
Abstract
Angiotensin (Ang)-(1-7) ameliorates vascular injury by increasing nitric oxide (NO) bioavailability. Evidence that Ang-(1-7) attenuates the development of atherosclerosis through a NO-dependent mechanism is still missing. Moreover, it has been postulated that Ang-(1-7) may mediate its effects by other mechanisms than Mas receptor activation. To investigate Ang-(1-7)-dependent Mas receptor function, we treated apoE-KO and apoE/Mas-KO mice chronically with Ang-(1-7) (82 μg/kg per hour) or saline for 6 weeks. Flow-mediated dilation (FMD), a measure for NO-dependent vasodilation and the most accepted prognostic marker for the development of atherosclerosis, was measured in vivo. Chronic Ang-(1-7) treatment improved FMD and attenuated the development of atherosclerosis in apolipoproteinE (apoE)-KO but not in apoE/Mas-KO mice. These effects were accompanied by increased aortic nitrite and cGMP levels. To test whether Ang-(1-7) modulates atherosclerosis through a NO-dependent mechanism, apoE-KO mice were treated with the NO synthase inhibitor L-NAME (20 mg/kg/day) in the presence or absence of Ang-(1-7). L-NAME treatment reduced aortic nitrite content and increased blood pressure and exaggerated atherosclerosis compared to untreated apoE-KO mice. In L-NAME-treated apoE-KO mice, chronic Ang-(1-7) treatment did not increase aortic nitrite content and consequently showed no effect on blood pressure and the development of atherosclerosis. The present study proves that Ang-(1-7) mediates its protective vascular effects through Mas receptor activation. Moreover, Ang-(1-7)-mediated NO generation is essential for improving vascular function and prevents atherosclerosis in apoE-KO mice.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is one of the leading causes for cardiovascular mortality worldwide. Endothelial dysfunction, characterized by decreased nitric oxide (NO) bioavailability, is considered as a key event in the early stage of atherosclerosis. Animal and human studies provide profound evidence that activation of the angiotensin (Ang) II type 1 receptor by Ang II exaggerates vascular injury by stimulating the recruitment of immune cells and vascular smooth muscle cell proliferation as well as impairing endothelial dysfunction [9]. In contrast, Ang-(1-7), recently recognized as another active metabolite of the renin angiotensin system which is cleaved from Ang I and Ang II by several endopeptidases and metalloproteinases like neprilysin and angiotensin-converting enzyme 2 (ACE2), has been shown to counter-regulate the pathophysiological effects of Ang II in cardiovascular diseases by activating its own receptor Mas [4, 26]. Thus, chronic Ang-(1-7) infusion improved vascular function and attenuated the development of atherosclerosis by increasing NO bioavailability, reducing vascular inflammation, immune cell response, and VSMC proliferation [6, 10, 16,17,18, 28]. Accordingly, deletion of the Mas receptor leads to endothelial dysfunction and vascular inflammation [25]. Although many studies have indicated a beneficial effect of Ang-(1-7) on vascular function, it is still unknown whether the Ang-(1-7)-mediated increase in NO bioavailability affects directly the development of atherosclerosis. Moreover, recent studies have indicated that Ang-(1-7) mediates its effects not solely via the Mas receptor but also via the Ang II type 2 receptor [18, 22]. To investigate this mechanism and to clarify the role of Mas as natural receptor of Ang-(1-7) in the development of atherosclerosis, we generated apolipoproteinE/Mas receptor double-deficient (apoE/Mas-KO) mice and fed high-fat, cholesterol-enriched western-type diet (WD) for 12 weeks.
Materials and methods
Animals
All animal experimental investigations were in accordance with the federal state authority (Landesamt fuer Natur-, Umwelt- und Verbraucherschutz Nordrhein Westfalen; reference: AZ. 8.87-50.10.34.08.216 and AZ 84–02.04.2012.A250) and performed according to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Mice were inspected daily for living environment and animation. The apoE-KO and apoE/Mas-KO mice were obtained from an in-house breed at the local animal care facility at Heinrich-Heine-Universität Düsseldorf. All mice were on a C57Bl/6 background. Littermates were used as controls. The animals were housed at 20 to 22 °C temperature and a 12-h day and night cycle with free access to water and food.
Diet and drug administration
Six-week-old apoE-KO and apoE/Mas-KO mice on a C57BL/6 background were fed WD food (Ssniff, Soest, Germany) (42% fat, 0.15% cholesterol) and allowed free access to tap water. After 6 weeks, mice were treated with either Ang-(1-7) (82 μg/kg/h) or saline via osmotic minipump (Alzet Model 1004) over 6 weeks as described previously [17]. The osmotic minipumps were replaced after 3 weeks.
To examine whether Ang-(1-7) attenuates the development of atherosclerosis through a NO-dependent mechanism, apoE-KO mice were treated in the presence or absence of chronic Ang-(1-7) infusion with the NO synthase inhibitor L-NAME (20 mg/kg/d) adapted after a protocol of Krowles et al. [7]. In detail, 8-week-old apoE-KO mice were divided into three groups. Mice were fed WD or WD + L-NAME (20 mg/kg/d) or WD + L-NAME + Ang-(1-7) (82 μg/kg/h) via an osmotic minipump (Alzet Model 1004) for additional 8 weeks. L-NAME was dissolved in water, and water was refreshed twice per week.
Flow-mediated dilation
Flow-mediated dilation (FMD) was measured on the day before termination. The details were described previously [15]. Mice were anesthetized with 2 vol% isoflurane. Body temperature was kept at 37 °C by using a heated examination table that was also equipped with ECG electrodes. Images of the femoral arteries (FA) in mice were obtained using the Visual Sonics imaging platform Vevo 2100 and a 30–70 MHz linear array micro scan transducer (Visual Sonics). Using duplex ultrasound mode, the artery and the arterial blood flow are identified by pulsed-wave Doppler (PWD). During FMD assessment, a vascular occluder was placed at the left limb to induce occlusion of the distal hind limb as an ischemic trigger. Inflation occlusion (5 min) was carried out manually. Following hind limb ischemia, the cuff was deflated and femoral artery diameter measurements were continuously recorded for 3 min at 30 s intervals. The heart rate and FA blood flow velocity were monitored by PWD. Quantification of the arterial lumen diameter was performed from recorded videos using an automated edge detection software (Brachial Analyzer, MIA, Iowa City) [15]. Percentage of FMD was calculated using the following formula: [(diameter post-ischemic – diameter baseline)/diameter baseline] × 100.
Measurement of vascular function in aortic rings
Endothelium-dependent vasorelaxation was measured in aortic rings from apoE-KO and apoE/Mas-KO mice treated either with Ang-(1-7) or saline. Aortic rings were mounted in a wire myography (Multi Myograph Model 610 M, Danish Myo Technology, Denmark) as described previously [3]. After equilibration, aortic rings were contracted with norepinephrine (1 μM; Sigma Aldrich) and dose–response curves to carbachol were recorded. Isometric forces are expressed as a percentage of the maximal response to norepinephrine.
Oil Red O staining and quantification
To quantify the extent of atherosclerotic lesions, the aortic arch was dissected from the mice as described previously [6]. The aortas were then incubated in 4% paraformaldehyde overnight. After removal of adventitia, tissues were stained with Oil Red O solution (ORO; 5 g ORO powder in 1 L methanol) histopathology staining method. Cleaned aortas were pretreated in 78% methanol for 5 min and then transferred to the fresh NaOH-ORO solution (1 ml NaOH: 3.5 ml ORO) for 90 min. After ORO staining, aortas were incubated in 78% methanol for 15 min and preserved in 1X PBS. To obtain photographs, aortas were pinned and photographed under a microscope and digital camera. The photos were captured by a camera (Coopix 4500, Nikon, Tokyo, Japan) with Carl Zeiss lens (426126 Carl Zeiss, Jena, Germany) under a Leica microscope (Leica MZ6, Wetzlar, Germany). Fiji ImageJ software (Fiji Version 2.0.0) was used to measure the atherosclerotic lesions.
Aortic cGMP measurement
Immediately after termination of the experiment, aortic slices were cut and snap-frozen in liquid nitrogen. To extract cGMP, samples were homogenized in 70% (v/v) ice-cold ethanol using a glass/glass homogenizer and then centrifuged (14000×g, 15 min, 4 °C). Supernatants were dried at 95 °C and the cGMP content was measured by RIA. To standardize the different samples, protein pellets were dissolved in 0.1 M NaOH/0.1% sodium dodecyl sulfate (SDS) and protein content was determined using the bicinchoninic acid method (Uptima).
Blood pressure measurement
Systolic blood pressure (SBP) was measured in conscious mice by tail-cuff method (BP-98A; Softron Co.) as described previously [20]. For habituation, mice were trained daily for 5 consecutive days prior to the experiment. Thereafter, ten measurements per mouse were recorded every day over a period of 8 weeks.
Determination of aortic nitrite levels
The descending aorta was excised, cleaned from adipose tissue, and immediately snap-frozen. Samples were weighed and homogenized in ice-cold sodium chloride (0.9%). Nitrite concentration in aortic tissue homogenates was assessed as previously described using HPLC (ENO20, Eicom, Dublin, Ireland) [21].
Statistical analysis
Data have been expressed as mean ± SEM (n = number of animals or samples). Version 5.0 of GraphPad Prism (San Diego, USA) was used for data analysis. The values for each parameter within a group are expressed as mean ± SEM. All data were analyzed by either Student’s t test or two-way ANOVA, followed by Bonferroni’s multiple comparison post-hoc test, or Mann–Whitney U test after testing for normal distribution. Results were considered statistically significant for P < 0.05.
Results
Ang-(1-7) improved endothelial-dependent vasorelaxation via Mas receptor activation
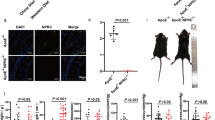
Six-week-old male apoE-KO and apoE/Mas-KO mice were fed WD for 12 weeks. After 6 weeks, apoE-KO and apoE/Mas-KO mice were treated either with Ang-(1-7) (82 μg/kg per hour) or with saline via osmotic minipumps for additional 6 weeks. To investigate the role of Ang-(1-7)-induced Mas receptor activation on endothelial-dependent vasodilation in apoE-KO and apoE/Mas-KO mice, vascular function was measured by FMD in vivo and ex vivo in aortic wire myograph. Chronic Ang-(1-7) infusion significantly increased FMD (FMD 17.5 ± 0.6 vs. 8.7 ± 0.4%, n = 5–11) and improved endothelial-dependent vasorelaxation in aortic rings of apoE-KO but not of apoE/Mas-KO mice (FMD 9.0 ± 1.2 vs. 8.0 ± 0.7%, n = 5; aortic rings) (Fig. 1a, b). This improvement was caused by an increase in Ang-(1-7)-mediated vascular NO bioavailability as nitrite (3.5 ± 0.4 vs. 2.3 ± 0.4 μM, n = 7–8) and cGMP content (3.0 ± 0.4 vs. 1.4 ± 0.2 pmol/l, n = 7–8); both markers for NO generation were significantly enhanced in aortas of Ang-(1-7)-treated apoE-KO mice (Fig. 2a, b).

Ang-(1-7) improves endothelial-dependent vasorelaxation through Mas receptor activation. a Ang-(1-7) increased flow-mediated dilation (FMD) in apoE-KO but not in apoE/Mas-KO. *P < 0.05, **P < 0.01, ***P < 0.001. Results are represented as mean ± SEM. b Ang-(1-7) improved endothelial-dependent vasorelaxation to carbachol in aortic rings of apoE-KO but not of apoE/Mas-KO mice. ApoE-KO (n = 11) vs. apoE/Mas-KO (n = 11), *P < 0.05, **P < 0.01, ***P < 0.001; apoE-KO (n = 11) vs. apoE-KO vs. apoE-KO[Ang-(1-7)] (N = 6), #P < 0.05; apoE-KO vs. apoE/mas-KO[Ang-(1-7)] + P < 0.05, ++P < 0.01; apoE/Mas-KO vs. apoE/mas-KO[Ang-(1-7)], P = not significant. Results are represented as mean ± SEM

Ang-(1-7) improves NO bioavailability in apoE-KO mice. a, b Chronic Ang-(1-7) treatment increased aortic nitrite concentration and cGMP levels in apoE-KO. *P < 0.05, **P < 0.01. Results are represented as mean ± SEM
Chronic Ang-(1-7) treatment attenuates the development of atherosclerosis in apoE-KO but not in apoE/Mas-KO mice
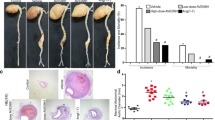
To investigate whether improved endothelial vascular function was also associated with reduced atherosclerosis, we analyzed atherosclerotic lesions in the aortic arch. Accordingly, Ang-(1-7) reduced atherosclerosis in apoE-KO (relative lesion area 11.2 ± 1.0 vs. 24.3 ± 2.1%; specific lesion area 2.9 ± 0.3 vs. 6.6 ± 0.8 mm2, n = 6–10) but not in apoE/Mas-KO mice (relative lesion area 37.0 ± 4.5 vs. 37.0 ± 2.9%; specific lesion area 12.3 ± 1.5 vs. 11.5 ± 1.1 mm2, n = 9) (Fig. 3). These results demonstrated that Ang-(1-7) mediates its protective vascular effects solely through the Mas receptor activation.

Ang-(1-7) attenuates the development of atherosclerosis through Mas receptor activation. Ang-(1-7) decreased the relative and specific atherosclerotic lesion area in apoE-KO but not in apoE/Mas-KO mice. Representative pictures of Oil Red O-stained aortic arches are shown. The distance between the digital calipers is 7 mm. **P < 0.01, ***P < 0.001. Results are represented as mean ± SEM
Ang-(1-7) attenuated atherosclerosis through a nitric oxide-mediated mechanism
To examine whether Ang-(1-7) attenuates the development of atherosclerosis through a NO-dependent mechanism, apoE-KO mice were treated in the presence or absence of chronic Ang-(1-7) infusion with the NO synthase inhibitor L-NAME (20 mg/kg/d) similar to a protocol of Krowles et al. [7]. As expected, chronic L-NAME treatment increased systolic blood pressure (apoE-KO 118 ± 2 mmHg vs. apoE-KO + L-NAME 125 ± 3 mmHg vs. apoE-KO + L-NAME + Ang-(1-7) 127 ± 2 mmHg, n = 7–9) and reduced aortic nitrite levels significantly (1.4 ± 0.2 vs. 0.8 ± 0.1, n = 6) (Fig. 4a, b). Ang-(1-7) infusion did not affect blood pressure and aortic nitrite levels in L-NAME-treated apoE-KO mice (0.8 ± 0.1 vs. 0.9 ± 0.1, n = 6). Moreover, chronic Ang-(1-7) infusion did not reduce atherosclerotic plaques in L-NAME-treated apoE-KO mice (relative lesion area 6.5 ± 1.4 vs. 11.8 ± 1.3 vs. 10.9 ± 1.2%; specific lesion area 1.5 ± 0.3 vs. 3.1 ± 0.4 vs. 3.1 ± 0.4 mm2, n = 7–9) demonstrating an essential contribution of NO on the beneficial effects of Ang-(1-7) in atherosclerosis (Fig. 4c).

Ang-(1-7) attenuates atherosclerosis through a NO-dependent mechanism. a, b L-NAME increased SBP and decreased aortic nitrite levels in apoE-KO. During L-NAME treatment, Ang-(1-7) failed to increase aortic nitrite levels. c Chronic L-NAME treatment accelerated atherosclerotic plaques. During L-NAME treatment, Ang-(1-7) failed to reduce atherosclerotic lesions. *P < 0.05. Results are represented as mean ± SEM
Discussion
In the present study, we show for the first time that Ang-(1-7) improves vascular function and attenuates the development of atherosclerosis via Mas receptor activation. Recently, it has been demonstrated that ACE2 deficiency, one of the key enzymes in Ang II degradation and an important member of the ACE2/Ang-(1–7)/Mas axis, causes vascular dysfunction and exaggerates the development of atherosclerosis [19]. However, since ACE2 deficiency also leads to an accumulation of the proatherogenic metabolite Ang II, the role of Ang-(1–7) and the Mas receptor in the regulation of these effects was still unclear [5]. Vascular function was measured in vivo by FMD, the most widely used and accepted prognostic marker for the development of atherosclerosis in humans and ex vivo by aortic ring myography [23]. And indeed, administration of Ang-(1-7) not only improved endothelial dysfunction, but also attenuated atherosclerosis in apoE-KO but not in apoE/Mas-KO mice. Although there are several reports suggesting non-specific actions of Ang-(1-7), here, we clearly demonstrate that Ang-(1-7) mediates its vasoprotective effects solely via Mas receptor activation [10, 17, 18, 27].
In humans, FMD is a widely accepted measure for NO bioavailability and NO-dependent vasodilation [12]. By using an additional ex vivo method for measuring NO-dependent vasodilation, we confirmed that the improvement observed in FMD by Ang-(1-7) reflects an improvement in NO-dependent endothelial vasorelaxation in Ang-(1-7)-treated apoE-KO mice. Moreover, we also show that Ang-(1-7) increased NO-dependent cGMP generation in aortas of apoE-KO mice underlining the important function of the Ang-(1-7)/Mas receptor axis in preventing vascular injury [17, 18]. In this regard, several studies have shown that Ang-(1-7)-mediated Mas receptor activation induces NO generation through a phosphoinositide 3 kinase (PI3K)/AKT-dependent mechanism leading to eNOS phosphorylation at serine 1177 [1, 2, 14, 17, 24]. In addition, it has been shown that Ang-(1-7) decreased NO degradation by reducing ROS production. Ang-(1-7) decreases the expression levels of NOX2 and p47phox and inhibits MAK kinase signaling leading to increased NO bioavailability and improved vascular function [8, 10, 13, 24, 28].
Based on the observation that reduced endothelial NO generation accelerated atherosclerosis, we tested the impact of Ang-(1-7)-mediated NO generation on the development of atherosclerosis [7]. Here, we demonstrate that Ang-(1-7) attenuates the development of atherosclerosis through an NO-dependent mechanism, as inhibition of NO synthase by L-NAME inhibits the beneficial effects of Ang-(1-7). These results are in accordance to previous studies showing that Mas receptor deletion accelerates endothelial dysfunction by reducing NO bioavailability [11, 25].
Taken together, the present study clearly demonstrates that Mas receptor-induced NO generation plays a key role in the improvement of vascular function and protects against the development of atherosclerosis. As recent studies have also shown a substantial effect of the Mas receptor on immune cell function and vascular inflammation, further studies need to evaluate whether these effects are also mediated through a NO-dependent mechanism [6].
References
Bader M (2013) ACE2, angiotensin-(1-7), and Mas: the other side of the coin. Pflugers Archiv : Eur J Physiol 465:79–85
Bader M, Alenina N, Andrade-Navarro MA, Santos RA (2014) MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol Rev 66:1080–1105. https://doi.org/10.1124/pr.113.008136
Broekmans K, Stegbauer J, Potthoff SA, Russwurm M, Koesling D, Mergia E (2016) Angiotensin II-induced hypertension is attenuated by reduction of sympathetic output in NO-sensitive guanylyl cyclase 1 knockout mice. J Pharmacol Exp Ther 356:191–199. https://doi.org/10.1124/jpet.115.227728
Domenig O, Manzel A, Grobe N, Konigshausen E, Kaltenecker CC, Kovarik JJ, Stegbauer J, Gurley SB, van Oyen D, Antlanger M, Bader M, Motta-Santos D, Santos RA, Elased KM, Saemann MD, Linker RA, Poglitsch M (2016) Neprilysin is a mediator of alternative renin-angiotensin-system activation in the murine and human kidney. Sci Rep 6:33678. https://doi.org/10.1038/srep33678
Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM (2006) Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest 116:2218–2225. https://doi.org/10.1172/JCI16980
Hammer A, Yang G, Friedrich J, Kovacs A, Lee DH, Grave K, Jorg S, Alenina N, Grosch J, Winkler J, Gold R, Bader M, Manzel A, Rump LC, Muller DN, Linker RA, Stegbauer J (2016) Role of the receptor Mas in macrophage-mediated inflammation in vivo. Proc Natl Acad Sci U S A 113:14109–14114. https://doi.org/10.1073/pnas.1612668113
Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N (2000) Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J Clin Invest 105:451–458. https://doi.org/10.1172/JCI8376
Liang B, Wang X, Zhang N, Yang H, Bai R, Liu M, Bian Y, Xiao C, Yang Z (2015) Angiotensin-(1-7) attenuates angiotensin II-induced ICAM-1, VCAM-1, and MCP-1 expression via the MAS receptor through suppression of P38 and NF-kappaB pathways in HUVECs. Cell Physiol Biochem: Int J Exp Cell Physiol Biochem Pharmacol 35:2472–2482. https://doi.org/10.1159/000374047
Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM (2014) Angiotensin II and vascular injury. Curr Hypertens Rep 16:431. https://doi.org/10.1007/s11906-014-0431-2
Potthoff SA, Fahling M, Clasen T, Mende S, Ishak B, Suvorava T, Stamer S, Thieme M, Sivritas SH, Kojda G, Patzak A, Rump LC, Stegbauer J (2014) Angiotensin-(1-7) modulates renal vascular resistance through inhibition of p38 mitogen-activated protein kinase in apolipoprotein E-deficient mice. Hypertension 63:265–272. https://doi.org/10.1161/HYPERTENSIONAHA.113.02289
Rabelo LA, Xu P, Todiras M, Sampaio WO, Buttgereit J, Bader M, Santos RA, Alenina N (2008) Ablation of angiotensin (1-7) receptor Mas in C57Bl/6 mice causes endothelial dysfunction. J Am Soc Hypertens: JASH 2:418–424. https://doi.org/10.1016/j.jash.2008.05.003
Rassaf T, Rammos C, Hendgen-Cotta UB, Heiss C, Kleophas W, Dellanna F, Floege J, Hetzel GR, Kelm M (2016) Vasculoprotective effects of dietary cocoa flavanols in patients on hemodialysis: a double-blind, randomized, placebo-controlled trial. Clin J Am Soc Nephrol : CJASN 11:108–118. https://doi.org/10.2215/CJN.05560515
Sampaio WO, Henrique de Castro C, Santos RA, Schiffrin EL, Touyz RM (2007) Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension 50:1093–1098. https://doi.org/10.1161/HYPERTENSIONAHA.106.084848
Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM (2007) Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 49:185–192. https://doi.org/10.1161/01.HYP.0000251865.35728.2f
Schuler D, Sansone R, Freudenberger T, Rodriguez-Mateos A, Weber G, Momma TY, Goy C, Altschmied J, Haendeler J, Fischer JW, Kelm M, Heiss C (2014) Measurement of endothelium-dependent vasodilation in mice—brief report. Arterioscler Thromb Vasc Biol 34:2651–2657. https://doi.org/10.1161/atvbaha.114.304699
Skiba DS, Nosalski R, Mikolajczyk TP, Siedlinski M, Rios FJ, Montezano AC, Jawien J, Olszanecki R, Korbut R, Czesnikiewicz-Guzik M, Touyz RM, Guzik TJ (2017) Anti-atherosclerotic effect of the angiotensin 1-7 mimetic AVE0991 is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br J Pharmacol 174:4055–4069. https://doi.org/10.1111/bph.13685
Stegbauer J, Potthoff SA, Quack I, Mergia E, Clasen T, Friedrich S, Vonend O, Woznowski M, Konigshausen E, Sellin L, Rump LC (2011) Chronic treatment with angiotensin-(1-7) improves renal endothelial dysfunction in apolipoproteinE-deficient mice. Br J Pharmacol 163:974–983. https://doi.org/10.1111/j.1476-5381.2011.01295.x
Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE (2010) Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 30:1606–1613. https://doi.org/10.1161/ATVBAHA.110.204453
Thatcher SE, Zhang X, Howatt DA, Lu H, Gurley SB, Daugherty A, Cassis LA (2011) Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor-/- mice. Arterioscler Thromb Vasc Biol 31:758–765. https://doi.org/10.1161/ATVBAHA.110.221614
Thieme M, Sivritas SH, Mergia E, Potthoff SA, Yang G, Hering L, Grave K, Hoch H, Rump LC, Stegbauer J (2017) Phosphodiesterase 5 inhibition ameliorates angiotensin II-dependent hypertension and renal vascular dysfunction. Am J Physiol Ren Physiol 312:F474–F481. https://doi.org/10.1152/ajprenal.00376.2016
Totzeck M, Hendgen-Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff HJ, Semmler D, Shiva S, Williams D, Kipar A, Gladwin MT, Schrader J, Kelm M, Cossins AR, Rassaf T (2012) Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 126:325–334. https://doi.org/10.1161/CIRCULATIONAHA.111.087155
Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, Unger T, Bader M, Santos RA, Sumners C, Steckelings UM (2015) Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin Sci 128:227–234. https://doi.org/10.1042/CS20130515
Widlansky ME, Gokce N, Keaney JF Jr, Vita JA (2003) The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42:1149–1160
Xiao X, Zhang C, Ma X, Miao H, Wang J, Liu L, Chen S, Zeng R, Chen Y, Bihl JC (2015) Angiotensin-(1-7) counteracts angiotensin II-induced dysfunction in cerebral endothelial cells via modulating Nox2/ROS and PI3K/NO pathways. Exp Cell Res 336:58–65. https://doi.org/10.1016/j.yexcr.2015.06.010
Xu P, Costa-Goncalves AC, Todiras M, Rabelo LA, Sampaio WO, Moura MM, Santos SS, Luft FC, Bader M, Gross V (2008) Endothelial dysfunction and elevated blood pressure in MAS gene-deleted mice. Hypertension 51:574–580
Yang G, Chu PL, Rump LC, Le TH, Stegbauer J (2017) ACE2 and the homolog collectrin in the modulation of nitric oxide and oxidative stress in blood pressure homeostasis and vascular injury. Antioxid Redox Signal. https://doi.org/10.1089/ars.2016.6950
Yang JM, Dong M, Meng X, Zhao YX, Yang XY, Liu XL, Hao PP, Li JJ, Wang XP, Zhang K, Gao F, Zhao XQ, Zhang MX, Zhang Y, Zhang C (2013) Angiotensin-(1-7) dose-dependently inhibits atherosclerotic lesion formation and enhances plaque stability by targeting vascular cells. Arterioscler Thromb Vasc Biol 33:1978–1985. https://doi.org/10.1161/ATVBAHA.113.301320
Zhang F, Ren X, Zhao M, Zhou B, Han Y (2016) Angiotensin-(1-7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci Rep 6:34621. https://doi.org/10.1038/srep34621
Acknowledgements
We thank Blanka Duvnjak and Christina Schwandt for their excellent technical assistance.
Funding
This work was supported by DFG (IRTG 1902) to G. Yang, M. Yakoub, J. Stegbauer, L.C. Rump, and M. Grandoch.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experimental investigations were in accordance with the federal state authority (Landesamt fuer Natur-, Umwelt- und Verbraucherschutz Nordrhein Westfalen; reference: AZ. 8.87-50.10.34.08.216 and AZ 84–02.04.2012.A250) and performed according to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Yang, G., Istas, G., Höges, S. et al. Angiotensin-(1-7)-induced Mas receptor activation attenuates atherosclerosis through a nitric oxide-dependent mechanism in apolipoproteinE-KO mice. Pflugers Arch - Eur J Physiol 470, 661–667 (2018). https://doi.org/10.1007/s00424-018-2108-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2108-1