
Abstract
The classic clinical definition of hypertrophic cardiomyopathy (HCM) as originally described by Teare is deceptively simple, “left ventricular hypertrophy in the absence of any identifiable cause.” Longitudinal studies, however, including a seminal study performed by Frank and Braunwald in 1968, clearly described the disorder much as we know it today, a complex, progressive, and highly variable cardiomyopathy affecting ~ 1/500 individuals worldwide. Subsequent genetic linkage studies in the early 1990s identified mutations in virtually all of the protein components of the cardiac sarcomere as the primary molecular cause of HCM. In addition, a substantial proportion of inherited dilated cardiomyopathy (DCM) has also been linked to sarcomeric protein mutations. Despite our deep understanding of the overall function of the sarcomere as the primary driver of cardiac contractility, the ability to use genotype in patient management remains elusive. A persistent challenge in the field from both the biophysical and clinical standpoints is how to rigorously link high-resolution protein dynamics and mechanics to the long-term cardiovascular remodeling process that characterizes these complex disorders. In this review, we will explore the depth of the problem from both the standpoint of a multi-subunit, highly conserved and dynamic “machine” to the resultant clinical and structural human phenotype with an emphasis on new, integrative approaches that can be widely applied to identify both novel disease mechanisms and new therapeutic targets for these primary biophysical disorders of the cardiac sarcomere.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Current challenges in our understanding of sarcomeric cardiomyopathies
Mutations in the genes that encode the protein components of the cardiac thin filament were first linked to clinical cardiomyopathies in 1994 [85]. Mutations in the regulatory thin filament comprise ~ 7–10% of hypertrophic cardiomyopathy (HCM) cases and represent a distinct and complex subset of the disorder with marked mutation-specific phenotypes [11, 84]. Despite 25 years of basic research, many questions remain regarding the clinical care of these complex patients who suffer from a disorder that is inherently genetic and affects 1/500 individuals worldwide [15, 21]. One of the most significant current limitations in the care of patients with sarcomeric cardiomyopathies (both hypertrophic and dilated) is the inability to use genotype to guide management. This persists despite extensive research by many groups over the past 25 years to probe the primary mechanisms of this inherently biophysical “disease of the cardiac sarcomere” [84]. While the root cause of this limitation is multifactorial, an overriding factor is the timescale of pathologic cardiac remodeling observed in patients with sarcomeric cardiomyopathies. Given that cardiac remodeling in HCM can occur over decades, establishing the mechanistic link between a mutation that alters a biophysical property of the cardiac sarcomere and the often variable whole-organ phenotype raises an important question regarding how to identify earlier or more “proximal” molecular phenotypes. Of particular note, reversing cardiac remodeling, especially in the later stages of disease is a formidable challenge, thus early interventions are more likely to be efficacious.
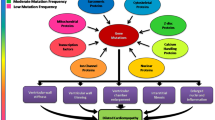
This basic limitation has led to renewed efforts to define the earliest stages of myocellular pathogenesis with a primary goal of developing better linkage between changes in sarcomeric function and the molecular responses that define the preclinical stages of disease [34]. A patient with “preclinical HCM” for example, carries a known pathogenic mutation in the absence of a clinical phenotype (genotype+, phenotype– or “G+P-”). A recently published cross-sectional multicenter observational study by Ho et al. directly addressed this central question of how to better identify and classify the earliest stages of disease in preclinical G+P- HCM cohorts [36]. In this study, “phenotypic burden” was assigned as the cumulative number of 7 parameters that were chosen based on the ability to discriminate between either G+/P+ versus G+/- siblings. Primary results demonstrated a significant increase in early phenotypes among G+/- individuals versus controls (G-P-). This “burden score” is cumulative and likely represents the earliest stages of myocellular and molecular remodeling that occurs in direct response to the primary biophysical insult. These myocellular responses are likely compensatory attempts by the myocyte to overcome the stress on the system induced or “triggered” by sarcomeric dysfunction. As shown in Fig. 1, the link(s) between the biophysical insult (trigger) and the initial compensatory response are proximal and thus both likely represent important therapeutic targets before the onset of “irreversible” cascades of pathogenic remodeling. This molecular compensatory phase correlates to patients with “preclinical disease” who are defined as carrying a pathogenic gene mutation (phenotype) yet remain in the preclinical state (phenotype) or G(+) P (-) [35, 47, 92]. Given the early stage of remodeling, these patients have the greatest potential benefit from novel therapeutic interventions. Targeting these initial molecular mechanisms before downstream signaling cascades have been activated could potentially alter the natural progression of the complex cardiomyopathy and represents a significant, novel approach to patient management.

Pathogenesis of sarcomeric cardiomyopathies. Timecourse and proposed trajectory of cardiac remodeling caused by pathogenic sarcomere gene mutations. G+P- refers to genotype-positive patients without evidence of pathogenic remodeling and G+P+ refers to genotype-positive patients with more advanced disease and overt ventricular remodeling
Sarcomeric cardiomyopathies and degrees of freedom
As noted above, the current limitations to using patient genotype in the management of clinical disease represent a core concern in the care of patients with sarcomeric cardiomyopathies. And while the complexity of the progressive cardiac remodeling and incomplete penetrance observed for many mutations leads to a vast array of end phenotypes, the growing number of identified mutations linked to these disorders raises the issue of how to couple these large genetic and phenotypic datasets in a meaningful and tractable manner. It is illustrative to consider the evolution of prior attempts to group or “bin” cardiomyopathies in order to decrease the overall degrees of freedom of this complex question. Initial approaches (pre genetic linkage) utilized end state morphology and led to the basic subsets in use today, namely hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and restrictive cardiomyopathy (RCM) [75]. While still useful, the combination of genetic linkage and the broader use of genetic testing in large multi-generational families soon revealed an unexpected degree of variable phenotypes among patients with the same gene mutation. For example, while the I79N mutation in cardiac TnT (cTnT) was initially linked to HCM, subsequent clinical phenotyping identified DCM and RCM among genotype positive family members [59]. Similar degrees of phenotypic variability were observed in patients carrying other cardiac troponin mutations including cTnI Δ183K [44] and cTnT R92L/Q/W [19, 88]. More recently, longitudinal clinical studies of genotyped populations have clearly shown that disease progression can be transitory in that patients with end-stage DCM may have first presented with HCM or even experienced non-linear remodeling patterns [36]. Thus, the complexity of the natural history of ventricular remodeling in sarcomeric cardiomyopathies coupled to the continuing reliance on end-stage classification of the disease into HCM, DCM, and RCM is a significant limitation to achieving the goal of utilizing genotype in clinical management and eventually developing effective strategies for novel therapeutics [2, 41].
In order to develop new approaches to these complex and highly clinically relevant issues, it is first necessary to develop more robust high resolution insight regarding the molecular pathogenesis of sarcomeric cardiomyopathies. Specifically, to incorporate structure, protein dynamics and function to develop a useful framework for linking alterations in biophysics to early molecular remodeling. The key component of this approach is to utilize both more complete structural information and greater biologic complexity in vitro such that mutations can be grouped or “binned” into relevant subsets for eventual targeting strategies. These robust mutational “bins” can then be coupled to the rapidly growing, genotyped patient cohort datasets that now incorporate early and longitudinal phenotypes. In this review, we will provide a framework for an integrated approach to this important clinical goal with a focus on the cardiac thin filament.
Thin filament structure and function
The central dogma of structural biology is that function is determined by structure. The thin filament is composed of filamentous actin (F-actin), tropomyosin (Tm), and the troponin (Tn) complex in a 7:1:1 M ratio (Fig. 2). F-actin is a double helical structure composed of polymerized globular actin (G-actin) [43]. Within the two grooves of F-actin lies Tm, a coiled-coil structure of two coiled α-helical Tm monomers that overlaps with adjacent Tm dimers in a head to tail formation to form a continuous Tm strand [32]. Tm provides the thin filament with stability, flexibility, and cooperativity. The Tn complex anchors Tm to F-actin, by the Tn complex tail region extending over the Tm C-terminus of the Tm-Tm overlap region [22]. The Tn complex is composed of troponin C (cTnC), troponin I (cTnI), and troponin T (cTnT) in a 1:1:1 M ratio, representing the Ca2+ regulatory binding protein, the inhibitory subunit of the Tn complex, and the Tm-binding domain, respectively [22].

Full atomistic model of the human cardiac thin filament. Actin is represented in gray. Tropomyosin dimers are represented in green and orange. Cardiac TnT is depicted in yellow, cTnI is shown in blue, and cTnC is represented in red
The basic function of the thin filament is to transduce chemical signals throughout the myofilament protein complex and directly regulate the conversion of energy to mechanical work via the actomyosin crossbridge cycle [10, 82]. Specifically, Ca2+ released from the sarcoplasmic reticulum binds to site II of the regulatory N-terminal domain of cTnC, leading to allosteric changes that release the cTnI inhibitory domain and favors the actin-Tm binding domains [42, 48]. The three-state molecular model of thin filament activation describes the azimuthal shift of Tm along the outer domain to the inner domain of F-actin to expose myosin binding sites [58]. The states in this model include a blocked state (B-state) where crossbridge formation is largely sterically blocked; a closed state (C-state) where weak crossbridge formation occurs in the presence of Ca2+ and no force is produced; and an open state (M-state) where in the presence of myosin and Ca2+, strong crossbridge formation occurs and strong force is generated [23]. Deactivation occurs via the reversal of this process whereby calcium dissociates from cTnC, strongly bound crossbridges detach in an ATP-dependent manner, and Tm returns to its initial position (B-state) [42, 58]. Thus, the thin filament is a highly cooperative and dynamic machine and even slight alterations to its molecular structure can significantly disrupt its function.
The regulatory thin filament
Current approaches used to classify the effects of a mutation on sarcomeric protein function for clinical assessment of pathogenicity are largely limited to computational tools (for example, SIFT and PolyPhen2) that are largely based on sequence homology and machine learning [1, 46]. These tools are of rather limited use for thin filament (or sarcomeric mutations in general), because they do not incorporate the effects of a mutation on inter- and intra-molecular interactions in ternary and quaternary structures. Indeed, it is not uncommon for the two algorithms to disagree with each other. In the main, these limitations remain despite numerous attempts by multiple groups to establish a robust predictive system to assign prognosis to de novo sarcomeric gene mutations. Put simply, new, integrative approaches are needed to address this significant roadblock to using genotype in the management of hypertrophic cardiomyopathy.
More recently, multiple groups have shown that allosteric effects must be considered in the context of evaluating the biophysical effects of single point mutations in thin filament proteins, especially with regard to linking alterations in “local” structure to biological function [16, 25, 62]. Limitations remain, however, in coupling these atom-level observations to steady-state in vitro measurements. While steady-state studies provide physiologically relevant information regarding alterations in myofilament function caused by thin filament mutations, they do not provide sufficient resolution to allow for precise, mutation-specific, mechanistic insight with regard to expected patient phenotypes. In particular, it has become clear that, in many cases, the oft-cited “binary” classifications of disease mechanism in cardiomyopathies (e.g., hypercontractility vs hypocontractility and calcium sensitization vs desensitization) insufficiently describe the heterogeneity of disease presentation in patients, thus limiting their predictive or translational power.
To directly address this challenge, our lab and others have turned to computational modeling to investigate allosteric effects of mutations [7, 14, 16, 25, 49, 50, 70, 90]. These models allow for the characterization of a broad array of structural and dynamic effects, including, but not limited to: myofilament Ca2+ kinetics, phosphorylation potential, cooperativity/flexibility, and a variety of high-resolution, structural changes. The long-term goal of these approaches is to define “computational clinical tests” that are based on robust molecular observations and, in many cases, directly coupled to in vitro experiments [25, 55, 56]. When linked to the recent advances in disease phenotyping and the availability of large clinical registries such as SHaRe, these approaches will hopefully facilitate the use of these computational models as a predictive tool, rather than a solely retrospective mechanistic probe [37]. While there are many approaches to address this important issue, a reasonable starting point is to evaluate multiple potential molecular mechanisms caused by known mutations, for example, those that affect structure (local and distant), mechanics, kinetics, cooperativity, and cooperativity. From our perspective, going forward, a key component of these studies will require effective strategies to incorporate computation coupled to rigorous in vitro methodologies that complement and validate these observations. In addition, to investigate these different molecular mechanisms, a wide variety of experimental approaches will need to be utilized with the recognition that no single assay will provide significant resolution to be predictive of phenotype. The resultant integrated definition of molecular phenotype (trigger) will facilitate the ability to group or “bin” a given mutation irrespective of individual protein, such that more informative sets can both be coupled to early in vivo compensatory responses and more robust strategies for therapeutic options focused on the earliest stages of cardiac remodeling can be developed.
An integrative approach to mutations in the cTnT extended flexible linker and the tropomyosin overlap domains
Cardiac troponin T is classically described as two chymotrypsin-defined fragments: first, TNT1 (comprised of the N-terminal hypervariable and extended Tm-binding domains) and second, the cTnI, cTnT, and Tm-binding TNT2 “core” [89]. Utilizing crystallized partial complexes, circular dichroism, and EM reconstruction, cTnT was revealed to be predominantly α-helical from residues 3–159 and 203–272 [18, 31, 74, 83, 91]. Within TNT1, there is an extendible and highly flexible linker domain (residues 158 and 203) comprised of a highly charged hinge region (residues 158–166) and a flexible region (residues 166–203) [55]. To date, the inherent flexibility of this domain has precluded successful high-resolution, three-dimensional structural determination. [83].
The C-terminus of TNT1 (residues 140–200) has been shown to be highly conserved across vertebrate species [64]. Within this region, several mutations have been identified and fall into two phenotypic groups: HCM causative, Δ160E; E163K; E163R; S179F and DCM causative, A172S; R173Q; R173W. These mutations are highly penetrant, clinically severe, and are also notable for a clearly progressive pattern of pathological cardiac remodeling [5, 60, 64, 80, 87, 88]. They have been shown to have significant differences in their molecular phenotypes across patients, including differences in severity between the women and men [79]. Given the high degree of conservation and differential phenotypes caused by mutations in close proximity (HCM vs DCM) and the resultant highly penetrant, severe clinical outcomes, the extended flexible domain of cTnT likely plays a central role in the regulatory function of the thin filament. Matching biophysical mechanisms to cardiac phenotypes, however, is complicated by the lack of known structure across this domain. Based on the data above, among the many plausible mechanisms for pathogenic mutations in this domain, two stand out: first, the primary structure and inherent flexibility of the flexible linker domain are altered, and/or second, calcium-dependent myofilament function is altered as a secondary effect. As shown in Fig. 3, the flexible linker domain is immediately distal to the extended Tm-cTnT binding domain that acts to stabilize the thin filament [86]. Additionally, this interaction plays an important role in determining the position of the Tn-Tm complex on actin, potentially altering efficient crossbridge cycling [76]. Therefore, alterations in the structure, position and flexibility of this region could affect both inter- and intra-protein interactions and significantly alter myofilament activation.

C-terminal TNT1 extended linker domain. Left panel (box) refers to position of extended linker in the context of the thin filament. Right panel shows the locations of the 160E (HCM) and R173W (DCM) mutations within the extended linker domain. cTnT is shown in yellow and tropomyosin (orange) is shaded for clarity
While both DCM- and HCM-linked mutations have been shown to alter the Ca2+ sensitivity of myofilament activation, there have been significant differences in the magnitude and even direction of the observed changes. Moore et al. utilized the regulated in vitro motility (R-IVM) assay to show that cTnT mutations Δ160E, E163K, and E163R led to a differential disruption of weak electrostatic interactions [64]. Measurement of the Ca2+ sensitivity of filament activation required a reduction in the overall ionic strength of the system and revealed that while E163R and E163K slightly decreased Ca2+ sensitivity when compared to WT, in contrast, the Ca2+ sensitivity of sliding speed for Δ160E was increased [64]. These results were largely in agreement with previous studies of Δ160E and E163K in reconstituted skinned fibers [27, 81]. In contrast, Sommese et al. reported that E163K, like Δ160E, increased the Ca2+ sensitivity of ATPase activity [77]. Finally, Messer, et al. suggested an alternative mechanism for the pathogenicity of the Δ160E and S179F HCM mutations as they observed an “uncoupling” of the effect of cTnI phosphorylation on Ca2+ sensitivity in the R-IVM assay [61]. It is important to note, however, that there have been conflicting reports of the pathogenicity of S179F in the heterozygous state [33]. Perhaps not surprisingly, studies of the DCM-linked cTnT R173W mutation on Ca2+-dependent properties have also yielded complex results. In the original report, Sun et al. generated cardiomyocytes from induced pluripotent stem cells (CM-iPSCs) from a R173W patient [80]. The R173W CM-iPSCs exhibited decreased Ca2+ transients consistent with a predicted decrease in contractility as seen in previous AFM measurements, and in agreement with ATPase studies [77, 80]. The decrease in contractility was interpreted to predict that the R173W mutation would cause decreased Ca2+ sensitivity as seen in other DCM-linked cTnT mutations [30, 52, 66, 67]. This lack of clear consensus regarding the primary mechanism(s) of cardiomyopathy-linked mutations in the cTnT linker domain is a reflection of both the limitations in relying on Ca2+ sensitivity as a primary assay and our lack of high-resolution structure of this important region.
Interestingly, in vivo characterizations of cardiomyopathy-linked mutations in the extended linker domain have revealed significantly abnormal sarcomeric organization and disarray. While myocellular disarray is a common (and relatively nonspecific) finding in pathologic sections from patients with cardiomyopathies (of multiple etiologies), significant sarcomeric disarray is not, in general, a consistent finding in animal models of either HCM or DCM. Initial characterization of the DCM-linked R173W CM-iPSCs revealed an abnormal distribution of sarcomeric alpha-actinin consistent with a highly disorganized myofilament structure [80]. Moreover, sarcomeric disorganization in these cells was increased due to both β-agonist stimulation and cyclic stretch, demonstrating their susceptibility to mechanical stress. Similarly, analysis of isolated adult ventricular myocytes from transgenic mouse models carrying the HCM-linked Δ160E and E163R mutations showed significant sarcomeric disruption [17, 63]. Comparison of cardiac myocytes from animals with different levels of cTnT Δ160E (35% vs 70%) exhibited a clear dosage-dependence in the degree of disruption, consistent with the more severe effects on myofilament activation and cardiac remodeling observed in the Δ160E R-IVM studies [64]. Taken together, these studies support an important and likely structural role for the cTnT extended linker domain.
To begin to address the hypothesis that one of the primary mechanisms underlying differential pathologic remodeling observed in HCM- and DCM-linked cardiomyopathies is a mutation-specific change in the structure of the linker, we turned to the all-atom computational model of the cardiac thin filament [56, 90]. Preliminary simulations and analysis of average structures have revealed that the proximal portion of the extended linker (~ residues 155–180) contains two α-helical components separated by a short linker. Residues 155–164 are part of the existing cTnT-TM binding domain while residues 171–180 comprise the second α-helix. Note that cTnT Δ160E, E163K/R (HCM) mutations are located within the first α-helix and the cTnT R173W (DCM) mutation is within the second α-helix. Initial results suggest that the Δ160E mutation partially rotates the first α-helix and leads to an overall decrease in the root mean squared fluctuation (RMSF) for residues 130–170, consistent with a decrease in flexibility of the proximal extended linker (and the distal cTnT-Tm binding domain). Changes in the flexibility of the proximal linker would be predicted to alter the position and/or dynamics of the distal domain (residues 170–200). These predicted structural perturbations may affect myofilament assembly or stability, potentially accounting for the sarcomeric disarray noted above. In addition, these studies could be extended via additional simulations incorporating other extended linker mutations and correlated via TR-FRET in reconstituted thin filaments under varying conditions. The end result would be an integrated in silico–in vitro approach to identify potential biophysical triggers at high resolution. The next step would be to correlate the effects of these individual triggers to analysis of early molecular signaling cascades in matched murine models to provide an integrated system to study the earliest stages of pathogenic remodeling, a current central focus of clinical research in the field. Finally, the availability of robust computational models will facilitate relatively high-throughput screening of mutations to allow for functional “binning” of potential triggers based on structural or dynamic effects on the extended linker, thus providing new targets for intervention in the early disease process.
The regulation of cardiac muscle contractility is highly dependent on the cooperative activation of the thin filament that is largely modulated by tropomyosin (reviewed in [65]). The cardiac troponins (I, C, and T) are all comprised of largely α-helical domains connected by linkers of varying length that impart a high degree of flexibility to the individual proteins and the overall complex. A mutation in any of the cardiac thin filament proteins can allosterically impact the cooperativity of the system by, among other things, altering overall flexibility and disrupting the efficiency of myofilament activation. A central modulator of thin filament cooperativity is the tropomyosin overlap domain. This multi-subunit region contains the head-to-tail overlap of adjacent Tm dimers (Tm C-terminus residues 254–284, Tm N-terminus residues 1–24) and is stabilized by the α-helical Tm-binding domain in TNT1 (residues 80–110) [68, 72]. The complex interdigitation of five independent α-helices acts as a dynamic and highly flexible “molecular swivel” that plays a central role in modulating the cooperativity of myofilament activation [4, 8, 24, 68, 71]. Approximately 75% of the known cTnT and Tm cardiomyopathy-causing mutations occur within or flanking the overlap domain [56, 69]. Moreover, given the well-described propagation of structural effects caused by Tm mutations throughout the dimer, it can be argued that any Tm mutation has the capability of disrupting overlap function [53]. While significant questions remain, however, regarding the structure and dynamics of this complex domain, mutations within the overlap clearly represents a significant cause of pathogenic cardiac remodeling [16, 25, 31].
The structure of the overlap is reliant on the interactions between F-actin, Tm, and cTnT. Contacts between actin and Tm are governed by weak electrostatic interactions that aid in regulating crossbridge formation [22, 23, 38]. Tm’s surface is largely negative with acidic residue pockets for actin binding [3]. Additionally, the interdigitation of adjacent Tm dimers modulates both the affinity for actin and its cooperativity [4, 8, 73]. Thus, one possibility is that cardiomyopathy-associated mutations in this region lead to alterations in these weak electrostatic interactions by altering the structure and dynamics of the Tm overlap. Support for this hypothesis has been provided by many groups including our own and have revealed that mutations in both Tm and cTnT cause structural changes that alter Tm stability, Tm and cTnT affinity for actin and Tm overlap flexibility [6, 20, 29, 39, 45, 51, 54, 57]. It is important to note that since Tm dimers form a contiguous chain along the entire length of the thin filament, mutations that affect overlap structure have the potential to profoundly disrupt thin filament function.
To investigate possible differential Tm-linked pathogenic mechanisms, Chang et al. examined a subset of known mutations (DCM: E40K, E54K and HCM: E62Q, L185R) at multiple levels of biologic complexity [6]. While a full discussion of the complex findings of this study are beyond the scope of the current review, several observations were particularly revealing. For example, while thermal denaturation studies of isolated Tm dimers suggested that independent mutations affected melting transitions to varying degrees (without altering overall helicity), no clear pattern regarding these differences at this level of experimental resolution could directly account for the observed phenotypic differences in patients. As noted by the authors, this was likely due to changes in inter- and- intra-coil interactions that would likely transmit their effects distally in the context of a reconstituted thin filament and was largely consistent with their ATPase results. The lack of any change in overall Tm helicity in this study is concordant with the results from a number of groups including our own and is not surprising, in that mutations in thin filament proteins by and large do not “destroy” function, instead, the resultant effect at the biophysical level is more likely to be modulatory. Given the inherent cooperativity of the complex, the structural disruption of a single functional unit could affect the entire thin filament and would not be tolerated in vivo. In the same study, computational modeling was performed on Tm fragments carrying the same mutations and compared to WT to assess the local effects on inter-dimer sidechain interactions. While some high-resolution mechanistic insight was obtained in comparisons to the thermal denaturation results (especially for E62Q where thermal stability was predicted to increase), the lack of time dependent simulations and the sole use of fragment-based modeling in a multiprotein complex somewhat limited the translational power of these findings. Similarly, Gupte et al. explored potential mechanisms whereby Tm mutations (located along the full length of the protein) caused differential patterns of cardiomyopathic remodeling [26]. In this well-controlled study, results of basic biophysical approaches (CD, co-sedimentation and ATPase activity) were compared to complexes incorporating cTnC labeled with an environmentally sensitive fluorescent probe (ANS) as a measure of subtle conformational changes within the thin filament complex in a fully reconstituted system. Again, a broad range of potential mechanistic triggers were observed, with some mutation-specific effects on overall Tm stability, actin binding and allosterically mediated changes in Tn conformation. One of the unique goals of this study was to evaluate the use of the ANS assay as an in vitro screen for small molecule modulators of thin filament function. Interestingly, as the authors noted, the Ca2+ sensitivity shifts in the context of the ANS assays did not correlate well with results from their standard thin filament activated ATPase assay and they concluded that the latter was more informative in the context of the “binary” hypothesis of cardiomyopathy. Another interpretation of their results is that instead of supporting the conclusion that there are multiple “mechanistic routes to a very limited set of cardiac pathologies”, perhaps the focus on end-stage remodeling as the representative phenotype is not sufficient to provide translational power.
As the above studies have shown, the unique structure of Tm both as a single dimer and as part of a continuous strand of molecules aligned along the regulatory thin filament coupled to the complex timescale of pathogenic remodeling in patients with Tm mutations represents a significant challenge to understanding basic mechanism. More recently, we have applied our integrated in silico–in vitro–in vivo approach to address the question of how mutations in close proximity to the overlap and located in different thin filament proteins (cTnT R92L–HCM, Tm D230N–DCM) cause differential cardiac remodeling (Fig. 4) [57]. Our approaches have both similarities and differences when compared to previous studies. We again employed the all atom Schwartz model of the cardiac thin filament (that allows for interrogation of the 5-helix overlap domain and actin), in this case the advantage is that we could account for both local and distant effects of mutations. We coupled the computational results (average structure and RMSF) to two independent methodologies (TR-FRET and DSC) utilizing fully reconstituted cardiac thin filaments. Finally, we developed robust transgenic mouse models for each mutation that exhibited strong phenocopies of the human disease. The use of in vivo models is important for two reasons. First, replicating the reported human phenotype in an animal model is a strong validation of mutation-specific remodeling (fidelity). Second, the availability of a strong phenocopy allows for longitudinal studies and, in particular, an intact system for investigating the most proximal molecular responses to the biophysical and structural affects of mutations. Initial skinned fiber mechanics on samples from the cTnT R92L (HCM) and Tm D230N (DCM) mouse models revealed a decrease in cooperative activation for R92L and a marked increase for D230N. TR-FRET (donor sites on cTnT, acceptor sites on TM) across the overlap domain revealed mutation-specific structural effects, specifically an increase in the distance between cTnT residue 100 and Tm 271, 279 for R92L, and a decrease for D230N. Average structures derived from all-atom simulations largely agreed with these results and supported the overall conclusion that the DCM-linked D230N mutation caused an overall “compaction” of the overlap while the HCM-linked R92L had the opposite effect. Regarding potential effects on the cooperativity of activation, compacting the overlap (D230N) would decrease the overall flexibility of this domain and thus increase cooperativity while increasing flexibility (R92L) would be predicted to decrease cooperativity, matching the original skinned fiber results. Finally, DSC performed on fully reconstituted R92L thin filaments revealed an increase in the full-width at half maximum of the dissociation of the Tm-Tn complex from actin and the opposite for D230N again consistent with a decrease in cooperativity for the cTnT HCM mutation and an increase for the Tm DCM. Taken together, the results of this integrative approach suggest that the overall flexibility of the Tm overlap can be finely tuned by independent mutations and that the direction (and perhaps magnitude) of that change can trigger differential patterns of cardiac remodeling. An important component of this potential mechanism is that it implies that there is a zone or gradient of flexibility that is tolerated and may account for the range of phenotypes for the many mutations and variants that have been mapped to this region. The next step is to leverage murine models with strong phenocopies and perform transcriptome and proteome-based pathway analysis on early stages of cardiac remodeling. While human tissue obtained from late stage disease may not be very informative, it may well be possible, in time, to leverage human iPSC-derived myocyte technologies to provide additional translational power to these important questions.

Tropomyosin overlap domain. Left panel (box) shows the position of the overlap domain in the context of the thin filament. Right panel depicts the position of the R92L substitution (HCM) in cTnT (yellow) and the D230N substitution (DCM) in tropomyosin (orange)
Conclusions/summary/going forward
While great strides have been made over the past 25 years, both in our understanding of basic thin filament biology and the pathogenesis of thin filament-linked cardiomyopathies, integration between these two fields remains challenging. These unanswered questions that will require novel techniques coupled to a more “modern” understanding of the natural history of sarcomeric cardiomyopathies. While the longstanding focus on binary hypotheses (hypercontractile vs hypocontractile, Ca2+ hyposensitivity vs Ca2+ hypersensitivity, etc) has proven useful in some instances, for example, small molecule screening for new therapeutic options, there are significant limitations to this approach. The need for higher resolution mechanistic insight was recently illustrated by the negative results of the trimetazidine clinical trial [9]. This trial was based on significant evidence from both patients and animal models that HCM-causing mutations led to an increase in tension cost and energetic inefficiency that was posited to affect exercise tolerance [12, 28, 40, 78]. While the direct mechanistic links between myofilament mutations and abnormalities in myocellular energetics remain unclear, the decision to increase mitochondrial glucose utilization and reduce fatty acid uptake via an existing and well-tolerated direct inhibitor of β-oxidation (trimetazidine dihydrochloride) was well posed. Interestingly, the trial cohort was comprised of nonobstructive HCM patients, a heterogeneous and clinically challenging patient subset that does not fall into a classic HCM vs DCM binary grouping. The trial did not demonstrate any improvement in exercise capacity for patients in the treatment arm and the authors concluded that a “therapeutic role for metabolic modulators” was not confirmed for nonobstructive HCM. This result, however, should not be interpreted as a failure of the overall hypothesis that myofilament mutations that increase tension cost influence disease pathogenesis. The accompanying editorial goes to great length to discuss the complexity of the clinical disorder including many issues raised in the current review as possible contributors to the negative outcome [13]. It is also highly likely that improved mechanistic insight at the myofilament level (as detailed in the current review) coupled to early stage animal models would result in improved translational power as compared to targeting mitochondrial pathways. From the experimental standpoint, the way forward is to improve our ability to couple high resolution structural and functional measurements (including computation) to robust, biologically “intact” in vitro systems and rigorously link these results to the early activation of myocellular signaling cascades in animal models. It is equally important, however, to accept and acknowledge the complexity of the human disorder and to avoid over-simplifying disease pathogenesis with an over reliance on end-stage phenotypes. These studies are perfectly suited to robust collaborative efforts between HCM centers and scientists and should be aggressively pursued.
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. https://doi.org/10.1038/nmeth0410-248
Axelsson A, Iversen K, Vejlstrup N, Ho C, Norsk J, Langhoff L, Ahtarovski K, Corell P, Havndrup O, Jensen M, Bundgaard H (2015) Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 3:123–131. https://doi.org/10.1016/S2213-8587(14)70241-4
Barua B, Pamula MC, Hitchcock-DeGregori SE (2011) Evolutionarily conserved surface residues constitute actin binding sites of tropomyosin. Proc Natl Acad Sci U S A 108:10150–10155. https://doi.org/10.1073/pnas.1101221108
Brown JH, Kim KH, Jun G, Greenfield NJ, Dominguez R, Volkmann N, Hitchcock-DeGregori SE, Cohen C (2001) Deciphering the design of the tropomyosin molecule. Proc Natl Acad Sci U S A 98:8496–8501. https://doi.org/10.1073/pnas.131219198
Campbell N, Sinagra G, Jones KL, Slavov D, Gowan K, Merlo M, Carniel E, Fain PR, Aragona P, Di Lenarda A, Mestroni L, Taylor MRG (2013) Whole exome sequencing identifies a troponin T mutation hot spot in familial dilated cardiomyopathy. PLoS One 8:e78104. https://doi.org/10.1371/journal.pone.0078104
Chang AN, Greenfield NJ, Singh A, Potter JD, Pinto JR (2014) Structural and protein interaction effects of hypertrophic and dilated cardiomyopathic mutations in alpha-tropomyosin. Front Physiol 5:460. https://doi.org/10.3389/fphys.2014.00460
Cheng Y, Lindert S, Kekenes-Huskey P, Rao Vijay S, Solaro RJ, Rosevear Paul R, Amaro R, McCulloch Andrew D, McCammon JA, Regnier M (2014) Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys J 107:1675–1685. https://doi.org/10.1016/j.bpj.2014.08.008
Cho YJ, Liu J, Hitchcock-DeGregori SE (1990) The amino terminus of muscle tropomyosin is a major determinant for function. J Biol Chem 265:538–545
Coats CJ, Pavlou M, Watkinson OT, Protonotarios A, Moss L, Hyland R, Rantell K, Pantazis AA, Tome M, McKenna WJ, Frenneaux MP, Omar R, Elliott PM (2019) Effect of trimetazidine dihydrochloride therapy on exercise capacity in patients with nonobstructive hypertrophic cardiomyopathy: a randomized clinical trial. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2018.4847
Cooke R (1997) Actomyosin interaction in striated muscle. Physiol Rev 77:671–697
Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, Mugelli A, Poggesi C, Tardiff J, Olivotto I (2014) Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol 64:2589–2600. https://doi.org/10.1016/j.jacc.2014.09.059
Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H (2003) Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 41:1776–1782
Day SM (2019) Nonobstructive hypertrophic cardiomyopathy-the high-hanging fruit. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2018.4953
Dewan S, McCabe KJ, Regnier M, McCulloch AD (2017) Insights and challenges of multi-scale modeling of sarcomere mechanics in cTn and tm DCM mutants—genotype to cellular phenotype. Front Physiol 8:151. https://doi.org/10.3389/fphys.2017.00151
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H, O'Mahony C, Zamorano JL, Achenbach S, Baumgartner H, Bax JJ, Bueno H, Dean V, Deaton C, Erol Ç, Fagard R, Ferrari R, Hasdai D, Hoes AW, Kirchhof P, Knuuti J, Kolh P, Lancellotti P, Linhart A, Nihoyannopoulos P, Piepoli MF, Ponikowski P, Sirnes PA, Tamargo JL, Tendera M, Torbicki A, Wijns W, Windecker S, Hasdai D, Ponikowski P, Achenbach S, Alfonso F, Basso C, Cardim NM, Gimeno JR, Heymans S, Holm PJ, Keren A, Kirchhof P, Kolh P, Lionis C, Muneretto C, Priori S, Salvador MJ, Wolpert C, Zamorano JL, Frick M, Aliyev F, Komissarova S, Mairesse G, Smajić E, Velchev V, Antoniades L, Linhart A, Bundgaard H, Heliö T, Leenhardt A, Katus HA, Efthymiadis G, Sepp R, Thor Gunnarsson G, Carasso S, Kerimkulova A, Kamzola G, Skouri H, Eldirsi G, Kavoliuniene A, Felice T, Michels M, Hermann Haugaa K, Lenarczyk R, Brito D, Apetrei E, Bokheria L, Lovic D, Hatala R, Garcia Pavía P, Eriksson M, Noble S, Srbinovska E, Özdemir M, Nesukay E, Sekhri N (2014) 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy. The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 35:2733–2779. https://doi.org/10.1093/eurheartj/ehu284
Ertz-Berger BR, He H, Dowell C, Factor SM, Haim TE, Nunez S, Schwartz SD, Ingwall JS, Tardiff JC (2005) Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc Natl Acad Sci U S A 102:18219–18224. https://doi.org/10.1073/pnas.0509181102
Ferrantini C, Coppini R, Pioner JM, Gentile F, Tosi B, Mazzoni L, Scellini B, Piroddi N, Laurino A, Santini L, Spinelli V, Sacconi L, De Tombe P, Moore R, Tardiff J, Mugelli A, Olivotto I, Cerbai E, Tesi C, Poggesi C (2017) Pathogenesis of hypertrophic cardiomyopathy is mutation rather than disease specific: a comparison of the cardiac troponin T E163R and R92Q mouse models. J Am Heart Assoc 6. https://doi.org/10.1161/JAHA.116.005407
Flicker PF, Phillips GN, Cohen C (1982) Troponin and its interactions with tropomyosin: an electron microscope study. J Mol Biol 162:495–501. https://doi.org/10.1016/0022-2836(82)90540-X
Forissier JF, Carrier L, Farza H, Bonne G, Bercovici J, Richard P, Hainque B, Townsend PJ, Yacoub MH, Fauré S, Dubourg O, Millaire A, Hagège A, Desnos M, Komajda M, Schwartz K (1996) Codon 102 of the cardiac troponin T gene is a putative hot spot for mutations in familial hypertrophic cardiomyopathy. Circulation 94. https://doi.org/10.1161/01.CIR.94.12.3069
Garfinkel AC, Seidman JG, Seidman CE (2018) Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail Clin 14:139–146. https://doi.org/10.1016/j.hfc.2017.12.004
Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW (2011) 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association task Force on Practice Guidelines. Circulation 124:e783–e831. https://doi.org/10.1161/CIR.0b013e318223e2bd
Gordon AM, Homsher E, Regnier M (2000) Regulation of contraction in striated muscle. Physiol Rev 80:853–924
Gordon AM, Regnier M, Homsher E (2001) Skeletal and cardiac muscle contractile activation: tropomyosin “rocks and rolls”. Physiology 16:49–55
Greenfield NJ, Huang YJ, Swapna GVT, Bhattacharya A, Rapp B, Singh A, Montelione GT, Hitchcock-DeGregori SE (2006) Solution NMR structure of the junction between tropomyosin molecules: implications for actin binding and regulation. J Mol Biol 364:80–96. https://doi.org/10.1016/j.jmb.2006.08.033
Guinto PJ, Manning EP, Schwartz SD, Tardiff JC (2007) Computational characterization of mutations in cardiac troponin T known to cause familial hypertrophic cardiomyopathy. J Theor Comput Chem 6:413–419. https://doi.org/10.1142/s0219633607003271
Gupte TM, Haque F, Gangadharan B, Sunitha MS, Mukherjee S, Anandhan S, Rani DS, Mukundan N, Jambekar A, Thangaraj K, Sowdhamini R, Sommese RF, Nag S, Spudich JA, Mercer JA (2015) Mechanistic heterogeneity in contractile properties of alpha-tropomyosin (TPM1) mutants associated with inherited cardiomyopathies. J Biol Chem 290:7003–7015. https://doi.org/10.1074/jbc.M114.596676
Harada K, Takahashi-Yanaga F, Minakami R, Morimoto S, Ohtsuki I (2000) Functional consequences of the deletion mutation ΔlGlul6O in human cardiac troponin T. J Biochem 127:263–268
He H, Javadpour MM, Latif F, Tardiff JC, Ingwall JS (2007) R-92L and R-92W mutations in cardiac troponin T lead to distinct energetic phenotypes in intact mouse hearts. Biophys J 93:1834–1844. https://doi.org/10.1529/biophysj.107.107557
Heller MJ, Nili M, Homsher E, Tobacman LS (2003) Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. J Biol Chem 278:41742–41748. https://doi.org/10.1074/jbc.M303408200
Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS, Potter JD (2009) Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2:306–313. https://doi.org/10.1161/circgenetics.108.846733
Hinkle A, Tobacman LS (2003) Folding and function of the troponin tail domain: effects of cardiomyopathic troponin t mutations. J Biol Chem 278:506–513. https://doi.org/10.1074/jbc.M209194200
Hitchcock-DeGregori SE (2008) Tropomyosin: function follows structure. In: Gunning P (ed) Tropomyosin. Springer New York, New York, pp 60–72. https://doi.org/10.1007/978-0-387-85766-4_5
Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE (2000) Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation 102:1950–1955
Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y (2015) Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res 105:397–408. https://doi.org/10.1093/cvr/cvv025
Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, Kwong RY, Antman EM, Semsarian C, Gonzalez A, Lopez B, Diez J, Orav EJ, Colan SD, Seidman CE (2015) Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail 3:180–188. https://doi.org/10.1016/j.jchf.2014.08.003
Ho CY, Day SM, Colan SD, Russell MW, Towbin JA, Sherrid MV, Canter CE, Jefferies JL, Murphy AM, Cirino AL, Abraham TP, Taylor M, Mestroni L, Bluemke DA, Jarolim P, Shi L, Sleeper LA, Seidman CE, Orav EJ, Investigators HC (2017) The burden of early phenotypes and the influence of wall thickness in hypertrophic cardiomyopathy mutation carriers: findings from the HCMNet study. JAMA Cardiol 2:419–428. https://doi.org/10.1001/jamacardio.2016.5670
Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I (2018) Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 138:1387–1398. https://doi.org/10.1161/CIRCULATIONAHA.117.033200
Holmes KC, Lehman W (2008) Gestalt-binding of tropomyosin to actin filaments. J Muscle Res Cell Motil 29:213–219. https://doi.org/10.1007/s10974-008-9157-6
Janco M, Kalyva A, Scellini B, Piroddi N, Tesi C, Poggesi C, Geeves MA (2012) α-Tropomyosin with a D175N or E180G mutation in only one chain differs from tropomyosin with mutations in both chains. Biochemistry 51:9880–9890. https://doi.org/10.1021/bi301323n
Javadpour MM, Tardiff JC, Pinz I, Ingwall JS (2003) Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. J Clin Invest 112:768–775. https://doi.org/10.1172/JCI15967
Jiang J, Wakimoto H, Seidman JG, Seidman CE (2013) Allele-specific silencing of mutant Myh6 allele in mice suppresses hypertrophic cardiomyopathy. Science (New York, NY) 342:111–114. https://doi.org/10.1126/science.1236921
Kobayashi T, Solaro RJ (2005) Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol 67:39–67. https://doi.org/10.1146/annurev.physiol.67.040403.114025
Kobayashi T, Jin L, de Tombe PP (2008) Cardiac thin filament regulation. Pflugers Arch - Eur J Physiol 457:37–46. https://doi.org/10.1007/s00424-008-0511-8
Kokado H, Shimizu M, Yoshio H, Ino H, Okeie K, Emoto Y, Matsuyama T, Yamaguchi M, Yasuda T, Fujino N, Ito H, D H M (2000) Clinical features of hypertrophic cardiomyopathy caused by a Lys183 deletion mutation in the cardiac troponin I Gene. Circulation 102. https://doi.org/10.1161/01.CIR.102.6.663
Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves MA, Levitsky DI (2004) Effects of two familial hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys J 87:3922–3933. https://doi.org/10.1529/biophysj.104.048793
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. https://doi.org/10.1038/nprot.2009.86
Lakdawala NK, Thune JJ, Colan SD, Cirino AL, Farrohi F, Rivero J, McDonough B, Sparks E, Orav EJ, Seidman JG, Seidman CE, Ho CY (2012) Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ Cardiovasc Genet 5:503–510. https://doi.org/10.1161/CIRCGENETICS.112.962761
Li MX, Wang X, Sykes BD (2004) Structural based insights into the role of troponin in cardiac muscle pathophysiology. J Muscle Res Cell Motil 25:559–579. https://doi.org/10.1007/s10974-004-5879-2
Li XE, Orzechowski M, Lehman W, Fischer S (2014) Structure and flexibility of the tropomyosin overlap junction. Biochem Biophys Res Commun 446:304–308. https://doi.org/10.1016/j.bbrc.2014.02.097
Lindert S, Cheng Y, Kekenes-Huskey P, Regnier M, McCammon JA (2015) Effects of HCM cTnI mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys J 108:395–407. https://doi.org/10.1016/j.bpj.2014.11.3461
Loong CKP, Zhou H-X, Bryant Chase P (2012) Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac α-tropomyosin. FEBS Lett 586:3503–3507. https://doi.org/10.1016/j.febslet.2012.08.005
Lu Q-W, Morimoto S, Harada K, Du C-K, Takahashi-Yanaga F, Miwa Y, Sasaguri T, Ohtsuki I (2003) Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T–tropomyosin interaction and causes a Ca2+ desensitization. J Mol Cell Cardiol 35:1421–1427. https://doi.org/10.1016/j.yjmcc.2003.09.003
Ly S, Lehrer SS (2012) Long-range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry 51:6413–6420. https://doi.org/10.1021/bi3006835
Lynn ML, Tal Grinspan L, Holeman TA, Jimenez J, Strom J, Tardiff JC (2017) The structural basis of alpha-tropomyosin linked (Asp230Asn) familial dilated cardiomyopathy. J Mol Cell Cardiol 108:127–137. https://doi.org/10.1016/j.yjmcc.2017.06.001
Manning EP, Tardiff JC, Schwartz SD (2011) A model of calcium activation of the cardiac thin filament. Biochemistry 50:7405–7413. https://doi.org/10.1021/bi200506k
Manning EP, Tardiff JC, Schwartz SD (2012) Molecular effects of familial hypertrophic cardiomyopathy-related mutations in the TNT1 domain of cTnT. J Mol Biol 421:54–66. https://doi.org/10.1016/j.jmb.2012.05.008
McConnell M, Tal Grinspan L, Williams MR, Lynn ML, Schwartz BA, Fass OZ, Schwartz SD, Tardiff JC (2017) Clinically divergent mutation effects on the structure and function of the human cardiac tropomyosin overlap. Biochemistry 56:3403–3413. https://doi.org/10.1021/acs.biochem.7b00266
McKillop DF, Geeves MA (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J 65:693–701
Menon S, Michels V, Pellikka P, Ballew J, Karst M, Herron K, Nelson S, Rodeheffer R, Olson T (2008) Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet 74:445–454. https://doi.org/10.1111/j.1399-0004.2008.01062.x
Merlo M, Sinagra G, Carniel E, Slavov D, Zhu X, Barbati G, Spezzacatene A, Ramani F, Salcedo E, Di Lenarda A, Mestroni L, Taylor MRG, on behalf of the Familial Cardiomyopathy R (2013) Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci 6:424–428. https://doi.org/10.1111/cts.12116
Messer AE, Bayliss CR, El-Mezgueldi M, Redwood CS, Ward DG, Leung M-C, Papadaki M, dos Remedios C, Marston SB (2016) Mutations in troponin T associated with hypertrophic cardiomyopathy increase Ca2+−sensitivity and suppress the modulation of Ca2+−sensitivity by troponin I phosphorylation. Arch Biochem Biophys 601:113–120. https://doi.org/10.1016/j.abb.2016.03.027
Moore JR, Li X, Nirody J, Fischer S, Lehman W (2011) Structural implications of conserved aspartate residues located in tropomyosin’s coiled-coil core. Bioarchitecture 1:250–255. https://doi.org/10.4161/bioa.18117
Moore RK, Grinspan LT, Jimenez J, Guinto PJ, Ertz-Berger B, Tardiff JC (2013) HCM-linked 160E cardiac troponin T mutation causes unique progressive structural and molecular ventricular remodeling in transgenic mice. J Mol Cell Cardiol 58:188–198. https://doi.org/10.1016/j.yjmcc.2013.02.004
Moore RK, Abdullah S, Tardiff JC (2014) Allosteric effects of cardiac troponin TNT1 mutations on actomyosin binding: a novel pathogenic mechanism for hypertrophic cardiomyopathy. Arch Biochem Biophys 552-553:21–28. https://doi.org/10.1016/j.abb.2014.01.016
Moore JR, Campbell SG, Lehman W (2016) Structural determinants of muscle thin filament cooperativity. Arch Biochem Biophys 594:8–17. https://doi.org/10.1016/j.abb.2016.02.016
Morimoto S (2007) Molecular pathogenic mechanisms of cardiomyopathies caused by mutations in cardiac troponin T. Adv Exp Med Biol 592:227–239. https://doi.org/10.1007/978-4-431-38453-3_19
Morimoto S, Lu Q-W, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M, Sasaguri T, Ohtsuki I (2002) Ca2+−desensitizing effect of a deletion mutation ΔK210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci 99:913–918. https://doi.org/10.1073/pnas.022628899
Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, Shirakihara Y, Wakatsuki S, Yasunaga T, Wakabayashi T (2008) Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc Natl Acad Sci U S A 105:7200–7205. https://doi.org/10.1073/pnas.0801950105
Olson TM, Kishimoto NY, Whitby FG, Michels VV (2001) Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol 33:723–732. https://doi.org/10.1006/jmcc.2000.1339
Orzechowski M, Moore JR, Fischer S, Lehman W (2014) Tropomyosin movement on F-actin during muscle activation explained by energy landscapes. Arch Biochem Biophys 545:63–68. https://doi.org/10.1016/j.abb.2014.01.001
Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ (2001) Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys J 81:2827–2837. https://doi.org/10.1016/s0006-3495(01)75924-3
Palm T, Greenfield NJ, Hitchcock-DeGregori SE (2003) Tropomyosin ends determine the stability and functionality of overlap and troponin T complexes. Biophys J 84:3181–3189. https://doi.org/10.1016/s0006-3495(03)70042-3
Phillips GN, Fillers JP, Cohen C (1986) Tropomyosin crystal structure and muscle regulation. J Mol Biol 192:111–127. https://doi.org/10.1016/0022-2836(86)90468-7
Pirani A, Vinogradova MV, Curmi PM, King WA, Fletterick RJ, Craig R, Tobacman LS, Xu C, Hatch V, Lehman W (2006) An atomic model of the thin filament in the relaxed and Ca2+−activated states. J Mol Biol 357:707–717. https://doi.org/10.1016/j.jmb.2005.12.050
Richardson P, Mckenna W, Bristow M, Maisch B, Mautner B, O’Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 93:841–842. https://doi.org/10.1161/01.CIR.93.5.841
Schwartz K, Mercadier J-J (2003) Cardiac troponin T and familial hypertrophic cardiomyopathy: an energetic affair. J Clin Investig 112:652–654. https://doi.org/10.1172/JCI200319632
Sommese RF, Nag S, Sutton S, Miller SM, Spudich JA, Ruppel KM (2013) Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human β-cardiac myosin. PLoS One 8:e83403. https://doi.org/10.1371/journal.pone.0083403
Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS (1998) Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest 101:1775–1783. https://doi.org/10.1172/JCI1940
Stefanelli CB, Rosenthal A, Borisov AB, Ensing GJ, Russell MW (2004) Novel troponin T mutation in familial dilated cardiomyopathy with gender-dependant severity. Mol Genet Metab 83:188–196. https://doi.org/10.1016/j.ymgme.2004.04.013
Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC (2012) Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med 4:130ra147. https://doi.org/10.1126/scitranslmed.3003552
Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD (2000) Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. J Biol Chem 275:624–630. https://doi.org/10.1074/jbc.275.1.624
Takano M, Terada TP, Sasai M (2010) Unidirectional Brownian motion observed in an in silico single molecule experiment of an actomyosin motor. Proc Natl Acad Sci U S A 107:7769–7774. https://doi.org/10.1073/pnas.0911830107
Takeda S, Yamashita A, Maeda K, Maeda Y (2003) Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature 424:35–41. https://doi.org/10.1038/nature01780
Tardiff JC (2011) Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res 108:765–782. https://doi.org/10.1161/CIRCRESAHA.110.224170
Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg H-P, Seldman JG, Seidman CE (1994) α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77:701–712. https://doi.org/10.1016/0092-8674(94)90054-X
Tobacman LS, Lin D, Butters C, Landis C, Back N, Pavlov D, Homsher E (1999) Functional consequences of troponin T mutations found in hypertrophic cardiomyopathy. J Biol Chem 274:28363–28370. https://doi.org/10.1074/jbc.274.40.28363
Van Acker H, De Sutter J, Vandekerckhove K, de Ravel TJ, Verhaaren H, De Backer J (2010) Dilated cardiomyopathy caused by a novel TNNT2 mutation-added value of genetic testing in the correct identification of affected subjects. Int J Cardiol 144:307–309. https://doi.org/10.1016/j.ijcard.2009.03.003
Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE (1995) Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 332:1058–1065. https://doi.org/10.1056/nejm199504203321603
Wei B, Jin JP (2011) Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch Biochem Biophys 505:144–154. https://doi.org/10.1016/j.abb.2010.10.013
Williams MR, Lehman SJ, Tardiff JC, Schwartz SD (2016) Atomic resolution probe for allostery in the regulatory thin filament. Proc Natl Acad Sci U S A 113:3257–3262. https://doi.org/10.1073/pnas.1519541113
Yang S, Barbu-Tudoran L, Orzechowski M, Craig R, Trinick J, White H, Lehman W (2014) Three-dimensional organization of troponin on cardiac muscle thin filaments in the relaxed state. Biophys J 106:855–864. https://doi.org/10.1016/j.bpj.2014.01.007
Yiu KH, Atsma DE, Delgado V, Ng ACT, Witkowski TG, Ewe SH, Auger D, Holman ER, van Mil AM, Breuning MH, Tse HF, Bax JJ, Schalij MJ, Marsan NA (2012) Myocardial structural alteration and systolic dysfunction in preclinical hypertrophic cardiomyopathy mutation carriers. PLoS One 7:e36115. https://doi.org/10.1371/journal.pone.0036115
Acknowledgements
The authors would like to thank Anthony Baldo for assistance with figures.
Funding
This work was supported by NHLBI Training Grant 5T32HL007955-19 to A.E. Deranek and T32-HL07249 to M.M. Klass. Additional funding sources include NIH HL HL075619 and HL107046 and the Steven M. Gootter Foundation to J.C. Tardiff.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Sarcomeric Mutations in Pflügers Archiv—European Journal of Physiology
Rights and permissions
About this article

Cite this article
Deranek, A.E., Klass, M.M. & Tardiff, J.C. Moving beyond simple answers to complex disorders in sarcomeric cardiomyopathies: the role of integrated systems. Pflugers Arch - Eur J Physiol 471, 661–671 (2019). https://doi.org/10.1007/s00424-019-02269-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-019-02269-0