
Abstract
As one of the most important second messengers, 3′,5′-cyclic adenosine monophosphate (cAMP) mediates various extracellular signals including hormones and neurotransmitters, and induces appropriate responses in diverse types of cells. Since cAMP was formerly believed to transmit signals through only two direct target molecules, protein kinase A and the cyclic nucleotide-gated channel, the sensational discovery in 1998 of another novel direct effecter of cAMP [exchange proteins directly activated by cAMP (Epac)] attracted a great deal of scientific interest in cAMP signaling. Numerous studies on Epac have since disclosed its important functions in various tissues in the body. Recently, observations of genetically manipulated mice in various pathogenic models have begun to reveal the in vivo significance of previous in vitro or cellular-level findings. Here, we focused on the function of Epac in the heart. Accumulating evidence has revealed that both Epac1 and Epac2 play important roles in the structure and function of the heart under physiological and pathological conditions. Accordingly, developing the ability to regulate cAMP-mediated signaling through Epac may lead to remarkable new therapies for the treatment of cardiac diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The appropriate regulation of cardiac function is critical in maintaining a proper metabolic state, and catecholamines, including norepinephrine and epinephrine, are among the most important hormones that regulate cardiac function. These catecholamines, which are released from the cardiac sympathetic nerve terminal or the adrenal medulla, bind to several subtypes of adrenergic receptors (AR) in the heart including β1-, β2-, β3-, and α1-AR. Among these, β1-AR is known to play a central role in catecholamines’ positive inotropic, lusitropic, and chronotropic effects [1].
The primary action of ligand-stimulated β1-AR is the activation of stimulatory G proteins (Gs), which activate adenylyl cyclase (AC). AC produces 3′,5′-cyclic adenosine monophosphate (cAMP) from ATP, leading to elevation of the intracellular concentration of cAMP [2]. cAMP is one of the most important second messengers in the body, as it mediates various extracellular signals including hormones and neurotransmitters, and induces responses in diverse types of cells.
cAMP was formerly believed to transmit its signals via only two direct target molecules, protein kinase A (PKA) and the cyclic nucleotide-gated channel. cAMP binds to the regulatory subunit of PKA, inhibiting it and thus causing conformational change resulting in the release of catalytic subunits. Consequently, PKA phosphorylates various important proteins, including L-type Ca2+ channel (LTCC), phospholamban (PLN), ryanodine receptor (RyR), troponin I, myosin-binding protein-C, and cAMP-response element binding protein (CREB), and thereby regulates cellular functions including cardiac contractility and gene transcription [1, 3]. Direct binding of cAMP to the cyclic nucleotide-gated channel, on the other hand, causes hyperpolarization-activated cation inward current (If), leading to a positive chronotropic effect. cAMP-induced responses of cardiomyocytes have been explained with reference to such mechanisms.
In 1998, however, a novel direct effecter of cAMP was discovered: exchange proteins directly activated by cAMP (Epac) [4, 5]. The discovery of Epac was remarkable in that it identified another novel pathway through which cAMP elevation signaling could be mediated and induce an original cellular response independent of PKA. This pathway may serve as a useful therapeutic target, because the modulation of Epac function is expected to enable more specific regulation of particular cAMP-mediated signals than is possible with therapies targeting β-AR and ACs. Specific regulation of signaling is required for the development of safe and useful therapies.
Numerous studies have clarified the important role of Epac as distinct from that of PKA in various tissues [6, 7]. In this review, we focused on Epac’s roles in the heart.
Epac
Structure
Epac1 and Epac2, the two identified Epac isoform proteins, have molecular masses of about 100 and 110 kDa (Epac2A), respectively [8]. They are encoded in distinct genes, namely, RAPGEF3 on chromosome 12 and RAPGEF4 on chromosome 2. Epac2 has three variants, Epac2A, Epac2B, and Epac2C, that stem from variations in the transcriptional start site and splicing manner [9].
Epac was initially identified as proteins in possession of both a cAMP-binding domain and motifs of guanine nucleotide exchange factors (GEFs) for the Ras superfamily of guanine nucleotide-binding proteins [4, 5]. Epac1 and Epac2 are also specific GEFs for both Rap1 and Rap2 [4, 10, 11], members of the Ras superfamily. Interacting with cAMP causes Epac to bind to Raps and to convert them to their activated forms by exchanging bound GDP for GTP.
Epac1, Epac2B and Epac2C all possess a cAMP-binding domain (cAMP-BD) called cAMP-BD-B, while Epac2A has a different type of domain known as cAMP-BD-A, which is believed to have no significant effect on Epac2 activity [11] but is thought to play an important role in localization to the plasma membrane [9] (Fig. 1a). In other major regions, Epac1 and Epac2 proteins are structurally similar. Their C-terminal side catalytic region contains three principal domains. The first of these is a cell division cycle 25 (Cdc 25) homology GEF domain, which is responsible for GEF activity of Epac and includes a nuclear pore targeting signal [12]. The second is a Ras-association (RA) domain, which is reported to play an important role in targeting Epac2 to the plasma membrane via interaction with activated Ras [13]. The third is a Ras-exchange motif (REM), which is believed to be involved in stabilization of the active conformation of Epac [14].

The structure of Epac. a Epac1, Epac2B, and Epac2C each contain one cAMP-binding domain-B (cAMP-BD-B). In contrast, Epac2A contains a different domain (cAMP-BD-A). In the other major region, Epac1 and Epac2 proteins share a common structure. The C-terminal side catalytic region contains three principle domains including a cell division cycle 25 (Cdc 25) homology GEF domain (CDC25-HD), Ras-association (RA) domain, and Ras-exchange motif (REM). The N-terminal side regulatory region includes a disheveled, Egl-10, pleckstrin (DEP) domain and cAMP-BD. b In the absence of cAMP (inactive state), Epac sterically auto-inhibits its GEF activity. Binding of cAMP to cAMP-BD-B induces a conformational change of Epac’s structure that promotes accessibility of Rap to the catalytic region of Epac (active state). Consequently, the GDP on Rap is exchanged to GTP by Epac, thereby facilitating its function
The N-terminal side regulatory region, meanwhile, includes a disheveled Egl-10 pleckstrin (DEP) domain and cAMP-BD. The DEP domain is reported to be necessary for the cAMP-induced translocation of Epac1 from the cytosol to the plasma membrane, where it activates Rap [15, 16]. The DEP domain is absent in Epac2C [17].
In the absence of cAMP (inactive state), Epac is reported to sterically auto-inhibit its GEF activity. The binding of cAMP to cAMP-BD-B is believed to induce a conformational change in Epac’s structure that promotes the accessibility of Rap to the catalytic region of Epac [18]. Consequently, the GDP on Rap is exchanged to GTP by Epac, thereby facilitating its function (Fig. 1b).
By taking advantage of this unique character of Epac, several Epac-based fluorescence resonance energy transfer biosensors have been developed to study cAMP compartmentation and dynamics in living cells [10, 19, 20]. Each probe consists of Epac and two fluorescent proteins, one tethered to Epac’s N terminus and the other to the C terminus. The conformational change that follows the binding of cAMP to the cAMP-BD of Epac causes an increase in the distance between these fluorescent proteins and a change in their relative orientation, which jointly inhibit fluorescent resonance energy transfer (FRET) between them. Consequently, we can detect elevated cAMP levels by looking for a reduction in the acceptor fluorescence intensity caused by FRET.
Epac pharmacological modulators
The identification of specific agonists and antagonists for Epac1 and/or Epac2 has facilitated the study of Epac-mediated signaling [21, 22]. The highly conserved glutamate residue that interacts with the 2-hydroxyl of the cAMP ribose group via hydrogen bonds is absent in the cAMP-binding domain of Epac, suggesting that the 2-hydroxyl is not necessary for binding between Epac and cAMP. For cAMP’s high affinity binding with PKA, on the other hand, the 2-hydroxyl of the cAMP ribose group is known to be required. Based on these findings, 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′-5′cyclic monophosphate (8-CPT) was identified as a selective activator for Epac [23]. Although 8-CPT activates both Epac1 and Epac2, a recent study clearly demonstrated that its effect on Epac1 is far more potent than that on Epac2 [24]. In an in vitro experiment, 8-CPT activated recombinant Epac1 about three times more potently than cAMP did, which is why 8-CPT is referred to as a superagonist of Epac1 [25, 26]. Its effect on Epac2, on the other hand, was about half of that caused by cAMP. Recently, Sp-8-BnT-cAMPS (S-220) was identified as an Epac2 selective potent activator. S-220 also appears to be a so-called superagonist of Epac2 [24]. To improve 8-CPT’s permeability through the cellular membrane, the acetoxymethyl (AM) ester of 8-CPT (8-CPT-AM) was synthesized [27]. When 8-CPT-AM comes in contact with cytosol, it is hydrolyzed by cellular esterases and turns into active 8-CPT. In human cells, 8-CPT-AM has been reported to activate Epac to a potency more than 100 times greater than normal. Yet several such cAMP analogues have been reported to inhibit phosphodiesterases (PDEs) as well [28]. Therefore, it should be considered that the consequent elevation of cAMP or cGMP may activate targets other than Epac, including PKA and PKG. Sulfonylurea (SU), a commonly used antidiabetic agent, is also suggested to be an Epac2 agonist [29]. However, a different study indicated that this property was not observed and that SU may instead activate Epac2 via elevation of intracellular cAMP [30].
Recently, several selective inhibitors of Epac have been reported. 5-cyano-6-oxo-1,6-dihydrocyclohexyl (ESI-08) and 3-(5-tert-butyl-isoxazol-3-yl)-2-[(3-chloro-phenyl)-hydrazno]-3-oxopropionitrile (ESI-09) were identified as inhibitors of Epac in a high-throughput screening assay [31, 32]. In addition, structure–activity relationship analysis revealed that 5-cyano-6-oxo-1,6-dihydrocyclopentyl (HJC0197) and 5-cyano-6-oxo-1,6-dihydrocyclopropyl (HJC0198) are potent Epac antagonists [32]. 5225554 and 5376753 were discovered as Epac inhibitors through computational screening of compounds that bind to the hinge region of Epac [33]. Epac mutation near the hinge region was reported to substantially affect its activity [34]. Several isoform-specific Epac inhibitors have been reported as well, including 5,7-dibromo-6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline-1-carbaldehyde (CE3F4) (Epac1-specific) [35], 4-methyl-2,4,6-trimethylphenylsulfone (ESI-05) (Epac2-specific), and ESI-07 (Epac2-specific) [36]. Because they were discovered relatively recently, there are fewer reports on these antagonists than there are on the agonists. Recently, ESI-09 is reported to exhibit a non-specific protein denaturing property at high concentrations [37, 38]. Their target-specificity and effectiveness in vitro and in vivo should be verified in future studies.
Expression
While Epac1 is reported to be expressed ubiquitously in various tissues, the expression patterns of the Epac2 isoforms are relatively tissue-specific [4]. Epac2A is detected most commonly in the brain and pancreatic islets. In contrast, Epac2B and Epac2C are typically expressed in the adrenal glands and liver, respectively [9, 39].
The mechanism by which Epac expression is regulated has not yet been well studied. HIF-1α directly binds to the hypoxia response element (HRE) in Epac1 promoter and up-regulates its transcription [40]. It was also suggested that the methylation state of Epac2 promoter was important for its activity [39]. In addition, microRNA-133, which has been suggested to play protective roles against cardiac dysfunction, fibrosis, and hypertrophy [41, 42], was reported to down-regulate Epac [43]. Further studies are required in order to clarify the molecular mechanisms involved.
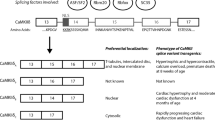
Both Epac1 and Epac2 mRNA are expressed in the mouse heart. Interestingly, it was suggested that Epac2 mRNA becomes predominant over Epac1 mRNA in the adult mouse heart [44]. Several reports indicate that the isoforms’ expression levels in the heart can change under pathological conditions, which is discussed in the following section. Greater mRNA expression of Epac1 than of Epac2 was observed in human hearts, especially in patients with failing hearts [45]. In contrast, Epac1 was down-regulated in cardiac fibroblasts after myocardial infarction or TGF-β stimulation [46].
Distribution and compartmentalization
cAMP is recognized as a critical second messenger of adrenergic stimuli. Yet not all signals that up-regulate cAMP production produce the same effects in the cell. For example, both of the major cardiac AC isoforms, type 5 (AC5) and type 6 (AC6), are expressed in cardiomyocytes, and overexpression of either AC gene causes significant elevation of AC activity [47]. Yet transgenic mice with cardiac-specific overexpression of AC5 or AC6 did not show similar phenotypes in response to cardiac stresses [48–51]. One possible explanation for such inconsistencies is the compartmentalization of signaling molecules in cardiomyocytes [52]. The molecules involved in the transduction of cardiac stress and adrenergic activation-induced signaling are not homogenously located in the cell. Traditional methods for evaluating the intracellular expression of certain molecules, including western blotting, RT-PCR, and ELISA using crude protein extracts from homogenates of cultured cells or tissues, cannot assess variation in molecular localization within the cell; this limitation has caused us to overlook such variation and prevents us from being able to explain several cellular responses. To overcome such difficulties, several biophysical techniques including fluorescent tags for protein labeling, biosensors of small molecules, and electrophysiological recordings using cyclic nucleotide-gated channels, etc., have been developed to study subcellular cAMP signaling. The isoform-specific distribution of each of several signaling molecules, including adrenergic receptors, ACs, PDEs, and A-kinase anchor proteins (AKAPs), has been recognized to play a critical role in the regulation of cells’ complicated responses to various stimuli and conditions. Through attempts to clarify the mechanisms underlying various cellular functions, the intracellular distribution and translocation of these molecules has come to be acknowledged as a very important factor. Here, we summarize the reports on the distribution, translocation, and compartmentalization of Epac1- and Epac2-mediated signaling.
The translocation and distribution of Epac have been reported for various cell types. In particular, the important roles of Epac-mediated signaling in the plasma membrane and the nuclear membrane have been identified. In HEK293 cells, for example, exogenously overexpressed Epac1 has been observed at the plasma membrane, in the cytosol, in the mitochondria, at the nuclear membrane, and inside the nucleus [53]. In cardiomyocytes, a recent report indicates a predominant distribution of Epac1 at the nuclear envelope, while Epac2 is concentrated at the T-tubules [54]. Forskolin- or ISO-induced up-regulation of cAMP and 8-CPT-induced Epac activation resulted in translocation of Epac1 from the cytosol toward the plasma membrane, thereby facilitating the activation of Rap at the membrane [15, 16] and the subsequent enhancement of Rap-induced adhesion of the cells [53]. The DEP domain and conformational change of Epac1 were demonstrated to play essential roles in this translocation [15]. In addition, Epac1 directly binds to phosphatidic acid (PA), a negatively charged phospholipid in the plasma membrane. This interaction was also demonstrated to be important in the translocation of Epac and in Epac1-induced Rap activation [16]. PA, which has been suggested to regulate various cellular functions, has also been reported to bind to important signaling molecules at the plasma membrane including PDE4 [55], Sos (a Ras GEF) [56], and mTOR [57]; the last of which plays a pivotal role in the heart. Thus, PA may function as a point for crosstalk between Epac and these signaling molecules. Interaction between the RA domain of Epac2 and activated Ras was reported to be critical in Ras-induced Epac2 translocation from the cytosol to the plasma membrane [13].
Moreover, in a thyroid cell line, N-terminal Epac1 was reported to bind to radixin [58], a member of the ezrin–radixin–moesin (ERM) family, a group of membrane-associated proteins that plays an important role in the regulation of cell structure and signal transduction [59]. The interaction between Epac1 and ERM was reported to be important in the activated-ERM-induced plasma membrane recruitment of Epac1 [60]. As radixin can also bind to PKA, the complex can serve as a point for crosstalk between PKA and Epac-mediated signaling.
Interaction between Epac1 and β-arrestin2, one of the scaffold proteins for β-AR-mediated signaling, was demonstrated in HEK-293 cells exogenously expressing those proteins [61]. In the same cells, β1-AR, but not β2-AR, induced Epac1 translocation from the cytosol to the plasma membrane via a β-arrestin2-dependent mechanism, resulting in activation of H-Ras through Rap2B, which induces hypertrophic signaling. Nevertheless, both β1- and β2-AR stimulation induced activation of Rap1, which induces non-hypertrophic signaling. β2-AR stimulation-induced competitive inhibition of interaction between β-arrestin2 and Epac1 by endogenous PDE4D5 was suggested as a mechanism for the elimination of β2-AR-induced Epac translocation. It has also been reported that β1-AR stimulation induces formation of a β-arrestin–CaMKII–Epac1 complex [62]. β-Arrestin has consistently been shown to play a pivotal role in ISO- or 8-CPT-induced Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation in the mouse heart. These mechanisms involved in the recruitment of Epac to the plasma membrane may play an important role in the regulation of Epac-mediated signaling.
Epac1 also binds to another small GTPase, Ran-GTP, via its RA domain [63]. Ran is known to exist at the nuclear envelope and to play an important role in nuclear-cytoplasmic transport. It has been demonstrated that Epac1 must interact with Ran at the nuclear membrane in order to activate Rap1. In addition, RanBP2, which binds to Ran, also interacts with Epac1 and recruits it to the nuclear membrane [64]. These interactions, along with H-Ras-mediated signaling [65], may be involved in the regulation by Epac1 of nuclear export of histone deacetylase (HDAC), which is reported to be a pivotal mechanism in Epac-induced cardiac hypertrophy. Phospholipase C (PLC) ε and inositol 1,3,5 triphosphate receptor (IP3R), both located at the nuclear membrane, are also thought to be involved in this mechanism [65, 66]. In cardiomyocytes, muscle-specific AKAP (mAKAP) at the nuclear membrane forms a complex with PKA, PDE4D3, Epac1, and ERK5. This has been suggested to be involved in the Epac1-induced negative regulation of the hypertrophic response [67]. These findings indicate that Epac1 plays an important role in the regulation of the cardiac hypertrophic response at the nuclear membrane.
A recent report evaluated the distribution of Epac in cardiomyocytes using a novel fluorescent cAMP derivate Epac ligand 8-[pharos-575]-2′-O-methyladenosine-3′,5′-cyclic monophosphate (Φ-O-Me-cAMP) [54]. As the probe binds to both Epac1 and Epac2, the authors evaluated the distribution of each isoform separately by using Epac1- or Epac2-deficient mice. In Epac2-deficient mouse hearts, Epac1 localizes at the nuclear envelope. On the other hand, in Epac1-knockout (Epac1 KO) mouse hearts, Epac2 appears to be concentrated around the T-tubules. These findings are compatible with those of other reports showing that Epac1 plays an important role in the induction of hypertrophic gene transcription [68], while Epac2 induces an arrhythmogenic SR Ca2+ leak [69].
Epac under physiological and pathological conditions
The role of Epac in the regulation of cardiac contractility
The first step in cardiac contraction is the transfer of an amount of Ca2+ through the L-type Ca2+ channel (LTCC) into cardiomyocytes in response to action potential-induced depolarization of the plasma membrane [70]. The resulting slight change in intracellular Ca2+ concentration triggers the release of a much larger amount of Ca2+ from the sarcoplasmic reticulum (SR) via ryanodine receptor 2 (RyR2), in a process called Ca2+-induced Ca2+ release (CICR). The consequent elevation of cytosolic Ca2+ concentration increases binding between Ca2+ and troponin C, thereby initiating sliding of muscle filaments. In the relaxation phase, the increased cytosolic Ca2+ is removed through the action of several Ca2+ handling proteins including SR Ca2+-ATPase, sarcolemmal Na+/Ca2+ exchanger, and sarcolemmal Ca2+-ATPase [71].
Like PKA, Epac has been indicated to enhance cardiac contractility (Fig. 2). In adult mouse ventricular myocytes (AMVM), 8-CPT treatment increased the amplitude of the Ca2+ transient via Rap-, PLCε-, protein kinase C (PKC) ε-, and CaMKII-mediated signaling [72, 73]. In addition, 8-CPT treatment enhanced phosphorylation of several CaMKII sites of Ca2+ handling proteins including RyR2 (Ser2815) and phospholamban (PLN) (Thr17), indicating that the consequent activation of those proteins may facilitate Ca2+ release from SR and its re-uptake. The critical role of PLCε in catecholamine-induced increases in Ca2+ transient amplitude and contractile function was demonstrated in an experiment using PLCε−/− mice [74]. Both of the major functions of PLCε, namely, PIP2 hydrolysis and Rap guanine nucleotide exchange (GEF), were demonstrated to be critical in the 8-CPT-induced elevation of Ca2+ transient amplitude [73]. As mentioned above, PLCε is known to be stimulated by Epac through Rap activation [75]. Therefore, following the activation of Epac, PLCε and Rap may presumably activate each other, thereby enhancing their signaling.

The role of Epac in cardiomyocytes. a Epac has been indicated to enhance cardiac contractility. Epac increased the amplitude of the Ca2+ transient in a process dependent on Rap-, PLCε-, PKCε-, and CaMKII-mediated signaling. Epac enhanced the phosphorylation of several Ca2+ handling proteins including RyR2 and PLN, thereby possibly facilitating Ca2+ release from SR and re-uptake. Epac also enhanced cardiac myofilament Ca2+ sensitivity, possibly through phosphorylation of cTnI and cMyBP-C via PKC- and CaMKII-mediated signaling. Adrenergic activation-induced hyperphosphorylation of PLN and RyR2 was reported to be involved in the development of HF and arrhythmias. b Epac has been indicated to regulate cardiac hypertrophy. Epac induced activation of the pro-hypertrophic transcription factors NFAT and MEF-2 in a CaMKII-dependent manner. Epac also induced nuclear export of HDAC4 and HDAC5, both of which inhibit MEF-2 activity in the nucleus The pro-hypertrophic kinase ERK5, which forms a complex with Epac and mAKAP at the nuclear membrane, was attenuated by Epac through Rap activation
In myocytes purified from 4-week-old, 8-CPT-treated rat hearts, an increased amplitude of Ca2+ transient and cell shortening were observed [76]. In this model, SR Ca2+ content was also elevated by 8-CPT treatment. In addition, 8-CPT treatment increased the L-type Ca2+ current in adult rat ventricular myocytes (ARVM). PLCε deficiency, on the other hand, caused no significant change in the current in AMVM [73].
Additionally, activation of Epac-mediated signaling was reported to induce an increase in cardiac myofilament Ca2+ sensitivity via PKC- and CaMKII-mediated signaling [77]. In AMVM, 8-CPT treatment induced phosphorylation of troponin I (cTnI) and cardiac myosin binding protein-C (cMyBP-C), both of which play critical roles in the regulation of Ca2+ sensitivity, independent of PKA.
An in vivo study suggested that Epac1 also has a promoting effect on cardiac contractility. We found that phosphorylation of PLN (Ser16), which has been reported as a major PKA-dependent phosphorylation site, is significantly attenuated in Epac1 KO mouse hearts [78] (Fig. 2). We did not observe any significant difference between WT and Epac1 KO mouse hearts in protein expression levels of molecules involved in catecholamine-mediated signaling, including β1- AR, β2-AR, β-AR kinase (βARK), Gsα, Giα, Gβ, Gq, type 5/6 AC, and PKA subunits. Ca2+ transient amplitude, however, was significantly smaller in the ARVM derived from Epac1 KO mice than in the wild type. The decay rate was also smaller and the decay time constant (τ) was significantly prolonged in Epac1 KO cardiomyocytes. Moreover, the amplitude of the caffeine-induced increase in cytoplasmic Ca2+ concentration, which represents the Ca2+ content in SR, was smaller in Epac1 KO cardiomyocytes. Consistently, left ventricle ejection fraction (LVEF) in the basal state was significantly decreased and LV end-systolic diameter (LVESD) was increased in Epac1 KO mice, indicating that Epac1 plays a critical role in the maintenance of contractility.
Interestingly, the responses of cardiac contractility to ISO stimulation were well preserved in Epac1 KO mice. During continuous intravenous infusion of ISO (0.13–0.40 µg/kg/min), the resulting increase in LVEF and decrease in LVESD reached similar levels in Epac1 KO mice and WT under the same treatment conditions [78], suggesting that Epac1 may not be involved in acute regulation of contractility in response to catecholamine signals. In Epac2 KO mouse hearts under PLN phosphorylation, no significant difference from WT mice was observed in Ser16, Thr17, LVEF, or LVESD in the basal state [78], indicating that the important role of Epac in maintenance of contractility may be mainly attributed to Epac1 rather than Epac2.
Nevertheless, several studies have failed to observe a promoting effect of Epac1 on cardiac contractility. For instance, a study using an alternate Epac KO mouse line showed that no significant difference in cardiac contractility in the basal state was associated with a deficiency of Epac1, Epac2, or both isoforms [69, 79]. The different genetic backgrounds of the different mouse lines might cause these discrepancies in the reported phenotypes of Epac1 KO mice. In addition, in some reports, Epac activation failed to increase, or even decreased, the amplitude of the Ca2+ transient. In ARVM, 8-CPT treatment decreased the amplitude of the Ca2+ transient [77, 80] and prolonged the decay time constant [80]. Differences in the species used and in the timing of Ca2+ transient measurement were suggested to be responsible for this inconsistency [81].
The role of Epac in the development of cardiac hypertrophy
As an adaptation to pressure- or volume-overload, cardiomyocytes increase the organization of sarcomere, leading to cardiac hypertrophy. The increase of cardiac wall thickness results in the alleviation of wall stress, thereby diminishing oxygen consumption [82]. Although the larger working structure created under these conditions generally supports the maintenance of cardiac function, in pathological conditions the vascular system may not develop sufficiently to meet those additional demands, reportedly leading to heart failure [83]. In addition, cardiac hypertrophy is demonstrated to be related to a poor prognosis and high risk for cardiovascular diseases [84]. Thus, prevention of cardiac hypertrophy is one of the major clinical issues in the management of heart diseases.
Epac is reported to be up-regulated in the hypertrophic heart. In a thoracic aortic constriction (TAC)-induced animal cardiac hypertrophy model, the elevation of Epac1 mRNA [44] and protein [45] expression were observed. In an ISO-induced mouse cardiac hypertrophy model, meanwhile, both Epac1 and Epac2 mRNA were up-regulated [44].
Epac has been reported to have both pro-hypertrophic and anti-hypertrophic effects (Fig. 2). Several small G proteins including Rap, Ras, and Rac have been reported to be involved in Epac-induced cardiac hypertrophy. Epac has been shown to stimulate PLCε, which plays an important role in the development of cardiac hypertrophy [85], through Rap activation [75]. In PLCε down-regulated NRVM, hypertrophic growth was drastically reduced. In addition, cardiomyocyte-specific PLCε knockout mice were reported to be resistant to stress-induced cardiac hypertrophy [66]. PLCε was shown to form a complex with Epac1, muscle-specific A-kinase anchoring protein (mAKAP), and the pro-hypertrophic kinases PKCε and PKD at the nuclear envelope. These findings suggest that Epac1 activation may lead to the acceleration of hypertrophic signals. In Epac1 KO mice, the attenuation of ISO-induced hypertrophic remodeling was observed [79]. Interestingly, in the same study, Epac was also reported to induce autophagy in NRVM via a Rap, Ca2+, Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ), AMP-dependent protein kinase (AMPK) pathway, thereby suppressing hypertrophy.
In ARVM, 8-CPT, an Epac-activating agent, induced hypertrophy through Ras-, calcineurin-, and CaMKII-mediated signaling independent of PKA and Rap1 activation [45]. Additionally, Epac is reported to activate H-Ras via PLC/IP3R-mediated signaling and elevation of intracellular Ca2+ in neonatal rat cardiomyocytes (NRVM) [65]. 8-CPT also induced activation of the pro-hypertrophic transcription factors nuclear factor of activated T cell (NFAT) and myocyte enhancer factor 2 (MEF-2) in a CaMKII-dependent manner. Consistently, after 8-CPT treatment, the nuclear export of histone deacetylase 4 (HDAC4) and HDAC5, which inhibit MEF-2 activity in the nucleus, was observed in COS cells [65] and ARVM [68]. Epac-induced elevation of nuclear Ca2+ was suggested to be involved in the mechanism controlling this process [68, 86]. The important role of Epac1 in the translocation of HDAC5 was also demonstrated in an experiment using Epac1-deficient mouse cardiomyocytes [54]. As mentioned above, an increased concentration of Epac1 in the nuclear membrane was observed in AMVM [54]. In addition, it was reported that β1-AR, but not β2-AR, induces Epac1 translocation from the cytosol to the plasma membrane via a β-arrestin2-dependent mechanism in HEK-293 cells, resulting in the activation of H-Ras through Rap2B, thereby inducing hypertrophic signaling [61].
In HEK-293 cells, Epac directly activates R-Ras and phospholipase D [87], both of which may play a role in the development of cardiac hypertrophy [88]. Another small G protein, Rac, is also reported to be involved in Epac-induced Ca2+- and NFAT-dependent cardiac hypertrophy in NRVM [89].
Anti-hypertrophic effects of Epac have also been suggested. It is reported that the activity of the pro-hypertrophic kinase ERK5 [90] is attenuated by Epac through Rap activation [67]. Both ERK5 and Epac are also components of the mAKAP complex, which localizes at the nuclear membrane of NRVM. In addition, neither our group nor another group observed any difference in heart size between Epac1 KO and WT mice at baseline or after pressure overload [69, 78]. In the ISO-induced hypertrophy model, Epac1 deficiency inhibited the development of hypertrophy after long-term ISO infusion [79], but not in another study with relatively short and high-dose treatment [78].
As mentioned above, the effect of Epac on the development of cardiac hypertrophy remains controversial. Further studies using tissue-specific conditional knockout animal models, as well as functional in vivo Epac specific ligands are required in order to confirm the role of Epac proteins in cardiac hypertrophy.
The role of Epac in the regulation of cardiomyocyte apoptosis
Cardiomyocyte apoptosis has been recognized as an important mechanism for the development of cardiac dysfunction [91–93]. The role of Epac in apoptosis has been reported in various studies. Both pro-apoptotic and anti-apoptotic roles of Epac have been suggested in different cell types and study conditions. In cardiomyocytes, the role of Epac remains controversial.
Several reports have indicated that Epac activation inhibits cardiomyocyte death. Glucagon-like peptide-1 receptors (GLP-1Rs), one of the G protein-coupled receptors (GPCR), have been reported to evoke cAMP-mediated signaling pathways. GLP-1R stimulation with exendin-4, a potent agonist for the receptor as well as an established agent for the treatment of type 2 diabetes, prevented H2O2-induced reactive oxygen species production and caspase-3 activation and apoptosis in cardiomyocytes. These effects were significantly attenuated by Epac1 silencing with siRNA in NRVMs [94]. Exendin-4 induced up-regulation of the anti-apoptotic protein Bcl-2, while antioxidant enzymes, including catalase, glutathione peroxidase-1, and manganese superoxide dismutase, were reduced by down-regulation of Epac1 with SiRNA. These findings indicate that Epac1 may play an important role in the protective effect of GLP-1R agonists against ischemia-induced cardiomyocyte apoptosis.
Phosphodiesterase (PDE) catalyzes the hydrolysis of cAMP, thereby down-regulating the intracellular cAMP concentration. Roflumilast, an inhibitor of the major cardiac PDE subtype PDE4, protects neonatal rat cardiomyocytes from NO-induced apoptosis in an Epac1-dependent manner [95]. Epac1-induced AKT phosphorylation was indicated to be involved in this effect.
In cultured adult feline ventricular myocytes (AFVMs), 8-CPT exerted a protective effect against high extracellular Ca2+-induced cell death in an ERK1/2 activation-dependent manner [96].
Several other studies, in contrast, have suggested a pro-apoptotic effect of Epac-mediated signaling. In vivo, pressure overload-induced up-regulation of Bax, a pro-apoptotic protein, and an increase in TUNEL-positive cardiomyocytes were significantly attenuated in Epac1 KO mouse hearts [78], indicating that the absence of Epac1 has a protective effect against apoptosis. Similarly, cardiomyocyte apoptosis induced by chronic catecholamine stress was also attenuated by Epac1 deficiency in vivo.
In addition, Epac activation induces apoptosis in mouse cortical neurons, but not in NRVMs. The absence of Bim expression in cardiomyocytes was suggested as a possible reason for this difference [97].
These inconsistencies indicate that further studies are necessary to clarify the role of Epac in the regulation of cardiomyocyte apoptosis.
The role of Epac in the development of heart failure
In keeping with the protective effect of Epac1 deficiency against the induction of cardiomyocyte apoptosis, pressure overload-induced cardiac dysfunction was significantly attenuated in Epac1 KO mice [78]. TAC-induced pressure overload did not cause significant cardiac dysfunction in Epac1 KO mice. Evaluated indices of cardiac function including LVEF, LVESD, maximum dp/dt, and minimum dp/dt did not change significantly 3 weeks after TAC in Epac1 KO mice, whereas in WT mice those indexes changed to indicate the onset of heart failure. Epac2 KO mice, on the other hand, exhibited significant attenuation of cardiac function similar to that seen in WT mice, indicating that Epac1 rather than Epac2 plays a critical role in the development of heart failure. In addition, in an aging [78] and chronic catecholamine stimulation [78, 79] induced mouse heart failure model, Epac1 deficiency exerts a protective effect against the development of cardiac dysfunction, indicating that Epac1 plays critical roles in stress-induced cardiac dysfunction. In Epac1 KO mouse hearts, we found that PLN phosphorylation at Ser16 was significantly attenuated in the basal state and after ISO stimulation. PLN is a negative regulator of SR Ca2+ transporting adenosine triphosphatase (SERCA2a) function. Thus, PLN attenuates both relaxation and contraction of hearts [98]. Phosphorylation of PLN at Ser16 has been reported to inhibit PLN function, thereby enhancing cardiac function. Although the role of PLN function in the development of heart failure remains controversial [99–102], the activated state of PLN in Epac1 KO mice may contribute to this genotype’s resistance to stress-induced heart failure (Fig. 2).
It has been indicated that harmful effects of β-AR signaling on the development of heart diseases may be mediated by AC5 [48, 103]. Recent report demonstrated that Epac1 deficiency attenuates AC5-mediated catecholamine stress-induced cardiac dysfunction, apoptosis, fibrosis and arrhythmogenesis [104].
Nevertheless, the role of Epac activation in the development of heart failure remains controversial, because experiments with another Epac1 KO mouse line showed no significant effect of Epac1 deficiency on the development of pressure overload-induced heart failure [69].
The role of Epac in the development of arrhythmia
Arrhythmias are among the most common problems seen in patients with cardiac diseases. In fact, severe ventricular arrhythmia is one of the main causes of sudden death. The presence of atrial fibrillation (AF) significantly increases the risk of stroke. Numerous studies have demonstrated that catecholamine stimulation plays a pivotal role in the development of these arrhythmias [105–107]. Accumulating evidence indicates that Epac-mediated signaling regulates several other signaling pathways that are involved in the development of arrhythmias, including calcium handling, K+ channel currency [108, 109], and Na+ channel function [110].
Epac activation has been suggested to have a pro-arrhythmic effect. Adrenergic receptor (AR) activation has been reported to induce SR Ca2+ leak and consequent spontaneous SR Ca2+ release (SCR) [111, 112]. Catecholamine-induced phosphorylation of RYR2 by protein kinase A or CaMKII was thought to be involved in this process. Diastolic SR Ca2+ leak is responsible for the development of delayed afterdepolarization (DAD), which is recognized as a major source of ectopic activity [105, 113]. Several reports indicate that Epac is involved in the arrhythmogenic effect of catecholamine (Fig. 2).
8-CPT treatment increased SCR and Ca2+ waves in ARVM [76]. In AMVM, the 8-CPT-induced increase in SCR was attenuated by either CaMKII inhibition via KN-93 treatment or inhibition of RYR2 phosphorylation at the CaMKII site (Ser2814), indicating that RYR2 phosphorylation by CaMKII is involved in this process. In addition, in perfused mouse hearts, 8-CPT infusion significantly increased the incidence of extrastimuli pacing-induced ventricular tachycardia (VT) [114]. Moreover, Epac2 deficiency completely abolished the 8-CPT-induced SCR increase in mouse cardiomyocytes [69] and decreased the ISO-induced increase in extrastimuli pacing-induced VT, while Epac1 KO did not affect the incidence of 8-CPT-induced SCR increase, indicating that Epac2 rather than Epac1 is involved in the arrhythmogenic effect. Consistently, Epac2 in AMVM was found to be concentrated in the T-tubules, where the calcium-handling proteins involved in CICR also occur [54].
On the other hand, the duration of pacing-induced AF was significantly shorter in Epac1 KO mice [78]. In addition, an Epac1-specific inhibitor attenuated 8-CPT-induced spontaneous Ca2+ waves in ARVM, indicating that Epac1 is also involved in the development of arrhythmias [115]. ISO-induced phosphorylation of RyR2 was attenuated at both the PKA site (Ser2808) and the CaMKII site (Ser2814) by silencing of the Epac1 gene with siRNA in NRVM.
In addition, it has been reported that 8-CPT treatment up-regulates transient receptor potential canonical channels isoform 3 (TRPC3) and 4 (TRPC4) and enhances store-operated Ca2+ entry (SOCE)-like activity in ARVM [115]. 8-CPT-induced spontaneous Ca2+ waves were inhibited by a TPPC3 antagonist. These findings indicate that Epac may cause a pro-arrhythmic effect through activation of TRPCs.
Prolongation of the action potential duration (APD) in the failing heart has been demonstrated in studies using animal models and human cardiomyocytes [116, 117]. APD prolongation has also been suggested to play an important role in arrhythmogenesis by increasing susceptibility to early afterdepolarizations and inducing dispersion of repolarization [118]. Sustained catecholamine stimulation induced down-regulation of potassium voltage-gated channel subfamily E member 1 (KCNE1), and the consequent repression of slow delayed-rectifier K+ current (I Ks) was suggested to be involved in this mechanism [108, 109]. In guinea pig ventricular cardiomyocytes, ISO treatment decreased mRNA and membrane protein expression of KCNE1 through β1-AR-, Epac1-, Ca2+-, calcineurin-, and NFAT-mediated signaling, independent of PKA activity. In vivo activation of Epac by 8-CPT infusion in guinea pig decreased KCNE1 expression and IKs density. 8-CPT-induced prolongation of APD was also observed in ARVM [109]. In that same study, the decrease in sustained K+ current was suggested to be responsible for the effect.
Cardiac Na current (I Na) has been also reported to play an important role in the development of arrhythmia [119, 120]. In addition, the enhancement of late I Na due to Na channel phosphorylation by CaMKII, which can be activated by Epac, has been suggested to be involved in the mechanism [121]. 8-CPT treatment enhanced late I Na, while inhibition of PKA with PKI did not affect the ISO-induced late I Na enhancement in AMVM, indicating that Epac is involved in catecholamine-induced regulation of late INa [110]. These findings indicate that Epac1 plays an important role in catecholamine-induced arrhythmogenesis in the failing heart.
Epac is also reported to be involved in the neoformation of gap junctions (GJ). Accumulation of Connexin43 (Cx43), a critical component of the GJ in cardiomyocytes, and N-cadherin at the cell–cell contacts [122] were induced by 8-CPT treatment in neonatal rat cardiomyocytes. PKC-dependent phosphorylation of Cx43 accompanied by enhanced gap junctional intercellular communication was also observed in 8-CPT-treated NRVM [123]. Since Cx43 deficiency has been demonstrated to cause arrhythmia [124, 125], Epac function may affect susceptibility to arrhythmia via regulation of GJ formation.
The role of Epac in the development of cardiac fibrosis
It has been reported that about 27 % of the cells in the adult murine heart are fibroblasts, while 56 % are cardiomyocytes [126]. Cardiac fibroblasts play an important role in the development of the extracellular matrix (ECM) of the myocardium [127]. The effect of Epac on the development of cardiac fibrosis remains controversial (Fig. 3).

The role of Epac in the development of cardiac fibrosis. Epac activation decreased collagen mRNA expression in cardiac fibroblasts. Moreover, pro-fibrotic factors, including TGFβ1 and Ang II, inhibited Epac1 expression. Ang II up-regulates PDE1A, thereby inducing collagen synthesis via the Epac1-mediated signaling pathway. Epac was reported to play a pivotal role in β-adrenergic receptor activation-induced production of interleukin-6 (IL-6), which is reported to mediate cardiac fibrosis
8-CPT treatment decreased collagen mRNA expression and DNA synthesis independent of Rap1 expression level in adult rat cardiac fibroblasts [46]. In addition, Epac overexpression in cardiac fibroblasts resulted in the repression of transforming growth factor β1 (TGFβ1)-induced collagen synthesis, indicating that Epac1-mediated signaling represses cardiac fibrogenesis. Moreover, pro-fibrotic factors, including TGFβ1 and angiotensin II (Ang II), inhibited the mRNA expression of Epac1 in cardiac fibroblasts. These factors are believed to play pivotal roles in the transformation of fibroblasts to myofibroblasts [127], the latter of which exhibit enhanced ECM protein synthesis activity. Consistently, the expression level of Epac1 was decreased in murine cardiac fibroblasts after myocardial infarction. In the adult rat cardiac fibroblast, Ang II-induced collagen synthesis was attenuated by 8-CPT treatment through phosphoinositol-3 kinase (PI3K) [128]. In the same study, Epac was thought to be involved in the anti-fibrotic effect of adenosine A2 receptor activation.
It has been indicated that Ang II induces collagen synthesis via regulation of a phosphodiesterase 1A (PDE1A)-, cAMP-, Epac1-, and Rap1-mediated signaling pathway in neonatal rat cardiac fibroblasts [129]. Ang II-induced PDE1A up-regulation, which could depress cAMP concentration, is suggested to be involved in the mechanism. PDE1A has also been demonstrated to be involved in the Ang II-induced transformation of fibroblasts to myofibroblasts.
Several other reports, in contrast, have failed to observe any anti-fibrotic effect of Epac. In neonatal mouse cardiac fibroblasts, Epac was reported to play a pivotal role in β-adrenergic receptor activation-induced production of interleukin-6 (IL-6) [130], which is reported to mediate cardiac fibrosis [131]. Activation of the PKCδ/p38 MAPK pathway by Epac was thought to be involved in this mechanism. In vivo, no difference in the severity of cardiac fibrosis was observed between WT and Epac1 KO at baseline [78]. In addition, the development of cardiac fibrosis induced by various stresses, including TAC, ISO infusion, and aging, was attenuated in Epac1 KO mice [78, 79]. However, based on these findings, we cannot conclude that Epac1 promotes pro-fibrotic signaling in cardiac fibroblasts, because these results were obtained in whole-body Epac1 KO mice. The loss of cardiomyocytes induces cardiac fibrosis as a means of replacing damaged cardiomyocytes with ECM to maintain cardiac structure. Therefore, Epac deficiency-induced differences in cardiomyocyte death may subsequently affect the amount of fibrosis in the heart. Further studies using tissue-specific conditional KO animals or in vivo Epac specific ligands will be important in clarifying the role of Epac in the development of cardiac fibrosis.
Conclusion
Numerous studies on Epac have disclosed its important functions in various tissues in the body. Recently, experiments using genetically manipulated mice in various pathogenic models have begun to reveal the in vivo significance of earlier findings that were observed in vitro or on a cellular level.
Interestingly, though the significance of Epac’s functions in various cells is widely accepted, systemic Epac1 and Epac2 double-knockout mice were reported to be viable and to show no significant differences from WT mice, at least at baseline, in heart structure or function [69]. This characteristic may make Epac a more desirable therapeutic target because it suggests that anti-Epac therapy will not result in very many or very severe side effects. Further in vivo investigations using tissue-specific and/or inducible conditional Epac-deficient animals or Epac pharmacological modulators will provide additional information regarding useful methods of temporal and spatial regulation of each Epac isoform’s activity for the treatment or prevention of cardiac diseases.
In addition, the relationships between Epac and other cAMP target molecules should be further clarified. PKA and Epac, for example, can work either cooperatively or in opposition to one another, depending on the cell type and conditions. Clarifying the roles of these molecules in each of their possible pathways and the detailed mechanisms of the crosstalk between them will aid in understanding their signaling. To achieve this, we must also come to understand the compartmentalization of each of these signaling molecules.
References
Lymperopoulos A, Rengo G, Koch WJ (2013) Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113(6):739–753. doi:10.1161/circresaha.113.300308
Ishikawa Y, Homcy CJ (1997) The adenylyl cyclases as integrators of transmembrane signal transduction. Circ Res 80(3):297–304
Sands WA, Palmer TM (2008) Regulating gene transcription in response to cyclic AMP elevation. Cell Signal 20(3):460–466. doi:10.1016/j.cellsig.2007.10.005
Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science (New York, NY) 282(5397):2275–2279
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396(6710):474–477. doi:10.1038/24884
Schmidt M, Dekker FJ, Maarsingh H (2013) Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev 65(2):670–709. doi:10.1124/pr.110.003707
Lezoualc’h F, Fazal L, Laudette M, Conte C (2016) Cyclic AMP sensor EPAC proteins and their role in cardiovascular function and disease. Circ Res 118(5):881–897. doi:10.1161/circresaha.115.306529
Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG (2006) Cell physiology of cAMP sensor Epac. J Physiol 577(Pt 1):5–15. doi:10.1113/jphysiol.2006.119644
Niimura M, Miki T, Shibasaki T, Fujimoto W, Iwanaga T, Seino S (2009) Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J Cell Physiol 219(3):652–658. doi:10.1002/jcp.21709
Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K (2004) Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep 5(12):1176–1180. doi:10.1038/sj.embor.7400290
de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL (2000) Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem 275(27):20829–20836. doi:10.1074/jbc.M001113200
Parnell E, Smith BO, Yarwood SJ (2015) The cAMP sensors, EPAC1 and EPAC2, display distinct subcellular distributions despite sharing a common nuclear pore localisation signal. Cell Signal 27(5):989–996. doi:10.1016/j.cellsig.2015.02.009
Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA (2006) The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem 281(5):2506–2514. doi:10.1074/jbc.M508165200
Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL (2008) Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 455(7209):124–127. doi:10.1038/nature07187
Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X (2002) Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem 277(29):26581–26586. doi:10.1074/jbc.M203571200
Consonni SV, Gloerich M, Spanjaard E, Bos JL (2012) cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc Natl Acad Sci USA 109(10):3814–3819. doi:10.1073/pnas.1117599109
Ueno H, Shibasaki T, Iwanaga T, Takahashi K, Yokoyama Y, Liu LM, Yokoi N, Ozaki N, Matsukura S, Yano H, Seino S (2001) Characterization of the gene EPAC2: structure, chromosomal localization, tissue expression, and identification of the liver-specific isoform. Genomics 78(1–2):91–98. doi:10.1006/geno.2001.6641
Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL (2006) Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 439(7076):625–628. doi:10.1038/nature04468
Salonikidis PS, Niebert M, Ullrich T, Bao G, Zeug A, Richter DW (2011) An ion-insensitive cAMP biosensor for long term quantitative ratiometric fluorescence resonance energy transfer (FRET) measurements under variable physiological conditions. J Biol Chem 286(26):23419–23431. doi:10.1074/jbc.M111.236869
Sprenger JU, Nikolaev VO (2013) Biophysical techniques for detection of cAMP and cGMP in living cells. Int J Mol Sci 14(4):8025–8046. doi:10.3390/ijms14048025
Chen H, Wild C, Zhou X, Ye N, Cheng X, Zhou J (2014) Recent advances in the discovery of small molecules targeting exchange proteins directly activated by cAMP (EPAC). J Med Chem 57(9):3651–3665. doi:10.1021/jm401425e
Parnell E, Palmer TM, Yarwood SJ (2015) The future of EPAC-targeted therapies: agonism versus antagonism. Trends Pharmacol Sci 36(4):203–214. doi:10.1016/j.tips.2015.02.003
Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL (2002) A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol 4(11):901–906. doi:10.1038/ncb874
Schwede F, Bertinetti D, Langerijs CN, Hadders MA, Wienk H, Ellenbroek JH, de Koning EJ, Bos JL, Herberg FW, Genieser HG, Janssen RA, Rehmann H (2015) Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol 13(1):e1002038. doi:10.1371/journal.pbio.1002038
Rehmann H, Schwede F, Doskeland SO, Wittinghofer A, Bos JL (2003) Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem 278(40):38548–38556. doi:10.1074/jbc.M306292200
Courilleau D, Bouyssou P, Fischmeister R, Lezoualc’h F, Blondeau JP (2013) The (R)-enantiomer of CE3F4 is a preferential inhibitor of human exchange protein directly activated by cyclic AMP isoform 1 (Epac1). Biochem Biophys Res Commun 440(3):443–448. doi:10.1016/j.bbrc.2013.09.107
Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H (2008) 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem Eur J Chem Biol 9(13):2052–2054. doi:10.1002/cbic.200800216
Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E (2008) Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods 5(4):277–278. doi:10.1038/nmeth0408-277
Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S (2009) The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science (New York, NY) 325(5940):607–610. doi:10.1126/science.1172256
Tsalkova T, Gribenko AV, Cheng X (2011) Exchange protein directly activated by cyclic AMP isoform 2 is not a direct target of sulfonylurea drugs. Assay Drug Dev Technol 9(1):88–91. doi:10.1089/adt.2010.0338
Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X (2013) A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol 83(1):122–128. doi:10.1124/mol.112.080689
Chen H, Tsalkova T, Mei FC, Hu Y, Cheng X, Zhou J (2012) 5-Cyano-6-oxo-1,6-dihydro-pyrimidines as potent antagonists targeting exchange proteins directly activated by cAMP. Bioorg Med Chem Lett 22(12):4038–4043. doi:10.1016/j.bmcl.2012.04.082
Brown LM, Rogers KE, Aroonsakool N, McCammon JA, Insel PA (2014) Allosteric inhibition of Epac: computational modeling and experimental validation to identify allosteric sites and inhibitors. J Biol Chem 289(42):29148–29157. doi:10.1074/jbc.M114.569319
Tsalkova T, Blumenthal DK, Mei FC, White MA, Cheng X (2009) Mechanism of Epac activation: structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J Biol Chem 284(35):23644–23651. doi:10.1074/jbc.M109.024950
Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, Blondeau JP, Lezoualc’h F (2012) Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J Biol Chem 287(53):44192–44202. doi:10.1074/jbc.M112.422956
Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL Jr, Cheng X (2012) Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci USA 109(45):18613–18618. doi:10.1073/pnas.1210209109
Rehmann H (2013) Epac-inhibitors: facts and artefacts. Sci Rep 3:3032. doi:10.1038/srep03032
Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X (2015) Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep 5:9344. doi:10.1038/srep09344
Hoivik EA, Witsoe SL, Bergheim IR, Xu Y, Jakobsson I, Tengholm A, Doskeland SO, Bakke M (2013) DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS One 8(7):e67925. doi:10.1371/journal.pone.0067925
Lai TW, Lin SZ, Lee HT, Fan JR, Hsu YH, Wang HJ, Yu YL, Shyu WC (2012) HIF-1alpha binding to the Epac1 promoter recruits hematopoietic stem cells to the ischemic brain following stroke. J Mol Cell Biol 4(3):184–187. doi:10.1093/jmcb/mjs009
Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW, 2nd (2010) MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res 106(1):166–175. doi:10.1161/circresaha.109.202176
Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13(5):613–618. doi:10.1038/nm1582
Castaldi A, Zaglia T, Di Mauro V, Carullo P, Viggiani G, Borile G, Di Stefano B, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G, Catalucci D (2014) MicroRNA-133 modulates the beta1-adrenergic receptor transduction cascade. Circ Res 115(2):273–283. doi:10.1161/circresaha.115.303252
Ulucan C, Wang X, Baljinnyam E, Bai Y, Okumura S, Sato M, Minamisawa S, Hirotani S, Ishikawa Y (2007) Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am J Physiol Heart Circ Physiol 293(3):H1662–H1672. doi:10.1152/ajpheart.00159.2007
Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F (2008) Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res 102(8):959–965. doi:10.1161/circresaha.107.164947
Yokoyama U, Patel HH, Lai NC, Aroonsakool N, Roth DM, Insel PA (2008) The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc Natl Acad Sci USA 105(17):6386–6391. doi:10.1073/pnas.0801490105
Iwatsubo K, Bravo C, Uechi M, Baljinnyam E, Nakamura T, Umemura M, Lai L, Gao S, Yan L, Zhao X, Park M, Qiu H, Okumura S, Iwatsubo M, Vatner DE, Vatner SF, Ishikawa Y (2012) Prevention of heart failure in mice by an antiviral agent that inhibits type 5 cardiac adenylyl cyclase. Am J Physiol Heart Circ Physiol 302(12):H2622–H2628. doi:10.1152/ajpheart.00190.2012
Vatner SF, Park M, Yan L, Lee GJ, Lai L, Iwatsubo K, Ishikawa Y, Pessin J, Vatner DE (2013) Adenylyl cyclase type 5 in cardiac disease, metabolism, and aging. Am J Physiol Heart Circ Physiol 305(1):H1–H8. doi:10.1152/ajpheart.00080.2013
Lai L, Yan L, Gao S, Hu CL, Ge H, Davidow A, Park M, Bravo C, Iwatsubo K, Ishikawa Y, Auwerx J, Sinclair DA, Vatner SF, Vatner DE (2013) Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation 127(16):1692–1701. doi:10.1161/circulationaha.112.001212
Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WY, Clopton P, Hammond HK (2006) Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 114(5):388–396. doi:10.1161/circulationaha.106.632513
Roth DM, Bayat H, Drumm JD, Gao MH, Swaney JS, Ander A, Hammond HK (2002) Adenylyl cyclase increases survival in cardiomyopathy. Circulation 105(16):1989–1994
Timofeyev V, Myers RE, Kim HJ, Woltz RL, Sirish P, Heiserman JP, Li N, Singapuri A, Tang T, Yarov-Yarovoy V, Yamoah EN, Hammond HK, Chiamvimonvat N (2013) Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circ Res 112(12):1567–1576. doi:10.1161/circresaha.112.300370
Ponsioen B, Gloerich M, Ritsma L, Rehmann H, Bos JL, Jalink K (2009) Direct spatial control of Epac1 by cyclic AMP. Mol Cell Biol 29(10):2521–2531. doi:10.1128/mcb.01630-08
Pereira L, Rehmann H, Lao DH, Erickson JR, Bossuyt J, Chen J, Bers DM (2015) Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc Natl Acad Sci USA 112(13):3991–3996. doi:10.1073/pnas.1416163112
Grange M, Sette C, Cuomo M, Conti M, Lagarde M, Prigent AF, Nemoz G (2000) The cAMP-specific phosphodiesterase PDE4D3 is regulated by phosphatidic acid binding. Consequences for cAMP signaling pathway and characterization of a phosphatidic acid binding site. J Biol Chem 275(43):33379–33387. doi:10.1074/jbc.M006329200
Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D (2007) Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol 9(6):706–712. doi:10.1038/ncb1594
Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science (New York, NY) 294(5548):1942–1945. doi:10.1126/science.1066015
Hochbaum D, Barila G, Ribeiro-Neto F, Altschuler DL (2011) Radixin assembles cAMP effectors Epac and PKA into a functional cAMP compartment: role in cAMP-dependent cell proliferation. J Biol Chem 286(1):859–866. doi:10.1074/jbc.M110.163816
Bretscher A, Edwards K, Fehon RG (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 3(8):586–599. doi:10.1038/nrm882
Gloerich M, Ponsioen B, Vliem MJ, Zhang Z, Zhao J, Kooistra MR, Price LS, Ritsma L, Zwartkruis FJ, Rehmann H, Jalink K, Bos JL (2010) Spatial regulation of cyclic AMP-Epac1 signaling in cell adhesion by ERM proteins. Mol Cell Biol 30(22):5421–5431. doi:10.1128/mcb.00463-10
Berthouze-Duquesnes M, Lucas A, Sauliere A, Sin YY, Laurent AC, Gales C, Baillie G, Lezoualc’h F (2013) Specific interactions between Epac1, beta-arrestin2 and PDE4D5 regulate beta-adrenergic receptor subtype differential effects on cardiac hypertrophic signaling. Cell Signal 25(4):970–980. doi:10.1016/j.cellsig.2012.12.007
Mangmool S, Shukla AK, Rockman HA (2010) beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol 189(3):573–587. doi:10.1083/jcb.200911047
Liu C, Takahashi M, Li Y, Dillon TJ, Kaech S, Stork PJ (2010) The interaction of Epac1 and Ran promotes Rap1 activation at the nuclear envelope. Mol Cell Biol 30(16):3956–3969. doi:10.1128/mcb.00242-10
Gloerich M, Vliem MJ, Prummel E, Meijer LA, Rensen MG, Rehmann H, Bos JL (2011) The nucleoporin RanBP2 tethers the cAMP effector Epac1 and inhibits its catalytic activity. J Cell Biol 193(6):1009–1020. doi:10.1083/jcb.201011126
Metrich M, Laurent AC, Breckler M, Duquesnes N, Hmitou I, Courillau D, Blondeau JP, Crozatier B, Lezoualc’h F, Morel E (2010) Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell Signal 22(10):1459–1468. doi:10.1016/j.cellsig.2010.05.014
Zhang L, Malik S, Pang J, Wang H, Park KM, Yule DI, Blaxall BC, Smrcka AV (2013) Phospholipase Cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 153(1):216–227. doi:10.1016/j.cell.2013.02.047
Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437(7058):574–578. doi:10.1038/nature03966
Pereira L, Ruiz-Hurtado G, Morel E, Laurent AC, Metrich M, Dominguez-Rodriguez A, Lauton-Santos S, Lucas A, Benitah JP, Bers DM, Lezoualc’h F, Gomez AM (2012) Epac enhances excitation-transcription coupling in cardiac myocytes. J Mol Cell Cardiol 52(1):283–291. doi:10.1016/j.yjmcc.2011.10.016
Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM (2013) Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 127(8):913–922. doi:10.1161/circulationaha.12.148619
Marks AR (2013) Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Investig 123(1):46–52. doi:10.1172/jci62834
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415(6868):198–205. doi:10.1038/415198a
Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV (2007) Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem 282(8):5488–5495. doi:10.1074/jbc.M608495200
Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV (2009) Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem 284(3):1514–1522. doi:10.1074/jbc.M806994200
Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV (2005) Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 97(12):1305–1313. doi:10.1161/01.RES.0000196578.15385.bb
Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, Lomasney JW, Jakobs KH (2001) A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol 3(11):1020–1024. doi:10.1038/ncb1101-1020
Ruiz-Hurtado G, Dominguez-Rodriguez A, Pereira L, Fernandez-Velasco M, Cassan C, Lezoualc’h F, Benitah JP, Gomez AM (2012) Sustained Epac activation induces calmodulin dependent positive inotropic effect in adult cardiomyocytes. J Mol Cell Cardiol 53(5):617–625. doi:10.1016/j.yjmcc.2012.08.004
Cazorla O, Lucas A, Poirier F, Lacampagne A, Lezoualc’h F (2009) The cAMP binding protein Epac regulates cardiac myofilament function. Proc Natl Acad Sci USA 106(33):14144–14149. doi:10.1073/pnas.0812536106
Okumura S, Fujita T, Cai W, Jin M, Namekata I, Mototani Y, Jin H, Ohnuki Y, Tsuneoka Y, Kurotani R, Suita K, Kawakami Y, Hamaguchi S, Abe T, Kiyonari H, Tsunematsu T, Bai Y, Suzuki S, Hidaka Y, Umemura M, Ichikawa Y, Yokoyama U, Sato M, Ishikawa F, Izumi-Nakaseko H, Adachi-Akahane S, Tanaka H, Ishikawa Y (2014) Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J Clin Investig 124(6):2785–2801. doi:10.1172/jci64784
Laurent AC, Bisserier M, Lucas A, Tortosa F, Roumieux M, De Regibus A, Swiader A, Sainte-Marie Y, Heymes C, Vindis C, Lezoualc’h F (2015) Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc Res 105(1):55–64. doi:10.1093/cvr/cvu242
Pereira L, Metrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, Lezoualc’h F, Gomez AM (2007) The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol 583(Pt 2):685–694. doi:10.1113/jphysiol.2007.133066
Smrcka AV, Oestreich EA, Blaxall BC, Dirksen RT (2007) EPAC regulation of cardiac EC coupling. J Physiol 584(Pt 3):1029–1031. doi:10.1113/jphysiol.2007.145037
Frey N, Katus HA, Olson EN, Hill JA (2004) Hypertrophy of the heart: a new therapeutic target? Circulation 109(13):1580–1589. doi:10.1161/01.cir.0000120390.68287.bb
Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Investig 115(8):2108–2118. doi:10.1172/jci24682
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990) Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322(22):1561–1566. doi:10.1056/nejm199005313222203
Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV (2011) Phospholipase C epsilon scaffolds to muscle-specific A kinase anchoring protein (mAKAPbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem 286(26):23012–23021. doi:10.1074/jbc.M111.231993
Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM (2006) Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Investig 116(3):675–682. doi:10.1172/jci27374
Lopez De Jesus M, Stope MB, Oude Weernink PA, Mahlke Y, Borgermann C, Ananaba VN, Rimmbach C, Rosskopf D, Michel MC, Jakobs KH, Schmidt M (2006) Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J Biol Chem 281(31):21837–21847. doi:10.1074/jbc.M604156200
Peivandi AA, Huhn A, Lehr HA, Jin S, Troost J, Salha S, Weismuller T, Loffelholz K (2005) Upregulation of phospholipase d expression and activation in ventricular pressure-overload hypertrophy. J Pharmacol Sci 98(3):244–254
Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc’h F (2005) cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res 97(12):1296–1304. doi:10.1161/01.res.0000194325.31359.86
Kimura TE, Jin J, Zi M, Prehar S, Liu W, Oceandy D, Abe J, Neyses L, Weston AH, Cartwright EJ, Wang X (2010) Targeted deletion of the extracellular signal-regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload-induced apoptosis in the heart. Circ Res 106(5):961–970. doi:10.1161/circresaha.109.209320
Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P (1997) Apoptosis in the failing human heart. N Engl J Med 336(16):1131–1141. doi:10.1056/nejm199704173361603
Fujita T, Ishikawa Y (2011) Apoptosis in heart failure. The role of the beta-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes. Circ J 75(8):1811–1818
Braunwald E (2013) Heart failure. JACC. Heart Fail 1(1):1–20. doi:10.1016/j.jchf.2012.10.002
Mangmool S, Hemplueksa P, Parichatikanond W, Chattipakorn N (2015) Epac is required for GLP-1R-mediated inhibition of oxidative stress and apoptosis in cardiomyocytes. Mol Endocrinol (Baltimore, Md) 29(4):583–596. doi:10.1210/me.2014-1346
Kwak HJ, Park KM, Choi HE, Chung KS, Lim HJ, Park HY (2008) PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal 20(5):803–814. doi:10.1016/j.cellsig.2007.12.011
Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X (2013) Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res 112(3):498–509. doi:10.1161/circresaha.112.273896
Suzuki S, Yokoyama U, Abe T, Kiyonari H, Yamashita N, Kato Y, Kurotani R, Sato M, Okumura S, Ishikawa Y (2010) Differential roles of Epac in regulating cell death in neuronal and myocardial cells. J Biol Chem 285(31):24248–24259. doi:10.1074/jbc.M109.094581
Koss KL, Kranias EG (1996) Phospholamban: a prominent regulator of myocardial contractility. Circ Res 79(6):1059–1063
Sato Y, Kiriazis H, Yatani A, Schmidt AG, Hahn H, Ferguson DG, Sako H, Mitarai S, Honda R, Mesnard-Rouiller L, Frank KF, Beyermann B, Wu G, Fujimori K, Dorn GW 2nd, Kranias EG (2001) Rescue of contractile parameters and myocyte hypertrophy in calsequestrin overexpressing myocardium by phospholamban ablation. J Biol Chem 276(12):9392–9399. doi:10.1074/jbc.M006889200
Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J Jr, Kranias EG, Giles WR, Chien KR (1999) Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99(3):313–322
Wittkopper K, Fabritz L, Neef S, Ort KR, Grefe C, Unsold B, Kirchhof P, Maier LS, Hasenfuss G, Dobrev D, Eschenhagen T, El-Armouche A (2010) Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Investig 120(2):617–626. doi:10.1172/jci40545
Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH (2010) Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res 106(2):354–362. doi:10.1161/circresaha.109.207423
Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, Ishikawa Y (2003) Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA 100(17):9986–9990. doi:10.1073/pnas.1733772100
Cai W, Fujita T, Hidaka Y, Jin H, Suita K, Prajapati R, Liang C, Umemura M, Yokoyama U, Sato M, Okumura S, Ishikawa Y (2016) Disruption of Epac1 protects the heart from adenylyl cyclase type 5-mediated cardiac dysfunction. Biochem Biophys Res Commun 475(1):1–7. doi:10.1016/j.bbrc.2016.04.123
Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S (2014) Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res 114(9):1500–1515. doi:10.1161/circresaha.114.303772
Foteinou PT, Greenstein JL, Winslow RL (2015) Mechanistic Investigation of the Arrhythmogenic Role of Oxidized CaMKII in the Heart. Biophys J 109(4):838–849. doi:10.1016/j.bpj.2015.06.064
Viatchenko-Karpinski S, Kornyeyev D, El-Bizri N, Budas G, Fan P, Jiang Z, Yang J, Anderson ME, Shryock JC, Chang CP, Belardinelli L, Yao L (2014) Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J Mol Cell Cardiol 76:247–256. doi:10.1016/j.yjmcc.2014.09.009
Aflaki M, Qi XY, Xiao L, Ordog B, Tadevosyan A, Luo X, Maguy A, Shi Y, Tardif JC, Nattel S (2014) Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained beta-adrenergic activation in guinea pig hearts. Circ Res 114(6):993–1003. doi:10.1161/circresaha.113.302982
Brette F, Blandin E, Simard C, Guinamard R, Salle L (2013) Epac activator critically regulates action potential duration by decreasing potassium current in rat adult ventricle. J Mol Cell Cardiol 57:96–105. doi:10.1016/j.yjmcc.2013.01.012
Dybkova N, Wagner S, Backs J, Hund TJ, Mohler PJ, Sowa T, Nikolaev VO, Maier LS (2014) Tubulin polymerization disrupts cardiac beta-adrenergic regulation of late INa. Cardiovasc Res 103(1):168–177. doi:10.1093/cvr/cvu120
Ogrodnik J, Niggli E (2010) Increased Ca(2+) leak and spatiotemporal coherence of Ca(2+) release in cardiomyocytes during beta-adrenergic stimulation. J Physiol 588(Pt 1):225–242. doi:10.1113/jphysiol.2009.181800
Suita K, Fujita T, Hasegawa N, Cai W, Jin H, Hidaka Y, Prajapati R, Umemura M, Yokoyama U, Sato M, Okumura S, Ishikawa Y (2015) Norepinephrine-induced adrenergic activation strikingly increased the atrial fibrillation duration through beta1- and alpha1-adrenergic receptor-mediated signaling in mice. PLoS One 10(7):e0133664. doi:10.1371/journal.pone.0133664
Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S (2011) Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Investig 121(8):2955–2968. doi:10.1172/jci46315
Hothi SS, Gurung IS, Heathcote JC, Zhang Y, Booth SW, Skepper JN, Grace AA, Huang CL (2008) Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Arch 457(2):253–270. doi:10.1007/s00424-008-0508-3
Dominguez-Rodriguez A, Ruiz-Hurtado G, Sabourin J, Gomez AM, Alvarez JL, Benitah JP (2015) Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J Mol Cell Cardiol 87:74–78. doi:10.1016/j.yjmcc.2015.07.002
Akar FG, Rosenbaum DS (2003) Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res 93(7):638–645. doi:10.1161/01.res.0000092248.59479.ae
Beuckelmann DJ, Nabauer M, Erdmann E (1993) Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73(2):379–385
Nattel S, Maguy A, Le Bouter S, Yeh YH (2007) Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87(2):425–456. doi:10.1152/physrev.00014.2006
Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS (2006) Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Investig 116(12):3127–3138. doi:10.1172/jci26620
Xie LH, Chen F, Karagueuzian HS, Weiss JN (2009) Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res 104(1):79–86. doi:10.1161/circresaha.108.183475
Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ (2012) Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126(17):2084–2094. doi:10.1161/circulationaha.112.105320
Somekawa S, Fukuhara S, Nakaoka Y, Fujita H, Saito Y, Mochizuki N (2005) Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes. Circ Res 97(7):655–662. doi:10.1161/01.RES.0000183880.49270.f9
Duquesnes N, Derangeon M, Metrich M, Lucas A, Mateo P, Li L, Morel E, Lezoualc’h F, Crozatier B (2010) Epac stimulation induces rapid increases in connexin43 phosphorylation and function without preconditioning effect. Pflugers Arch 460(4):731–741. doi:10.1007/s00424-010-0854-9
Morley GE, Danik SB, Bernstein S, Sun Y, Rosner G, Gutstein DE, Fishman GI (2005) Reduced intercellular coupling leads to paradoxical propagation across the Purkinje-ventricular junction and aberrant myocardial activation. Proc Natl Acad Sci USA 102(11):4126–4129. doi:10.1073/pnas.0500881102
Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, Gutstein DE (2004) Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res 95(10):1035–1041. doi:10.1161/01.RES.0000148664.33695.2a
Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA (2007) Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293(3):H1883–H1891. doi:10.1152/ajpheart.00514.2007
Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC (2012) Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 10(1):15–26. doi:10.1038/nrcardio.2012.158
Villarreal F, Epperson SA, Ramirez-Sanchez I, Yamazaki KG, Brunton LL (2009) Regulation of cardiac fibroblast collagen synthesis by adenosine: roles for Epac and PI3K. Am J Physiol Cell Physiol 296(5):C1178–C1184. doi:10.1152/ajpcell.00291.2008
Miller CL, Cai Y, Oikawa M, Thomas T, Dostmann WR, Zaccolo M, Fujiwara K, Yan C (2011) Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol 106(6):1023–1039. doi:10.1007/s00395-011-0228-2
Chen C, Du J, Feng W, Song Y, Lu Z, Xu M, Li Z, Zhang Y (2012) beta-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br J Pharmacol 166(2):676–688. doi:10.1111/j.1476-5381.2011.01785.x
Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL (2010) Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56(2):225–231. doi:10.1161/hypertensionaha.109.148635
Acknowledgments
This study has been supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants (25460296, 24390200, 25670131), The Ministry of Education, Culture, Sports, Science and Technology (MEXT) KAKENHI Grant (22136009), the New Energy and Industrial Technology Development Organization (NEDO) (60890021), the National Cerebral and Cardiovascular Center (NCVC) (22-2-3), the Japan Agency for Medical Research and Development (AMED) (66890005, 66890011, 66890001, 66890023), and a Grant for Strategic Research Promotion of Yokohama City University. The authors are grateful to Ms. Nana Fujita for improving the figures.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Fujita, T., Umemura, M., Yokoyama, U. et al. The role of Epac in the heart. Cell. Mol. Life Sci. 74, 591–606 (2017). https://doi.org/10.1007/s00018-016-2336-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2336-5