of rat pituitary tumor cells, Pflugers Arch. Eur J Phys 459:389–397. https://doi.org/10.1007/s00424-009-0737-0
Sitdikova GF, Fuchs R, Kainz V, Weiger TM, Hermann A (2014) Phosphorylation of BK channels modulates the sensitivity to hydrogen sulfide (H2S). Front Physiol 5:431. https://doi.org/10.3389/fphys.2014.00431
Stojilkovic SS, Tabak J, Bertram R (2010) Ion channels and signaling in the pituitary gland. Endocr Rev 31:845–915. https://doi.org/10.1210/er.2010-0005
Tollinger CD, Vreman HJ, Weiner MW (1979) Measurement of acetate in human blood by gas chromatography: effects of sample preparation, feeding, and various diseases. Clin Chem 25:1787–1790. https://doi.org/10.1093/clinchem/25.10.1787
Waniewski RA, Martin DL (1998) Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci 18:5225–5233. https://doi.org/10.1523/jneurosci.18-14-05225.1998
Weiger TM, Hermann A, Levitan IB (2002) Modulation of calcium-activated potassium channels. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 188:79–87. https://doi.org/10.1007/s00359-002-0281-2
Ye C, Tu BP (2018) Sink into the epigenome: histones as repositories that influence cellular metabolism. Trends Endocrinol Metab 29:626–637. https://doi.org/10.1016/j.tem.2018.06.002
Yoon MN, Kim MJ, Koong HS, Kim DK, Kim SH, Park HS (2017) Ethanol suppresses carbamylcholine-induced intracellular calcium oscillation in mouse pancreatic acinar cells. Alcohol. 63:53–59. https://doi.org/10.1016/j.alcohol.2017.03.006
Zhou Y, Xia XM, Lingle CJ (2018) BK channel inhibition by strong extracellular acidification. Elife. 7:e38060. https://doi.org/10.7554/eLife.38060
Zimatkin SM, Buben AI (2007) Ethanol oxidation in the living brain. Alcohol Alcohol 42(6):529–532. https://doi.org/10.1093/alcalc/agm059
Acknowledgments
We cordially thank Mag. Astrid Handlechner for her competent help in the cell culture lab.
Funding
Funded by Russian Foundation of Basic Research No. 19-315-90084 and the subsidy allocated to Kazan Federal University for the state assignment № 0671-2020-0059 in the sphere of scientific activities.
Author information
Authors and Affiliations
Contributions
I. Shaidullov: investigation, formal analysis. E.Ermakova: investigation, formal analysis, writing - original draft. A.Gaifullina: investigation, formal analysis. A. Mosshammer: investigation, formal analysis. A. Yakovlev: formal analysis, validation, visualization, writing - original draft. T.M. Weiger: conceptualization, methodology, writing - review and editing. A. Hermann: conceptualization, writing - original draft, writing - review and editing. G. Sitdikova: methodology, formal analysis, validation, visualization, writing - original draft, writing - review and editing, supervision, funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shaidullov, I., Ermakova, E., Gaifullina, A. et al. Alcohol metabolite acetic acid activates BK channels in a pH-dependent manner and decreases calcium oscillations and exocytosis of secretory granules in rat pituitary GH3 cells. Pflugers Arch - Eur J Physiol 473, 67–77 (2021). https://doi.org/10.1007/s00424-020-02484-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-020-02484-0