
Abstract
Estrogen and β2-adrenergic receptors (β2AR) play important roles in the processes that protect the heart. Here, we investigated how ovariectomy influenced the β2AR downstream pathways in the context of catecholaminergic stress. In vivo and in vitro stress models were developed in female Sprague-Dawley (SD) rats by epinephrine (Epi) treatments. The cardiac function was evaluated at in vivo and in vitro levels in terms of contraction, rhythm, and injury. We found that myocardial contractility was not significantly different between Sham and ovariectomized (OVX) group rats in the normal state. However, Epi pretreatment decreased the contractility and increased abnormal rhythms especially in OVX group, which were attributed to lack of estrogen. Inhibition of the β2AR-Gi-PI3K/p38MAPK pathway with ICI118,551, PTX or LY294002 increased contractility and aggravated Epi-induced injury on cardiomyocytes, decreased p38MAPK phosphorylation, and only increased arrhythmia in Sham group. These results indicated that OVX exacerbated cardiac injury and abnormal rhythms through β2AR-Gi-PI3K and β2AR-Gi-p38MAPK pathways, respectively. In normal state, the levels of activated Gi were similar in both groups, but those of cAMP and activated Gs were higher in OVX group. Epi treatment increased activated Gi (especially in Sham group) and activated Gs and cAMP in Sham group but decreased it in OVX group. These results suggested that estrogen increased the Gi activity in normal and stress states and Gs activity in stress state. These results indicated that lack of estrogen impaired the β2AR-Gs/Gi coupling during stress which compromised cardiac contractility and increased abnormal rhythms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Premenopausal women have a lower incidence of cardiovascular disease compared with age-matched men. However, this phenomenon fades away after menopause [23]. It is postulated that the gradual decline of estrogen increases the risks of heart disorders, but the mechanism involved is unclear. In our previous studies, we demonstrated that lack of estrogen increased the expression of β1-adrenoceptor (β1AR) and decreased the expression of β2-adrenoceptor (β2AR) in ovariectomized rats [17, 35]. In addition, we demonstrated that β2AR plays a protective role on the ischemia/reperfused hearts or hypoxia-cultured rat cardiomyocytes [36].
Both β1AR and β2AR couple to the Gs-adenylyl cyclase (AC)-cAMP-protein kinase A (PKA) pathway, which not only increases cardiac contractility and relaxation, and oxygen consumption but also increases the susceptibility to myocardial injury. The cardiac effects of the β2AR-Gs-cAMP pathway could be mediated by the exchange protein directly activated by cAMP (Epac), whose cardiovascular importance has been demonstrated in recent years [11, 18]. Unlike β1AR, β2AR couples to the Gi-phosphoinositide 3-kinase (PI3K)-Akt pathway. Phosphorylation of β2AR by PKA increases its coupling to the Gi-PI3K-Akt pathway, an effect that weakens the effect of the Gs-AC-cAMP-PKA pathway [22].
High catecholaminergic stress, especially of epinephrine (Epi), triggers β2AR stimulus trafficking from Gs to Gi pathway [22, 26, 34]. The β2AR-Gi pathway suppresses cardiac contraction and protects against myocardial apoptosis and injury. It has been reported that activation of β2AR-Gi pathway prevented the development of arrhythmia [6, 28]. Other studies showed that the occurrence of arrhythmia may be linked to the p38 mitogen-activated protein kinase (p38MAPK), which is a downstream effector of the β2AR-Gi pathway [1, 5, 29]. Based on the observations above, we postulated that estrogen may protect against arrhythmia by regulating the β2AR-Gi-p38 MAPK. Indeed, there is functional synergism between the molecular pathways of estrogen and beta-adrenergic receptors in cardiac cells [16].
Excessive activation of the β2AR-Gi pathway has been linked to Takotsubo cardiomyopathy (TCM), a clinical syndrome characterized by acute and severe but reversible apical ventricular dysfunction [3, 13, 15]. In our previous research, we showed that estrogen increased the activity of the β2AR-Gs pathway and resisted TCM in the rats in vivo [10]. In this study, we postulated that at high concentration of catecholamines, estrogen may alter the activities of β2AR-Gs and Gi pathways, to satisfy the need for more blood supply to the body tissues at the same time offer optimal myocardium protection. Here, we evaluated the status of the β2AR-Gs/Gi pathways in the context of catecholaminergic stress and estrogen environment. The aim of this study was to search for the mechanisms through which estrogen confers cardioprotection in terms of myocardium contractility, arrhythmia, and injury.
Methods
Animals
All studies complied with the Animal Ethics Committee of Xuzhou Medical University (permit number: xz11-12540) and with the Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of health (NIH Publication, 8th Edition, 2011). Female Sprague-Dawley (SD) 8 to 12-week-old rats purchased from Xuzhou Medical University (China) were used for all experiments. They were group-housed in standard shoebox cages at a temperature of 23 ± 2 °C and humidity of 45–55% controlled environment with a 12-h/12-h dark-light cycle. Rats were fed on pelleted food and water ad libitum.
Rats were randomly selected and subjected to sham operation (Sham group) and ovariectomy (OVX group). All rats selected were those in the same menstrual phase through vaginal mucus examination. For OVX group rats, after anesthetization, the ovaries were exteriorized, ligated, and removed via bilateral paraspinal incisions, which were then closed with sterile absorbable sutures. For Sham group rats, the ovaries were only exposed through incisions into the abdominal cavity and then closure of the wounds. After surgery, ketamine (10 mg/kg, I.P.) was used for the postoperative pain management.
Materials
Epi (8.56 × 10−8 mol/100 g in vivo, 100 nmol/L in vitro), isoprenaline (ISO, 100 nmol/L), and the adenylyl cyclase inhibitor SQ22, 536(50 μg/mL) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The β2AR selective antagonist ICI118,551 (55 × 10−9 mol/L) and the Gi-specific inhibitor pertussis toxin (PTX, 10−4 μg/μl) were purchased from Tocris (Bristol, UK). The PI3k inhibitor LY294002 and the p38MAPK inhibitor SB203580 (10−6 mol/L) were purchased from Selleck (USA). Rat lactic dehydrogenase (LDH), brain natriuretic peptide (BNP), and cAMP ELISA kits were purchased from West Tang Biotechnology (Shanghai, China). Gαi and Gαs activation assay kits were purchased from NewEast Biosciences (Malvern, PA).
In vivo stress model and echocardiography
Six weeks after ovariectomy, rats were anesthetized and Epi (8.56 × 10−8 mol/100 g) was injected via the tail vein [10, 26]. Transthoracic echocardiography was performed immediately following the injection (Philips, iE33) with a 12 MHz transducer (3 cm depth with fundamental and harmonic imaging). Diastolic and systolic diameters (cm) were measured by the M-mode echocardiography. The left ventricular internal diameters at end-diastolic and end-systolic dimension are represented as abbreviations LVIDd and LVIDs, respectively. The left ventricular ejection fraction %ΔEF was calculated as (LVIDd − LVIDs) / LVIDd × 100%.
Measurement of arrhythmia in the in vivo stress model
Rats were anesthetized and fixed in a dorsal position. A thermostatically controlled warming plate was used to keep body temperature at 37 ± 0.5 °C throughout the experiment. The electrodes were inserted into the subcutaneous tissue of limbs to record the electrocardiogram (ECG) using LabChart 7.0 software. After stabilization of the cardiac function for 15 min, the right jugular vein was dissected to allow insertion of a catheter for Epi (8.56 × 10−8 mol/100 g) bolus injection [10]. ECG changes were recorded continuously for 6 min by the PowerLab data acquisition system. The types of arrhythmia were classified into ventricular premature beat (VPB), ventricular tachycardia (VT), and ventricular fibrillation (VF). According to the standard of Lambeth conference, VPB is defined as a ventricular electrical complex that is different in shape from the preceding (non-VPB) ventricular complex and is premature in relation to the preceding ventricular complex. VT is defined as a sequence of a minimum of four consecutive ventricular complexes, while VF is defined as a sequence of a minimum of four consecutive ventricular complexes without intervening diastolic pauses, in which the intrinsic shape, the peak-peak interval, and the height vary, and the variation between each is non-progressive [7]. The arrhythmia scores according to Lambeth conference were used to assess the severity of arrhythmia in this study [9] (Table 1).
Isolation of myocardial cells
Six weeks after OVX, rats were intraperitoneally injected with heparin (5000 U/kg) followed by anesthesia injection after 15 min. The thoracic cavity was exposed; hearts were quickly isolated and rinsed in ice-cold Ca2+-free buffer. Ventricular cardiomyocytes were isolated from the Sham and OVX rat hearts using the same methods we described before with minor adjustments [19, 35]. For this study, once the digestion was complete, the heart was cut and evenly divided into three sections on the left ventricular long axis, i.e., apical, middle, and basal parts of the left ventricle. The isolated cardiomyocytes were suspended in Dulbecco’s minimal essential medium (DMEM) at a density of 2 × 104 cells/well in 12-well culture dishes. Finally, the cardiomyocytes were allowed to settle in a 95% O2 and 5% CO2 incubator at 37 °C for 1 h before use.
Development of the in vitro stress model in cardiomyocytes
Based on the method used by Paur et al., an in vitro stress model was developed in cardiomyocytes by pretreatment with Epi (100 nmol/L) for 20 min [26].
Measurement of cardiomyocyte shortening amplitude
The fresh ventricular cardiomyocytes were superfused with a Krebs-Henseleit solution of pH 7.4 and equilibrated with mixed air (95% O2 and 5% CO2) at 37 °C. In brief, few drops of medium containing cardiomyocytes were added to the superfusion chamber on the stage of an inverted microscope (Olympus, Tokyo, Japan). The cardiomyocytes were allowed to settle to the bottom of the chamber after which they were continuously superfused with Krebs-Henseleit buffer (containing 100 nmol/L ISO and 2.0 mmol/L Ca2+). The cardiomyocytes were electrically stimulated at 0.5 Hz. A video recorder (Panasonic, Japan) was used to record the amplitude shortening. At least ten regularly beating myocytes were recorded for each group. Only rod-shaped cardiomyocytes, quiescent, and with clear striations were used for this experiment. Analysis of the recorded data was done using Optical Measure software (presented by China’s National Defense University of Science and Technology).
In vitro measurement of the abnormal rhythm
Isolated cardiomyocytes were electrically stimulated and the data was recorded using a video camera attached to an inverted microscope (Panasonic, Japan). All the beating cardiomyocytes in each microscopic field were counted. The abnormal rhythm was calculated as the number of irregularly beating cardiomyocytes divided by the number of all the beating cardiomyocytes. The data regarding abnormal rhythm was collected by two observers simultaneously assessing the rhythm. A section of the data recording was carried out by an edge-detection method using the IonOptix software that simultaneously assessed the myocyte contraction and Ca2+ transients.
Fura-2 loading of isolated myocytes and determination of cell Ca2+ transients
Adult rat cardiomyocytes were loaded with 1 μM Fura-2 (2 mM, Beyotime Biotechnology, Beijing, China) in a dark room at 25 ± 1 °C for about 20 min placed in a chamber on an inverted microscope (Olympus IX-70), and allowed to adhere for 5 min. The cells were washed by superfusing at 3 ml/min with Krebs-Henseleit buffer (containing 100 nmol/L ISO and 2.0 mmol/L Ca2+) and then electrically stimulated at 1, 2, and 3 Hz. During stimulation, the cells were continuously superfused at 2 ml/min with Krebs-Henseleit buffer at 37 °C. Ultraviolet light dual-excitation at 340/380 nm connected to the microscope through an optical fiber and a video camera was attached to the side port of the inverted microscope. Cytosolic Ca2+ ([Ca2+]i), fura-2 fluorescence, and cell length changes were recorded simultaneously using a soft-edge software (IonOptix Corp., Milton, MA, USA). Signals from the video edge system were digitized and stored in a computer. All Ca2+ transient measurement data were calculated from 20 consecutive contractions after stabilization following stimulation. Cells that remained rod-shaped, without blebs or other visible morphological alterations, and that responded adequately to electrical stimulation were used for this experiment.
Measurement of the BNP and cAMP concentration and LDH activity
The supernatant of the culture medium in each group was collected and the concentrations of BNP and cAMP and the activity of LDH were assayed according to the instructions of ELISA kits. All samples were assayed in triplicate.
Identification of rod-shaped cells
Both rod-shaped and round-shaped cells were observed under a microscope. The former are viable cells and the latter are non-viable cells. We randomly took five micrographs for each sample and examined all cardiomyocytes in each field. Eight hundred to 1000 cells were observed for each condition. The percentage of rod-shaped cells of the total cells was calculated. The analysis of cardiomyocytes was done under blind conditions [31].
Western blot analysis
Whole-cell lysates: Cultured ventricular cardiomyocytes were treated with Epi (100 nM) for 20 min, lysed, and subjected to SDS-PAGE protocol. The following antibodies were used in western blot analyses: p38 MAPK polyclonal antibody (1:1000, Abcam), p-p38 monoclonal antibody (1:1000, Cell Signaling Technology), GAPDH polyclonal antibody (1:1000, Bio world). The membranes were incubated with the corresponding secondary antibodies (1:4000, Zhongshan) and visualized with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate. Membranes were scanned into the computer and the relative intensity of the bands was analyzed by Photoshop software (Adobe, San Jose, CA, USA).
Gαi and Gαs activation assays
The levels of activated GTP-bound Gαs and Gαi proteins were measured using their corresponding assay kits [2, 27]. Whole-cell lysates were incubated for 1 h with an anti-active Gαi antibody and protein A agarose beads. Activated Gαi was quantified by means of western blotting using an anti-Gαi antibody. As a control for sampling errors, total levels of the Gαs and Gαi proteins were also detected.
Statistical analysis
For each experimental series, data were shown as means ± SEM. Statistical analysis was performed with Graphpad Prism 5.0 (Graphpad Software, San Diego, CA, USA). Statistical significance (P < 0.05) for each variable was estimated by t test, one-way ANOVA, and two-way ANOVA followed by Bonferroni post hoc tests or Fisher exact test.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Results
The influence of estrogen on the cardiac function and arrhythmia in stress condition
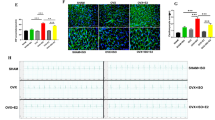
Transthoracic echocardiography was used to detect the cardiac function in vivo. In the normal state, the LVIDd, LVIDs, and EF% measurements were not significantly different between the Sham and OVX rats. Conversely, injection of Epi (8.56 × 108 mol/100 g) increased LVIDs and decreased LVIDd and EF% in both Sham and OVX rats in the first 3 to 4 min, after which their levels gradually returned towards the normal values. The increase in LVIDs and the decrease in EF% were more obvious in OVX + Epi group compared with the Sham + Epi (P < 0.05 vs Sham + Epi group) (Fig. 1a, b).

Lack of estrogen aggravated cardiac function and arrhythmia in stress condition. a Representative echocardiography from Sham and OVX rats treated with Epi. b The percentage of changes from baseline (untreated) of left ventricular internal diameter at end-diastole (%△LVIDd), end-systole (%△LVIDs), and ejection fraction (%△EF) after injection of Epi; Each value represents mean ± SEM. n = 5; *P < 0.05, **P < 0.01 vs baseline of Sham group, #P < 0.05, ##P < 0.01 vs baseline of OVX group; $P < 0.05 and $$P < 0.01, vs Sham + Epi group. The actual starting value (untreated): LVIDd: Sham (0.654 ± 0.017), OVX (0.638 ± 0.008); LVIDs: Sham (0.316 ± 0.011), OVX (0.306 ± 0.005); EF%: Sham (0.516 ± 0.007), OVX (0.520 ± 0.003). c Representative electrocardiograms (ECGs) from rats recorded by the PowerLab data acquisition system. VPB, ventricular premature beat; VT, ventricular tachycardia; VF, ventricular fibrillation. d The score of arrhythmia, VT duration and VT% of rats; n = 10 in each group; **P < 0.01 vs Sham group, ##P < 0.01vs OVX group, $P < 0.05 vs OVX + Epi group
In the normal state, the scores of arrhythmia, VT duration, and VT% were not significantly different between the Sham and OVX groups. In contrast, they all increased following Epi injection in OVX group (P < 0.01 vs OVX group), but only VT% increased in the Sham group (P < 0.01 vs Sham + Epi group). The score of arrhythmia was higher in OVX + Epi group than Sham + Epi group (P < 0.05 vs Sham + Epi group) (Fig. 1c, d).
Effect of estrogen on contraction and injury of myocytes through the β2AR-Gi-PI3K pathway
To observe the effect of estrogen on cardiomyocytes to avoid interference from the effects of other endogenous hormones and the nervous system, ventricular cardiomyocytes were isolated. It was reported that the ratio of β2AR/β1AR in the left ventricle was higher at the apex than at the base. For this reason, myocytes from the base and apex were investigated separately. The myocytes from the base and apex were treated with different doses of Epi for 20 min, after which, their response to ISO (100 nmol/L) was measured.
At low concentration of Epi, the contraction of the basal myocytes in response to ISO stimulation increased initially but returned near to basal level at a high concentration of Epi. In contrast, the contraction of the apical cardiomyocytes in response to ISO stimulation gradually decreased with increase in Epi concentration (Fig. 2a) (P < 0.01 vs basal line). Epi at 1000 nmol/L concentration caused arrhythmia or death in most of the cardiomyocytes. Thus, in the subsequent experiments, the Epi dose of 100 nmol/L was used to compare the response of apical myocytes in different groups because, at this concentration, there was no excessive arrhythmia or cell death.

Effects of estrogen on contraction and injury of myocytes mediated by β2AR-Gi-PI3K pathway. a Responses of ventricular cardiomyocytes treated with different concentration of Epi to ISO (100 nmol/L); Each value represents the mean percentage change in shortening amplitude (SA) from baseline (untreated, SA = 0), levels ± SEM, n = 6 hearts in each group. *P < 0.05 and **P < 0.01 vs baseline. The actual starting value of SA: apex (6.765 ± 0.4361), base (5.287 ± 0.5687). b Response time curves of ventricular myocytes treated with Epi (100 nmol/L) to ISO (100 nmol/L). Each value represents the mean percentage change in shortening amplitude (SA) from baseline (untreated) levels ± SEM, n = 6 hearts in each group. #P < 0.05 vs baseline of Sham apex SA = 0, *P < 0.05, ***P < 0.01 vs baseline of OVX apex SA = 0, $P < 0.05 vs baseline of OVX base SA = 0. The actual starting value of SA: Sham apex (6.765 ± 0.4361), OVX apex (10.06 ± 0.7802), Sham base (5.287 ± 0.5687), OVX base (4.324 ± 0.4316). c Shortening amplitudes of apical cardiomyocytes; d Release of LDH from apical myocytes; e Release of BNP from apical myocytes; f Percentage of rod-shaped apical myocytes. Each value represents the mean ± SEM, n = 6 hearts in each group. *P < 0.05 vs Sham group; #P < 0.05 and ##P < 0.01 vs OVX group; ▲P < 0.05 and ▲▲P < 0.01 vs Sham + Epi group; $P < 0.05 and $$P < 0.01 vs OVX + Epi group
The contractile response of apical myocytes in both Sham and OVX groups was inhibited by Epi in the first 20 min, while at 60 min, the values returned to the baseline level. The decline in contraction contractility was severe in OVX group (P < 0.05 vs Sham group). Moreover, the decrease in contraction in basal myocytes was lower in OVX group but was absent in Sham group (Fig. 2b). These results demonstrated that the inhibition in the apex was more severe, suggesting that β2AR played an important role in contraction during stress state.
In the normal condition, the response of cardiomyocytes to ISO was higher in OVX group than that of Sham group (Fig. 2c). However, when the myocytes were treated with Epi for 20 min, the response of cardiomyocytes to ISO decreased both in Sham and OVX groups. In addition, the decrease was more severe in the OVX group (P < 0.01 vs Sham group). To further examine the role of the β2AR-Gi-PI3K pathway, myocytes were assessed after treatment with the corresponding drugs. Pretreatment of the myocytes with ICI118,551 (β2AR antagonist), PTX (Gi inhibitor), or LY294002 (PI3K antagonist) abolished the difference in response to ISO between Sham and OVX groups and increased the shortening amplitude of the cardiomyocytes (Fig. 2c).
In the normal condition, there was no difference in LDH and BNP levels and the number of rod-shaped cells between Sham and OVX groups (Fig. 2d–f). In contrast, the Epi treatment induced a marked increase in LDH and BNP levels (Fig. 2d, e) and a significant decrease in the number of rod-shaped cells, especially in OVX group (Fig. 2f) (P < 0.05 vs Sham group). Pretreatment of the cardiomyocytes with PTX or LY294002 further aggravated the injury of cardiomyocytes and abolished the difference in LDH and BNP levels between Sham and OVX group.
The effect of estrogen on the myocyte rhythm through the β2AR-Gi-p38MAPK pathway
In order to investigate the occurrence of an abnormal rhythm, cardiomyocytes were electrically stimulated at different frequencies. The rhythm was recorded by a video recorder (Panasonic, Japan). The abnormal rhythm of cardiomyocytes increased with the increments in stimulation frequencies (Fig. 4a, video 1–4). The frequency of 4 Hz was too fast to clearly distinguish the contraction state of the cells. Therefore, the 3 Hz frequency was used to measure abnormal rhythms throughout the experiment. In order to investigate the characteristic of Ca2+ under different frequencies, the intercellular Ca2+ at resting time and the Ca2+transient amplitude of normal cells were recorded by an edge-detection method using IonOptix software. No significant difference in both the resting intracellular Ca2+ and the Ca2+ transient amplitudes was detected among all groups, only the resting intracellular Ca2+ tended to be high with the increase of stimulation frequency (Fig. 3a), and the Ca2+ transient amplitude tended to decrease with the increase of the frequency (Fig. 3b). All the normal cells’ Ca2+ transient was shown in accordance with the frequency (Fig. 3c) similar to videos. The several Ca2+ transient of abnormal cells was shown in Fig. 3d.

Parameters of Ca2+ transient. a The resting intercellular Ca2+ varied with different frequencies of stimulation. b The Ca2+ transient amplitude varied with different frequencies of stimulation. Each value was collected from the Sham groups’ normal rhythm cells and represents mean ± SEM, n = 3 hearts in each group. c Pictures of normal cells’ Ca2+ transient. d Pictures of abnormal cells’ Ca2+ transient
In the normal state, there was no difference in abnormal rhythms between Sham and OVX groups (Fig. 4b). Following Epi treatment for 20 min, abnormal rhythms increased in myocytes from OVX group (P < 0.01 vs OVX group). The p38MAPK, a downstream target of the β2AR-Gi pathway, has been shown to possess anti-arrhythmia effects [38]. Indeed, pretreatment with ICI118, 551 (antagonist of β2AR), PTX (inhibitor of Gi), or SB203580 (antagonist of p38MAPK) increased abnormal rhythms in Sham group, but did not in OVX group (Fig. 3b), suggesting that lack of estrogen enhanced the risk of arrhythmia through the β2AR-Gi-p38MAPK pathway in stress state.

The role of the β2AR-Gi-p38MAPK pathway as a mediator of estrogen protection against abnormal rhythm. a Abnormal rhythm induced by different frequencies of stimulation. Each value represents mean ± SEM, n = 6 hearts in each group. *P < 0.05 and **P < 0.01 vs 1 Hz group; b Effect of β2AR-Gi-p38MAPK on abnormal rhythm. Each value represents mean ± SEM, n = 6 hearts in each group. *P < 0.05 and **P < 0.01 vs Sham+ Epi group; ##P < 0.01 vs OVX group; c, d Effect of β2AR-Gi on phosphorylation of p38 MAPK in lack of estrogen state. Each value represents the mean ± SEM, n = 3. **P < 0.01 vs Sham; #P < 0.05 vs Sham + Epi group
In addition, there was no difference in p38MAPK phosphorylation state between the Sham and OVX groups in a normal state (Fig. 4c). Epi treatment increased the phosphorylation of p38 MAPK in Sham group (P < 0.01 vs Sham group), but not in OVX group. Pretreatment with ICI118,551 or PTX decreased the phosphorylation of p38MAPK in Sham group treated with Epi (P < 0.05 vs Sham + Epi group), but still it had no effect on OVX group (Fig. 4d). These results further indicated that estrogen increased the activity of p38MARK and protected against arrhythmia via the β2AR-Gi pathway.
Effects of estrogen on the activity of Gi and Gs, cAMP concentration, and myocyte injury
Apart from coupling to Gi, β2AR also couples to the Gs and activates Gs-cAMP-PKA pathway. Under normal condition, the activity of Gi protein was the same between the Sham and OVX groups, while the activity of Gs protein and the concentration of cAMP were higher in OVX group (P < 0.05 vs Sham group). At high Epi concentration, the activity of Gi increased in both groups (P < 0.01), especially in Sham group. Epi also increased the activity of Gs and the concentration of cAMP in Sham group (P < 0.01 vs Sham group) but decreased them in OVX group (P < 0.01 vs OVX group) (Fig. 5a–d).

Effects of estrogen on Gi and Gs activities, cAMP concentration, and myocytes injury. a Activity of Gi protein; b Activity of Gs protein; c Ratio of activated Gi to Gs; d Concentration of cAMP. Each value represents the mean ± SEM, n = 3.*P < 0.05 and ** P < 0.01 vs Sham group; $$P < 0.01 vs Sham + Epi group; ##P < 0.01 vs OVX group; e Percentage of rod-shaped myocytes; f, g Release of LDH and BNP. Each value represents the mean ± SEM, n = 6 hearts in each group. ▲▲P < 0.01 vs Sham + Epi group; $P < 0.05 and $$P < 0.01 vs OVX+ Epi group
Blocking adenylate cyclase (AC) with SQ22, 536 (antagonist of AC) increased the release of LDH and BNP and decreased the percentage of rod-shaped cells in Sham and OVX groups (P < 0.05), and the differences in these indicators between the two groups disappeared (Fig. 5e–g), suggesting that β2AR-Gs-cAMP-PKA pathway also contributed to the mechanism of estrogen cardioprotection.
Discussion
Firstly, this study demonstrated that lack of estrogen aggravated the Epi-induced arrhythmia and decrease in cardiac contractility. Secondly, in estrogen deficiency, the activity of the β2AR-Gs pathway was stronger in the normal state but weaker in stress state. Inhibition of the β2AR-Gs pathway enhanced the damage to the myocytes isolated from both Sham and OVX rats and abolished the differences in damage levels between the two groups in stress state. Thirdly, lack of estrogen weakened the activation of Gi in stress state. Blockade of the β2AR-Gi-PI3K pathway enhanced contraction exacerbated the damage to the myocytes from Sham and OVX rats and canceled the differences of these parameters between the two groups. Fourthly, lack of estrogen weakened the activation of p38MAPK in stress state. Inhibition of the β2AR-Gi-p38MAPK increased the susceptibility to arrhythmia only in Sham myocytes and offset the difference in arrhythmia patterns between the Sham and OVX myocytes. To our knowledge, this is the first study that demonstrates that OVX may cause abnormal coupling of the β2AR-Gi and β2AR-Gs pathways and promote arrhythmia and cardiac injury under stress state.
For a long time, the discrepancies from reports regarding the effectiveness of estrogen in preventing or treating cardiovascular diseases have been confusing to doctors and researchers. Many factors may influence the success of estrogen supplementation therapy, such as the optimal time points for administration, dose duration, and underlying diseases. Therefore, the present study contributes to the understanding of the mechanism of action of estrogen, which may lead to the discovery of estrogenic drugs and inform its clinical application.
It has been reported that elevated levels of catecholamines during stress state, particularly Epi [25], can suppress cardiac contractility, arrhythmia, or even heart failure [4, 30, 32, 37]. In this study, cardiac stress and injury model was modeled by an injection of a high dose of Epi to rats. Epi at the concentration of 8.56 × 108 mol/100 g suppressed cardiac contraction which was detected by echocardiography, and that was consistent with our previous study results which were reflected by hemodynamics [10]. The results showed that the decline in cardiac contractility and high rate of arrhythmia caused by Epi were severe in OVX hearts both in vivo and in vitro. The results clearly demonstrated that estrogen conferred cardioprotection in stress state. Subsequently, the mechanism that underlies the susceptibility to heart injury and arrhythmia in estrogen deficiency in stress state was explored in the study.
Epi has a high affinity for β2AR, which mediates signals through two pathways: Gs-AC-cAMP pathway and Gi-PI3K-Akt or p38MARK cascades. Considering the effects of these two pathways, we considered that estrogen may act as a regulator of the β2AR-Gs and Gi signals to optimize energy conservation and utilization at the same time match energy metabolism with the functions of the body. Estrogen decreases the plasma levels of Epi and norepinephrine by inhibiting the sympathetic activity and decreasing the catecholamine in plasma [10, 12]. Therefore, in order to study the direct protective effect of estrogen on cardiac muscle while avoiding the endogenous effects of estrogen on the sympathetic nervous system, isolated cardiomyocytes were used for this purpose. To confirm whether β2AR plays a pivotal role in the cardioprotection of estrogen, we preliminarily investigated the effect of estrogen on myocytes from base and apex of hearts separately. This maneuver was based on previous reports that there is an imbalanced distribution of β2AR in the heart, being more in the apex than in the base of the left ventricle [21]. Initially, the contraction of myocytes was inhibited by Epi (100 nmol/L) followed by gradual recovery towards the baseline levels. These observations demonstrated that Epi induced TCM-like feature in cardiomyocytes [10]. The level of the inhibition was greatest at the apex, especially in the OVX rats. This is in line with our speculation that β2AR mediated the cardioprotection coffered by estrogen, as the high density of β2AR in the apex translates to relatively higher β2AR-Gi signals. These results are similar to those of Paur et al. who observed that high doses of Epi induced cardiac hypokinesia in vivo followed by gradual recovery [26], which was interpreted as a form of myocardial hibernation [14]. However, we note that the differences observed between the apex and base of left ventricle myocytes may not be limited to the β2AR signaling pathway. The possibility that variation in expression pattern of estrogen receptors in the left ventricle may be linked to the observed differences cannot be ruled out. In addition, the effects of Epac pathway on cardiomyocyte contraction as an alternative pathway that may be activated by β2AR-Gs-cAMP were not investigated. Nevertheless, this study showed that estrogen deficiency aggravated and prolonged the decline in cardiac contractility at high levels of Epi, suggesting that estrogen may improve the myocyte contraction in stress state.
In the normal state, β2AR couples to Gi which suppresses contraction and this pathway is postulated to mediate cardioprotection [8, 10, 21]. Previously, we demonstrated that long-term lack of estrogen decreased the expression of β2AR [17]. Therefore, in this study, lack of estrogen may have reduced the β2AR-Gi coupling in the heart. This premise may explain why the heart contraction is weaker in normal rats than in OVX rats, in women than in man in the normal state, and women are less susceptible to heart diseases before menopause. Our results showed that the damage to the myocytes caused by high concentration of Epi was severe in those isolated from OVX rats. Inhibition of the Gi or PI3K in myocytes increased the damage in both groups and abolished the difference between Sham and OVX groups. The results implied that lack of estrogen reduced the activation of β2AR-Gi cascade, hence decreased the myocytes protection as evidenced by the exacerbated decline in shortening amplitude and damage to the myocytes in stress state. These in vitro results are in accordance with our previous observations in vivo [10], in which lack of estrogen decreased the phosphorylation of PKA (a key determinant of the β2AR-Gi pathway activation [26]), and induced cardiac dysfunction.
p38MAPK is a downstream target of β2AR-Gi cascade. It is regarded as a key mediator of cardioprotection against arrhythmia [1]. Intriguingly, it has been recognized that β2AR and Gi are also implicated in the suppression of arrhythmia [6, 28]. In order to examine the occurrence of abnormal rhythms in cardiomyocytes, we stimulated the myocytes at the frequency of 3 Hz. It is known that excitation of cardiomyocytes triggers an increase in intracellular Ca2+ by the entry of extracellular Ca2+ through the L-type Ca2+ channel and by the release of Ca2+ from sarcoplasmic reticulum. Adult cardiomyocytes have a long refractory period; therefore, the decrease in Ca2+ amplitude at high stimulation frequency could be a result of impaired Ca2+ regulation due to inadequate time for Ca2+ uptake and release. For the same reason, at high frequency, the intracellular Ca2+ levels are expected to be higher which might lead to Ca2+ overload. Indeed, our results showed that the resting intracellular Ca2+ levels tended to be high with a higher stimulation frequency. Studies have shown that cardiomyocyte Ca2+ overload increases the risk of arrhythmia [33]. This suggested that the cell under higher electrical stimulation was prone to arrhythmia. In the normal state, the rhythm was not significantly different between myocytes isolated from the Sham and OVX rats. However, when myocytes were treated with high concentration of Epi, the rate of abnormal rhythm increased in OVX group only. On the other hand, the levels of activated p38MAPK were the same in both groups in the normal state, but in a stress state, the activated p38MAPK increased in Sham group but not in OVX group. The results implied that estrogen deficiency increased the occurrence of the abnormal rhythms in cardiomyocytes in stress state. Furthermore, inhibition of β2AR, Gi, or p38 MAPK with ICI118, 551, PTX, or SB203580 augmented the occurrences of irregular contractions to the same level in Sham and OVX groups. At the same time, the levels of activated p38MAPK in Sham group decreased to the level of those in OVX group. In contrast, there was no obvious change in OVX group. Collectively, these results indicated that estrogen protected the myocytes against arrhythmia through β2AR-Gi-p38MARK cascade pathway in the stress state.
In the normal state, the levels of activated Gs were higher in OVX, while those of activated Gi were similar in Sham and OVX groups. This suggested that estrogen downregulated the activation of Gs, a mechanism that would keep the metabolic rate and oxygen consumption at a low level. Under the stress state, both the activated Gs and Gi increased in Sham group. Surprisingly, the level of activated Gs reduced while those of activated Gi increased in OVX group, although to a lesser extent compared with to that of Sham group. Therefore, these results imply that estrogen increased the activation of Gs and Gi in a stress state, an effect that offered better protection on the myocardium.
In the stress state, the ratio of activated Gi to Gs was nearer to that of a normal state, and it was higher in OVX group, although the activated Gi was higher in the Sham group. Hence, myocytes of OVX group were vulnerable to Epi-induced contractile dysfunction and injury. Taken together, the results demonstrated that during stress, estrogen strengthened and balanced the β2AR-Gs and Gi pathways. The relatively higher activation of Gi proteins may be a compensatory strategy to protect the heart against catecholaminergic stress. In this experiment, we found that lack of estrogen suppressed myocytes contraction in a stress state, which simulated the cardiac depression observed in TCM, a syndrome that occurs predominantly in postmenopausal women. Hyperactivation of the β2AR-Gs pathway increases energy consumption and production of metabolites and oxygen-free radicals, which may increase cytotoxicity and impair myocytes contractility. High Epi concentration is known to trigger the signal trafficking from β2AR-Gs to β2AR-Gi through a mechanism that involves β2AR phosphorylation by PKA [26]. Therefore, these results indicated that estrogen optimized the ratio of Gs to Gi and balanced the β2AR coupling of from Gs to Gi in stress state. The concentration of cAMP matched with the levels of activated Gs. Blocking adenylate cyclase (AC) with SQ22,536 aggravated the damage induced by Epi and matched the extent of damage between the two groups. This observation was unexpected since Gs-cAMP pathway is thought to mediate cardiac injury. However, it should be noted that the β2AR-Gs to β2AR-Gi signal trafficking is partially dependent on the PKA, which is produced downstream of the cAMP pathway [26]. Therefore, the observation that the myocyte injury increased when the Gs-cAMP pathway was blocked may be due to inhibition of β2AR-Gs to β2AR-Gi stimulus trafficking. These results demonstrated that estrogen increased β2AR-Gs pathway and protected cardiomyocytes in the stress state.
Pharmacological implications
The clinical use of β-blockers is based on their ability to suppress the effects of catecholamines primarily on the β-AR-Gs pathway. However, there have been clinical uncertainties regarding their use, as they have been associated with adverse effects and mortality [24]. Moreover, their efficacy may vary with gender [20]. The observations, in the present study, that inhibition of the AC-PKA pathway aggravated the cardiac injury provides a mechanism that may explain the adverse effects of β-blockers in certain clinical contexts.
In conclusion, this study demonstrated that in the normal state, estrogen deficiency elevated the contractile response by upregulating the activity of β2AR-Gs signal pathway. During acute stress, the activities of β2AR-Gi/Gs pathways were suppressed in OVX group, which led to the loss of protection against arrhythmia, myocyte injury, and weakened the strength of the myocardium contraction. Based on these results, it can be concluded that estrogen may lower the occurrence of TCM-like diseases and arrhythmia induced by high concentration of Epi via increasing the activities of both β2AR-Gs and Gi signal pathway and optimizing the rate of β2AR-Gs to β2AR-Gi coupling. These results provide a novel mechanism that orchestrates cardioprotection and the experimental basis upon which drugs for preventing arrhythmia may be developed. Further studies are required to uncover the details on how estrogen regulates the activities and balances the β2AR-Gs/Gi pathways.
Abbreviations
- AC:
-
adenylyl cyclase
- BNP:
-
brain natriuretic peptide
- β1AR:
-
β1-adrenoceptor
- β2AR:
-
β2-adrenoceptor
- Epi:
-
epinephrine
- EF:
-
left ventricular ejection fraction
- Gs:
-
stimulatory G proteins
- Gi:
-
inhibitory G proteins
- ISO:
-
isoprenaline
- LDH:
-
lactic dehydrogenase
- LVIDd:
-
left ventricular internal diameters at End-diastolic dimension
- LVIDs:
-
left ventricular internal diameters at End-systolic dimension
- OVX:
-
ovariectomy
- p38 MAPK:
-
p38 mitogen-activated protein kinase
- PKA:
-
protein kinase A
- PI3K:
-
phosphoinositide 3-kinase
- TCM:
-
Takotsubo cardiomyopathy
- VPB:
-
ventricular premature beat
- VT:
-
ventricular tachycardia
- VF:
-
ventricular fibrillation
- Epac :
-
exchange protein directly activated by cAMP
References
Ai X, Yan J, Carrillo E, Ding W (2016) The stress-response MAP kinase signaling in cardiac arrhythmias. Rev Physiol Biochem Pharmacol 172:77–100. https://doi.org/10.1007/112_2016_8
Anastasaki C, Gutmann DH (2014) Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet 23(25):6712–6721. https://doi.org/10.1093/hmg/ddu389
Akashi YJ, Nef HM, Lyon AR (2015) Epidemiology and pathophysiology of Takotsubo syndrome. Nat Rev Cardiol 12(7):387–397. https://doi.org/10.1038/nrcardio.2015.39.
Akashi YJ, Nef HM, Mollmann H, Ueyama T (2010) Stress cardiomyopathy. Annu Rev Med 61(1):271–286. https://doi.org/10.1146/annurev.med.041908.191750
Aschar-Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang FH, Yang W, Dorian D, Simpson JA, Tuomi JM, Jones DL, Nanthakumar K, Cox B, Wehrens XHT, Dorian P, Backx PH (2015) Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNF-alpha. Nat Commun 6:6018. https://doi.org/10.1038/ncomms7018.
Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, Jaques K, Lai EW, Pacak K, Zhu WZ, Xiao R, Tomaselli GF, Kass D (2009) Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation 119(9):1231–1240. https://doi.org/10.1161/CIRCULATIONAHA.108.774752
Curtis MJ, Hancox JC, Farkas A, Wainwright CL, Stables CL, Saint D, Clements-Jewery H, Lambiase PD, Billman GE, Janse MJ, Pugsley MK, Ng GA, Roden DM, Camm AJ, Walker MJA (2013) The Lambeth conventions (II): guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol Ther 139(2):213–248. https://doi.org/10.1016/j.pharmthera.2013.04.008
Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, Crow MT (2000) The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3′-kinase. Circ Res 87(12):1172–1179. https://doi.org/10.1161/01.RES.87.12.1172.
Curtis MJ, Walker MJ (1988) Quantification of arrhythmias using scoring systems: an examination of seven scores in an in vivo model of regional myocardial ischemia. Cardiovasc Res 22(9):656–665. https://doi.org/10.1093/cvrese/22.9.656.
Cao X, Zhou C, Chong J, Fu L, Zhang L, Sun D, Hou H, Zhang Y, Li D, Sun H (2015) Estrogen resisted stress-induced cardiomyopathy through increasing the activity of beta (2) AR-G alpha signal pathway in female rats. Int J Cardiol 187:377–386. https://doi.org/10.1016/j.ijcard.2015.02.113
Fujita T, Umemura M, Yokoyama U, Okumura S, Ishikawa Y (2017) The role of Epac in the heart. Cell Mol Life Sci 74(4):591–606. https://doi.org/10.1007/s00018-016-2336-5
Gomes HL, Graceli JB, Goncalves WLS et al (2012) Influence of gender and estrous cycle on plasma and renal catecholamine levels in rats. Can J Physiol Pharmacol 90(1):75–82. https://doi.org/10.1139/y11-102
Garcia M, Mulvagh SL, Merz CNB, Buring JE, Manson JE (2016) Cardiovascular disease in women: clinical perspectives. Circ Res 118(8):1273–1293. https://doi.org/10.1161/CIRCRESAHA.116.307547
Heusch G, Sipido KR (2004) Myocardial hibernation: a double-edged sword. Circ Res 94(8):1005–1007. https://doi.org/10.1161/01.RES.0000128071.90075.e3
Huynh K (2015) Cardiomyopathies: clinical features of patients with Takotsubo syndrome. Nat Rev Cardiol 12(12):684. https://doi.org/10.1038/nrcardio.2015.153
Machuki JO, Zhang HY, Harding SE, Sun H (2017) Molecular pathways of oestrogen receptors and β-adrenergic receptors in cardiac cells: recognition of their similarities, interactions and therapeutic value. Acta Physiol 00:e12978. https://doi.org/10.1111/apha.12978
Kang S, Liu Y, Sun D, Zhou C, Liu A, Xu C, Hao Y, Li D, Yan C, Sun H (2012) Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS One 7(10):e48185. https://doi.org/10.1371/journal.pone.0048185
Lezoualc’h F, Fazal L, Laudette M, Conte C (2016) Cyclic AMP sensor EPAC proteins and their role in cardiovascular function and disease. Circ Res 118(5):881–897. https://doi.org/10.1161/CIRCRESAHA.115.306529
Liu A, Gao L, Kang S, Liu Y, Xu C, Sun H, Li D, Yan C (2012) Testosterone enhances estradiol’s cardioprotection in ovariectomized rats. J Endocrinol 212(1):61–69. https://doi.org/10.1530/JOE-11-0181
Ljungman C, Kahan T, Schioler L et al (2014) Gender differences in antihypertensive drug treatment: results from the Swedish Primary Care Cardiovascular Database (SPCCD). J Am Soc Hypertens 8(12):882–890. https://doi.org/10.1016/j.jash.2014.08.015
Lyon AR, Rees PSC, Prasad S, Poole-Wilson PA, Harding SE (2008) Stress (Takotsubo) cardiomyopathy—a novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat Clin Pract Cardiovasc Med 5(1):22–29. https://doi.org/10.1038/ncpcardio1066
Liu R, Ramani B, Soto D, De Arcangelis V, Xiang Y (2009) Agonist dose-dependent phosphorylation by protein kinase A and G protein-coupled receptor kinase regulates β2 adrenoceptor coupling to G(i) proteins in cardiomyocytes. J Biol Chem 284(47):32279–32287. https://doi.org/10.1074/jbc.M109.021428
Mikkola TS, Gissler M, Merikukka M, Tuomikoski P, Ylikorkala O (2013) Sex differences in age-related cardiovascular mortality. PLoS One 8(5):e63347. https://doi.org/10.1371/journal.pone.0063347
Jørgensen ME, Hlatky MA, Køber L, Sanders RD, Torp-Pedersen C, Gislason GH, Jensen PF, Andersson C (2015) β-blocker–associated risks in patients with uncomplicated hypertension undergoing noncardiac surgery. JAMA Intern Med 175(12):1923–1931. https://doi.org/10.1001/jamainternmed.2015.5346
Nef HM, Mollmann H, Akashi YJ, Hamm CW (2010) Mechanisms of stress (Takotsubo) cardiomyopathy. Nat Rev Cardiol 7(4):187–193. https://doi.org/10.1038/nrcardio.2010.16
Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O'Gara P, Stuckey DJ, Nikolaev VO, Diakonov I, Pannell L, Gong H, Sun H, Peters NS, Petrou M, Zheng Z, Gorelik J, Lyon AR, Harding SE (2012) High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation 126(6):697–706. https://doi.org/10.1161/CIRCULATIONAHA.112.111591
Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ (2010) Beta-arrestin-but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A 107(2):628–632. https://doi.org/10.1073/pnas.0912852107.
Rau T, Nose M, Remmers U et al Overexpression of wild-type Galpha(i)-2 suppresses beta-adrenergic signaling in cardiac myocytes. FASEB J 17:523–525. https://doi.org/10.1096/fj.02-0660fje
Surinkaew S, Kumphune S, Chattipakorn S, Chattipakorn N (2013) Inhibition of p38 MAPK during ischemia, but not reperfusion, effectively attenuates fatal arrhythmia in ischemia/reperfusion heart. J Cardiovasc Pharmacol 61(2):133–141. https://doi.org/10.1097/FJC.0b013e318279b7b1
Schwartz PJ, Volders PGA (2014) Sudden death by stress: how far under the nerves should we dig to find out why LQT1 patients die? J Am Coll Cardiol 63(8):828–830. https://doi.org/10.1016/j.jacc.2013.09.059
Sun H, Zhou F, Wang Y, Zhang Y, Chang A, Chen Q (2006) Effects of beta-adrenoceptors overexpression on cell survival are mediated by Bax/Bcl-2 pathway in rat cardiac myocytes. Pharmacology 78(2):98–104. https://doi.org/10.1159/000095785
Traynham CJ, Cannavo A, Zhou Y, Vouga A, Woodall BP, Hullmann JE, Ibetti J, Gold JI, Chuprun JK, Gao E, Koch WJ (2015) Differential role of G protein-coupled receptor kinase 5 in physiological versus pathological cardiac hypertrophy. Circ Res 117:1001–1012. https://doi.org/10.1161/CIRCRESAHA.115.306961
Thandroyen FT, Morris AC, Hagler HK, Ziman B, Pai L, Willerson JT, Buja LM (1991) Intracellular calcium transients and arrhythmia in isolated heart cells. Circ Res 69(3):810–819. https://doi.org/10.1161/01.RES.69.3.810
Wang Y, De Arcangelis V, Gao X, Ramani B, Jung Y, Xiang Y (2008) Norepinephrine- and epinephrine-induced distinct beta2-adrenoceptor signaling is dictated by GRK2 phosphorylation in cardiomyocytes. J Biol Chem 283(4):1799–1807. https://doi.org/10.1074/jbc.M705747200
Wu Q, Zhao Z, Sun H, Hao Y, Yan C, Gu S (2008) Oestrogen changed cardiomyocyte contraction and beta-adrenoceptor expression in rat hearts subjected to ischaemia-reperfusion. Exp Physiol 93(9):1034–1043. https://doi.org/10.1113/expphysiol.2007.041939a
Xu C, Liu A, Sun H, Sun Y, Wang G, Gao L, Hao Y, Yan C (2010) β2-Adrenoceptor confers cardioprotection against hypoxia in isolated ventricular myocytes and the effects depend on estrogenic environment. J Recept Signal Transduct 30(4):255–261. https://doi.org/10.3109/10799893.2010.488242.
Yarnell J (2008) Stress at work—an independent risk factor for coronary heart disease? Eur Heart J 29(5):579–580. https://doi.org/10.1093/eurheartj/ehm641
Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP (2001) Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A 98(4):1607–1612. https://doi.org/10.1073/pnas.98.4.1607
Funding
This work was supported by the National Natural Science Foundation of China (No.81370329, No.81461138036) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All studies complied with the Animal Ethics Committee of Xuzhou Medical University (permit number: xz11-12540) and with the Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of health (NIH Publication, 8th Edition, 2011).
Competing interests
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hou, H., Zhao, Z., Machuki, J.O. et al. Estrogen deficiency compromised the β2AR-Gs/Gi coupling: implications for arrhythmia and cardiac injury. Pflugers Arch - Eur J Physiol 470, 559–570 (2018). https://doi.org/10.1007/s00424-017-2098-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-2098-4