
Abstract
The ectopic overexpression of the voltage-dependent Eag1 (Kv10.1) K+ channel is associated with the cancerous phenotype in about 70% of human cancers and tumor cell lines. Recent reports showed that, compared with the canonical Shaker-related Kv family, Kv10.1 presents unique structural and functional properties. Herein, we report the interaction of the class III anti-arrhythmic compound amiodarone with Kv10.1. Using whole-cell patch clamp, we found that amiodarone inhibits Kv10.1 channel conductance with nanomolar affinity. Additionally, and interestingly, we also report that amiodarone inhibits the characteristic Cole-Moore shift of Eag1 channels. Our observations are interpreted considering the structural-functional characteristics of these channels. We conclude that amiodarone possibly binds with high affinity to the voltage sensor module, altering the gating of Kv10.1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human potassium channel Kv10.1, a member of the Eag family of ion channels [22, 38], is particularly notable because of its participation in the cancerous phenotype of several types of tumors. The undoubted, but not yet well understood, role of Kv10.1 in cancer pathology was first strikingly demonstrated by Walter Ster StStnd Luis Pardo, who found that CHO (or HEK) mammalian cells stably expressing Kv10.1, but not other K+ channels, acquire the hallmark characteristics of cancerous cells. Moreover, they demonstrated that transplantation of Kv10.1-expressing cells to immunosuppressed mouse leads to the rapid growth of massive tumors [24]. In agreement with the latter, it is now know that Kv10.1 is ectopically overexpressed in about 70% of human tumors and cancerous cell lines and that channel inhibition reduces tumor growth [12, 23, 25].
Potassium channels present the defining property of sharply selecting K+ over Na+, the most abundant cation in the extracellular medium. However, in spite of this unifying characteristic, K+ channels are highly variable regarding molecular structure and gating mechanisms [39]. In particular, recent reports demonstrate that the tumor-related, voltage-dependent Kv10.1 (Eag1) channel displays structural and functional characteristics that differ from those common to canonical KcsA and Shaker-related Kv channels. Some of these differences are as follows [40]: (a) Eag1 presents an extended and glycosylated extracellular S5–S6 connecting loop, where sugar chains surround the pore in positions that may hinder the binding of peptide toxins which, in contrast, commonly bind to canonical K+ channels; (b) along its full length, the Kv10.1 pore diameter is rather uniform and smaller than that of a hydrated K+ ion. In other words, Kv10.1 lacks the conspicuous widening, or central cavity, located below the selectivity filter of KcsA and related canonical K+ channels [7, 18, and Figure 5A of 40]. The central cavity is the place where many hydrophobic cations, as some anti-arrhythmic compounds, block the pore of canonical K+ channels; (c) even in the absence of a covalent link between voltage sensor and pore domains, Kv10.1 is capable of undergoing voltage-dependent activation gating [20, 35]. In summary, in Kv10.1, communication between voltage sensor movement and pore opening is different, and not yet well understood, as compared with canonical, Shaker-related, Kv channels.
Another well-known functional hallmark of Kv10.1 is the presence of both a conspicuous delay for current surge [4] and the development of a markedly sigmoidal time course of current flow, observed upon channel activation from hyperpolarized resting membrane potentials [e.g.,3, 28, 29, 31, 32]. The latter part of this combined kinetic phenomenon is attributed to the presence of a rate-limiting step linking the passage from deep-closed states, where channels dwell at hyperpolarized potentials, to states near the final open state [e.g., 29, 31, 32]. Although, as pointed out by Hoshi and Armstrong [14], the term Cole-Moore shift properly applies only to the initial lag of current activation [4], it is a widespread practice to call the above-mentioned combined kinetic phenomenon as the Cole-Moore shift of Kv10.1.
Considering the above, it is clear that pharmacological studies of Kv10.1 are a pertinent approach to point out particular functional characteristics that may arise from the structural particularities of Kv10.1, and to discover drugs that may be useful in the treatment of tumor development. Regarding the former, it is pertinent to consider that the structural constrains mentioned above may prevent binding of peptide toxins, for example, scorpion toxins to the Kv10.1 pore, and therefore, pharmacological studies carried out with non-peptide compounds of clinical use can become instrumental.
Herein, we report the effects of amiodarone [17], a class III anti-arrhythmic compound, on Kv10.1 channels. Amiodarone (hereafter referred to as Ad) exerts an overall high-affinity inhibitory effect on the Kv10.1 K+ conductance, at concentrations smaller than those employed clinically to treat cardiac arrhythmias [17]. Additionally, and interestingly, Ad also inhibits the so-called characteristic Cole-Moore shift of Kv10.1.
Materials and methods
Cell culture
HEK293 cells stably expressing the Kv10.1 channel [10] were kept in culture at 37 °C in a humidified 5% CO2 atmosphere in DMEM/F12 media supplemented with 10% FBS and containing zeocin (300 μatmosphere in DMEM/F12 media supplemented with 10% FBS and containing24 h after being plated on glass coverslips, as previously reported [10]. HEK293-Kv10.1 cells were kindly provided by Dr. Luis A. Pardo and Walter Stvided
Electrophysiological recordings
Macroscopic currents were recorded under whole-cell patch clamp with either Axopatch 1D (Axon Instruments) or HEKA-EPC10 (HEKA Instruments) amplifier. Currents were filtered with the built-in filters of the amplifiers and sampled with either a Digidata 1322A interface (Axon Instruments) or the built-in interface of the HEKA amplifier fulfilling the Nyquist criteria. Electrodes were made of borosilicate glass (KIMAX51) pulled to a 1–1.5-M.5silicate glass (KIMAX51) pulled to a 1 built-in interface of theensated with the built-in circuits of the amplifiers. Eighty percent series resistance compensation was always applied. Experiments were conducted at room temperature, as previously reported [10].
Solutions
The extracellular solution contained (in mM) the following: 5 KCl, 2 CaCl2, 157 NaCl, 10 HEPES-Na, pH 7.2. External solutions containing higher [K+] (> 5 mM) were prepared by iso-osmolarly replacing NaCl by KCl. The intracellular solution contained (in mM) the following: 90 KF, 30 KCl, 2 MgCl2, 10 EGTA, 10 HEPES-K, pH 7.2. Amiodarone (Sigma-Aldrich, St. Louis, MO) stocks were dissolved DMSO (dimethyl sulfoxide) (Sigma-Aldrich, St. Louis, MO) and kept in the freezer. Before the experiments, Ad was dissolved in the extracellular solution at the desired concentration. Final [DMSO] was always less than 0.01% (v/v). DMSO tested alone at the highest V/V ratio used in the study did not affect Kv10.1 channels (not shown). The external solution was applied with a gravity-driven perfusion system.
Data analysis
Results are expressed as mean ± SEM of at least four independent experiments at each condition. Currents analysis was carried out using Clampfit 10.6 (Axon Instruments) and Pulsefit (HEKA Instruments). Statistical significance was evaluated with either the unpaired t-test or the extra-sum-of-squares F-test, using GraphPad Prism version 5.00 (GraphPad Software). Curve fitting was carried out with Pulsefit and Sigmaplot 10.
The differential equations of the kinetic models in Fig. 4 were solved with the built-in Runge-Kutta numerical integration method and fitted to the experimental traces with the least squares algorithm, of Mathematica 9 (Wolfram).
Results
Inhibition of K+ conductance by Ad
Upon addition to the extracellular solution, Ad affects the K+ conductance of Kv10.1 channels, in a manner that depends of the time window under observation and the holding potential (HP), from which channels are activated. First, we will present a high-affinity Ad inhibition of the K+ conductance, which is observed applying depolarizing pulses long enough to fully activate the channels.
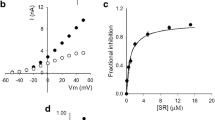
Ad inhibition of K+ conductance is illustrated in Fig. 1a, which compares K+ currents (I K) recorded in control conditions (left panel), against I K recorded after perfusion an extracellular solution containing 1 μrecorded after perfusion an extracI K was activated by 1-s pulses from − 50 to + 50 mV applied in 10-mV increments from the HP of − 70 mV. Notice that upon Ad addition, I K reaches maximum or peak amplitude, and thereafter decays exponentially to a final, steady-state current amplitude. The Ad-induced decay phase of I K during activation pulses will be referred to as apparent or induced inactivation. The plot of I K measured at pulse end as a function of voltage of the traces in panel a (Fig. 1b) indicates that inhibition by Ad is voltage-dependent, since the difference between I K in control and Ad-containing solution varies with voltage (see below).

Amiodarone inhibition of Kv10.1. a Left panel: control I K elicited by activating pulses from − 40 to + 50 mV, applied in 10-mV steps, HP = − 70 mV. Right panel: I K recorded after perfusing an extracellular solution containing 1 μrecordb I–V plot of the traces in (a); currents were measured at pulse end. c Fractional channel inhibition vs. [Ad]. I K fractional inhibition was assessed as f = 1 − (I Ad / I C), where I Ad is I K at the end of + 40-mV pulses, at the indicated [Ad], and I C is the corresponding control I K. The line is the fit of the points with a Hill equation with n H = 0.91, and Kd = 203 nM. Inset: double reciprocal plot of the points. The straight line is the least squares fit of the points (r = 0.98)
Figure 1c shows the concentration dependence of current inhibition by Ad at + 40 mV, a voltage that fully activates I K under our recording conditions (see later). The points are the average fractional current inhibition at pulse end, obtained from experiments as in Fig. 1a, at the indicated [Ad] (see figure legend). The line is the least squares fit of the data with a Hill equation with Hill number n H = 0.91 and Kd = 203 nM, a half-inhibition concentration about one order of magnitude lower than the therapeutic [Ad] (1–3 μM, a half-inhibition concentration ab [17]. The inset shows the expected linear (r = 0.98) double reciprocal plot of the points (inset). These results indicate either that Ad binds to a single site or that it binds to more than one site but in a non-cooperative, independent manner [5].
As noted above, a conspicuous feature of Ad inhibition is the appearance of a noticeable decay phase of I K during sustained depolarization, or apparent inactivation. A drug-induced inactivation was first analyzed in the hallmark observations of Clay Armstrong, regarding the block of the squid K+ channel by internally added TEA and TEA-derivative ions [1]. Armstrong concluded that the pore activation gate had an intracellular location and that TEA and derivative ions entered the pore, blocking K+ conduction only after the activation gate opened, hence producing, depending on their rate of association, a time-dependent reduction of I K, or apparent inactivation. The site of internal TEA ions open-pore slow blockage was later identified as the wide central cavity of the pore, and the activation gate as the S6 intracellular bundle crossing of canonical K+ channels [7, 13, 18].
Regarding the above, it is pertinent to recall that communication between voltage sensor and pore modules of Kv10.1 is different from that in canonical K+ channels and that additionally, the Kv10.1 structure does not show a pore widening, which could be identified as equivalent to the central cavity of canonical K+ channels [7, 18, 40]. Hence, the question arises of whether the decay phase of I K means that Ad acts as a slow open-pore blocker, similar to intracellular TEA derivatives on the squid K+ channel. The observations shown hereafter were aimed to gain insight into this question.
First, we compared the voltage dependencies of the fractional channel inhibition exerted by Ad and current activation. As shown in Fig. 2a, Ad inhibition at depolarized potentials presents a voltage dependency qualitatively similar to the channel’s chord conductance. This suggests that, in this voltage range, current inhibition is somehow coupled to or facilitated (see below) by the activation of the channels. Thereafter, to further characterize the relationship between Ad inhibition and channel activation, we compared the chord conductance of control vs. of Ad-modified channels. The data presented in Fig. 2b demonstrate that Ad shifts the conductance curve to the left (V 1/2(control) = − 2 mV; V 1/2(Ad) = − 19 mV; two-tailed t-test P = 0.688), inducing only a minor change on the slope of the curve (z(control) = + 1.7; z(Ad) = + 1.3; two-tailed t-test P < 0.05). Thus, somehow Ad shifts considerably the activation-gating equilibrium towards the open state, exerting only a minor change on the effective voltage sensitivity of the channels (see below and “Discussion”).

Voltage dependence of Kv10.1 inhibition. a Comparison of the voltage dependence of Ad fractional inhibition ([Ad] = 1 μr) vs. control chord conductance, as indicated. Ad fractional inhibition was calculated as in Fig. 1 (closed circles). The normalized chord conductance was assessed as G(Vm) / Gmax = I(Vm) / (Vm − V K), where I(Vm) is I K at Vm potential, V K is the K+ equilibrium potential (in this case −80 mV), and Gmax is the maximal G value. b Comparison of the normalized chord conductance of control (open circles) vs. Ad-modified channels (closed circles). In the case of Ad-modified channels, I(Vm) is the peak or maximal current at voltage Vm. The lines are the fit of the points with a Boltzmann equation: G/Gmax = 1/[1 + exp(−(zF/RT)*(Vm − V 1/2)], where F, R, and T have their usual meaning; V 1/2 is the half activation voltage: V 1/2(control) = − 2 mV; V 1/2(Ad) = −19 mV; and z is the apparent valence: z(control) = +1.7, z(Ad) = +1.3. [Ad] = 1 μ1
It is known that open-pore blocking compounds may hinder the closing of the activation gate of K+ channels [e.g., 1, 13]. Hence, in order to get further insight on whether Ad-induced inactivation arises from an open-pore block mechanism, we compared deactivation tail currents of control and Ad-modified channels. Taking into account the possibility that we could record tail currents only from channels that were not inhibited by Ad, we apply a high (10 μurrents of control and Ad-modified channels. Taking into account the possibility that we could record tail currents only from channels that were not inhibited by Ad, we apply a h
Figure 3a illustrates superimposed control I K elicited by 40-mV pulses of increasing duration applied from the HP of − 70 mV. Notice the inward tail currents recorded at the repolarization potential of − 140 mV. As expected, tail current kinetics of unmodified channels do not vary with the pulse duration. The traces in Fig. 3b illustrate a similar recording of Ad-modified channels on the same cell. The first activation pulse had duration short enough (20 ms) to allow the corresponding inward tail current to be recorded before a significant decay of outward I K took place. As expected, tail current amplitudes decrease, as the apparent open-sate inactivation develops during the activating pulse. More importantly, the tail current kinetics remains constant independently of the pulse duration, that is, regardless of the extent of apparent inactivation attained during the pulse and equal to that of control unmodified channels.

Deactivation tail currents in the presence of Ad. a Representative superimposed control I K recorded in the same cell, applying a + 40-mV pulse of varying duration. Repolarization potential at the end of each pulse was − 140 mV. b Superimposed I K recorded as in (a) but in the presence of 1 μbut inc Inward tail currents at − 140 mV recoded in the indicated conditions. HP = − 70 mV. d Deactivation time constants vs. Vm. Time constants were obtained by fitting a single exponential function to tail currents. There is no difference between control tails and those in the presence of Ad (F-test, P = 0.962)
The above is best illustrated in Fig. 3c which displays, superimposed, normalized tail currents recorded either under control conditions (after a 200-ms activating pulse) or in the presence of Ad (after the indicated pulse durations). Notice that Ad does not hinder current deactivation. The latter is quantified in Fig. 3d which shows deactivation time constants of both control and Ad-modified channels, obtained by fitting a single exponential curve to tail currents as a function of repolarization potential (see figure legend).
These observations suggest that apparent inactivation may not arise from an open-pore block of Kv10.1, but further information regarding inhibition onset and recovery from inhibition (inactivation) is needed in order to support this proposal (see below and “Discussion”).
Regarding the latter, we wondered if Ad inhibits the channels once the K+ permeation gate is open, as expected from a strict open-pore blockage mechanism. The strong inhibition seen shortly after delivering the activation pulses (Fig. 1a) already suggested that this may not be the case.
To gain some insight into this matter, we considered a simplified model which takes into account that I K activation from a depolarized holding potential (− 70 mV, as in Fig. 1a) is well fitted by a single exponential time course of the form I K = I K,max(1−exp(−t/Ta)), where I K,max is the maximal value of I K. This is illustrated in Fig. 4a which shows the fit of a normalized representative I K with a time constant Ta of 7.2 ms at + 40 mV (R 2 = 0.97). Operationally, this behaves as if there was a single closed state C in the activation pathway. This is because at – 70-mV resting potential, channels dwell in late closed states, which are not connected by rate-limiting constants [29], and when a sufficiently depolarized voltage is applied, they are traversed towards the final open state fast enough as to operationally behave as a single closed state C. This can be represented by the following simplified scheme:
with ra = 1 / Ta.

Assessment of Ad inhibition from the open state. a Normalized representative control I K at + 40 mV (noisy black trace, I K from Fig. 1), HP = − 70 mV. The red line is the least squares fit of I K with an exponential function I K = 1−exp(−t/Ta), as contained in the kinetic Scheme 1 (see text), Ta = 7.2 ms. b Comparison of I K at + 40 mV recorded in the presence of 1 μV recorded in the I K under the hypothesis that Ad-induced inactivation proceeds only from the open state (kinetic Scheme 2, text). The observed I K (noisy black trace, I K from Fig. 1) shows a peak value clearly smaller than that predicted by this hypothesis (red trace)
Under these conditions, the Ad-induced inhibition I, or apparent inactivation, follows a single exponential time course with time constant of 127 ms = 1 / ri (black trace in Fig. 4b), where ri is a pseudo-first-order rate constant. Therefore, if Ad inhibition proceeds only after the K+ permeation gate opens, the following simplified scheme should apply:
The fitted reverse constant rv (0.1 × 10−2 (1/ms)) allows the observed average steady-state I K inhibition of ~ 80% at pulse end.
Figure 4b compares I K recorded in the presence of Ad against I K predicted by Scheme 2 (see “ (see ted by hmethods”thodsLIN). Note that the observed peak I K (black trace) is significantly smaller than that predicted by Scheme 1 (red trace). This suggests that, in contrast to an open-pore blockage mechanism, Ad inhibition starts before the K+ conduction gate opens.
We tested the above prediction assessing the near steady-state Ad-induced inactivation elicited by long-lasting pre-pulses of varying amplitude, applying the standard two-pulse protocol, commonly employed to study the steady-state inactivation of channels.
Mechanism of amiodarone-induced apparent inactivation
Upon prolonged depolarizations, the current through wild-type Kv10.1 channels may just present just a subtle decrement (supplementary Fig. 1; “Discussion”) [e.g., 3, 9, 34, and references therein cited]. Therefore, we shall consider the results of the two-pulse protocol applied only to Ad-modified channels. Figure 5a shows representative I K recordings obtained by applying 1-s pre-pulses from − 140 to + 30 mV given in 10-mV increments, to activate and inhibit (inactivate) I K, followed by a test pulse to + 50 mV, to assess the state of the channels.

Ad inhibition following prolonged pre-pulses. a I K evoked by a constant + 50-mV/0.1-s pulse following 1-s pre-pulses from − 140 to + 50 mV, applied in 10-mV increments, as indicated. Time between pair of pulses was 20 s. HP = − 70 mV. b Non-inactivated (non-inhibited) fraction vs. pre-pulse potential, from four independent experiments as in a. The non-inhibited fraction was assessed as I(Vpre-pulse)/Imax, where I(Vpre-pulse) is the peak or maximal I K at the test + 50-mV pulse, following a 1-s pre-pulse of the indicated potentials, and Imax is the maximal I K. c Comparison of non-inhibited channel fraction (closed circles) vs. chord conductance (open circles) at the indicated Vm. Ad inhibition starts from potentials at which K+ permeation is closed
The average non-inactivated (non-inhibited) current fraction, as a function of the pre-pulse potential, from experiments as in panel a, is shown in Fig. 5b (see figure legend). Interestingly, notice that Ad inhibition starts at potentials negative to the Kv10.1 current activation threshold (~ − 50 mV). This is best seen in Fig. 5c, where the fraction of non-inhibited channels (closed symbols, data from Fig. 5b) is plotted along with the Kv10.1 control chord conductance (open symbols). This last observation is in agreement with the prediction of the simple model of Fig. 4 and shows that, contrary to a strict open-pore blockade mechanism, Ad somehow inhibits a fraction of the channels at voltages where the K+ conduction pathway is closed (see “Discussion”).
On the other hand, it is also clear (Fig. 5c) that the non-inhibited channel fraction drops more steeply at potentials positive to ~ − 50 mV (where K+ conductance activates), than at more negative potentials. That is, although Ad does inhibit Kv10.1 at voltages where K+ conduction pathway is closed, the activation of the K+ conductance further facilitates inhibition. This leaves the question open of whether the steeper inhibition observed at voltages positive to − 50 mV in Fig. 5b might arise from Ad block of the open K+ permeation pathway. To address this question, we studied the rate of recovery from Ad-promoted inactivation in near physiological (5 mM) as well as in elevated extracellular [K+] (50 mM).
Recovery from Ad-promoted inactivation was examined by applying a pair of + 40-mV pulses separated by a variable time interval during which Vm was held at either – 70- or – 140-mV recovery potentials. The representative traces in Fig. 6a, b show that unexpectedly, with 5 mM Ko+, the extent of recovery attained after a 2-s interval at − 140 mV was smaller than that attained at the recovery potential of − 70 mV. The latter is best seen in Fig. 6e which shows the time course of recovery at both potentials. Note that recovery is slower at − 140 mV. The lines are the fit of the data with single exponential functions with time constants Tr(− 70 mV, 5 Ko+) = 1 s, and Tr(− 140 mV, 5 Ko+) = 2 s.

Recovery from Ad-induced inactivation. a I K evoked by a pair of + 40-mV pulses separated by a 2-s interval at − 70 mV, with the cell bathed in 5 mM Ko+ solution containing 1 μsolution containing 1ed in 5ctivation. ch Kn (closed circles) vs. chord conductance (open circles) at the indicated Vm. Ad. There is no difference between control tails and those in the presence20 s. b As in (a) but the recovery potential was − 140 mV. c As in (a) (recovery potential = − 70 mV) but with the cell bathed in 50 mM Ko+ solution containing 1 μsolutid As in (c) but recovery potential was − 140 mV. e Extent of I K recovery in 5 mM Ko+ as a function of the time between pulses, from experiments as in (a) and (b), as indicated. The extent of I K recovery Rec was assessed as Rec = [Ipeak,2-Iend,1] / [Ipeak,1-Iend,1], where Ipeak,2 and Iend,2 are peaks I K and I K at pulse end, evoked by the second + 40 mV/0.25 s, respectively; and Ipeak,1 and Iend,1 are peaks I K and I K at pulse end, evoked by the first + 40-mV/0.5-s pulse. The lines are the least squares fit of the points with a function of the form Rec = 1−exp(−t/Tr), with time constants Tr(−70 mV, 5 Ko+) = 1 s, and Tr(−140 mV, 5 Ko+) = 2 s, respectively. f As in (e) but [Ko+] = 50 mM. The lines are the fit of the points with Tr(−70 mV, 50 Ko+) = 0.813 s, and Tr(−140 mV, 50 Ko+) = 2.1 s. g Comparison of recovery kinetics at either − 70 (open symbols) or − 140 mV (closed symbols), with either 5 or 50 mM [Ko+], as indicated (see text). Rising external K+ does not enhance recovery from Ad-induced inactivation (F-test P (− 70 mV, 5 vs. 50 Ko+) = 0.712; F-test P (− 140 mV; 5 vs. 50 Ko+) = 0.997)
The above result could be considered as suggestive of open-pore block as the mechanism of the increased inactivation observed at Vm↔− 50 mV in Fig. 5b, because if that was the case, an increase of the drug affinity at hyperpolarized potentials and/or a possible trapping of the drug within the pore could cause the slower recovery observed in Fig. 6e at − 140 mV.
In order to test the above possibility, we studied the effect of increasing the external [K+] on recovery rate. The traces in Fig. 6c vs. Fig. 6d illustrate that when cells are bathed in 50 mM Ko+, recovery after a 2-s interval at − 140 mV is again smaller than that at − 70 mV. The latter is best shown in Fig. 6f which compares the time course of recovery at both potentials. The lines are the single exponential fit of the data with time constants that, remarkably, are quite similar to those obtained with only 5 mM Ko+, namely, Tr(− 70 mV, 50 Ko+) = 0.9 s, and Tr(− 140 mV, 50 Ko+) = 2.1 s.
In summary, increasing ten times [Ko+] does not change the rate of recovery, neither at − 70 (P = 0.7122) nor at − 140 mV (P = 0.9265), and regardless of [Ko+], recovery is slower at the more negative potential, where the driving force for inward K+ current is bigger.
The slow rates of inactivation (Fig. 4) and recovery (Fig. 6), as compared to control deactivation rates (Fig. 3), bring out the implication that even in the case that inactivation was the result of pore block, this would not affect the channels deactivation rate assessed in the presence of Ad, as was shown in Fig. 4. However, the lack of effect of K+ on recovery from inhibition clearly supports the proposal that Ad-promoted inactivation is not the consequence of pore block [1].
In summary, the above results (a) demonstrate that Ad inhibition is reversible and (b) support the proposal that Ad inhibition of K+ conductance, that is Ad-induced or -facilitated inactivation, is not due to a pore blockage mechanism, because in that case recovery in 50 mM Ko+ should have been faster, not identical, than that in 5 mM Ko+, and it should have been accelerated, not slowed down, by the more negative recovery potential [1]. We conclude that collectively the data in Figs. 4, 5, and 6 are not compatible with an open-pore block as the underlying mechanism of the Ad-induced apparent inactivation.
Amiodarone inhibits the Cole-Moore shift of Kv10.1
Interestingly, besides its inhibition of the K+ conductance, we observed that Ad also inhibits the so-called characteristic Cole-Moore shift of Kv10.1 (“Introduction” and “Discussion”). This is illustrated in Fig. 7a, which shows a family of superposed I K recordings obtained by a test pulse to + 30 mV, from holding potentials (HP) ranging from − 70 to − 140 mV in steps of 10 mV. Notice that unmodified control Kv10.1 currents present their characteristic holding potential dependency; that is, instead of superimposing to each other, the initial activation surge of currents, evoked by the + 30 mV activation potential, but from different HPs, spans an easily perceptible time window, in which I K evoked from more negative HPs is shifted to the right and activates more slowly. In contrast, the time window over which the initial I K spreads is noticeably narrowed in Ad-modified channels (Fig. 7b). This demonstrates that Ad inhibits the holding potential dependency or Cole-Moore shift, of Kv10.1 currents.

Ad inhibition of the Cole-Moore shift of Kv10.1. Representative I K at + 40 mV evoked from the indicated membrane potentials in control (left panel) and Ad-modified channels (right panel), [Ad] = 1 μV evoked from th inhibition of the Cole-Moore shift of the channels
The reduction of the Cole-Moore shift by Ad yields a paradoxical result: in spite of its overall inhibitory effect on the K+ conductance, it also facilitates channel activation from hyperpolarized potentials. That is, the initial I K surge of Ad-modified channels may exceed that of unmodified channels, depending on the holding potential from which the currents are activated. This is illustrated in Fig. 8a. Here, I K, recorded from the same cell before (black traces) and in the presence of Ad (1 μ recorded from, is shown superimposed. I K was elicited by test pulses to + 30 mV from the indicated HPs, repolarization potential − 70 mV in all cases. Notice that when channels are activated from HPs more negative than ~ − 90 mV, I K of Ad-modified channels transiently exceeds control I K. This is quantified in Fig. 8b, where the time spent to reach 50% of the maximal current amplitude (T 1/2) is plotted against HP both for control (closed circles) and Ad-modified channels (open circles). Note the faster activation rate of Ad-modified Kv10.1 currents at all HPs more negative than − 90 mV.

I K initial surge of control vs. Ad-modified channels. a I K activated by a + 40-mV pulse applied from the indicated membrane potentials. Black traces: control I K; red traces: I K through Ad-modified channels. Membrane voltage was clamped at the indicated membrane potentials 2 s before delivering the + 40-mV activating pulse. In all cases, repolarization potential was − 70 mV. b Activation half-time (T 1/2) vs. pre-pulse potential in control vs. Ad-modified channels, as indicated, measured from experiments as in (a), [Ad] = 1 μ [ T 1/2 is the time it takes I K to reach 50% of its peak amplitude
Discussion
The recently reported structure of the tumor-related Kv10.1 (rKv10.1) channel shows that the structural array of the external pore vestibule may hinder the binding of peptide toxins, as scorpion toxins [34]; therefore, it is important to study Kv10.1 interaction with non-peptide compounds of clinical use, as amiodarone.
Amiodarone, as other class III anti-arrhythmic drugs, interacts with several K+ conductances [e.g., see 2, 16, 17, 19, 26, 27, 36, 37, 41]. Even for a particular conductance, the reported affinities for Ad vary amply among different authors, likely because of the variety of biological preparations and experimental conditions employed. In ventricular myocytes of rabbit, it was found that Ad inhibits I Kr (ERG) with a 50% inhibition concentration of 2.8 μ(ERG) with I Ks (KvLQT1/minK) was unaffected even at 300 μ(KvLQT1/minK) was unaffected even, the same authors found that Ad inhibited HERG with a lower 38-μthe same au [16]. Other authors have reported a 50% inhibition concentration for HERG channels expressed in HEK cells of 0.8 μ O [41] and 2.8 μ2. [19], and values as high as 9.8 μ a [19], and values as [30]. Additionally, Ad inhibits the KATP channel of sarcolemma with a 50% inhibition concentration of 0.35 μchannel of sarcolemma with a 50%ATP channel of mitochondria [27]. Besides, Ad also inhibits the small-conductance Ca++-activated channel SK2 with a 50% inhibition concentration of 2.7 μac [36].
In spite of this ample target spectrum, herein, we reported that Ad inhibits Kv10.1 with high nanomolar affinity; hence, Ad significantly affects Kv10.1 at concentrations smaller than the therapeutic plasma concentrations used to treat cardiac arrhythmias (1–3 μM) [17]. This suggests that either amiodarone or similar, structurally related, compounds have the potential of being useful drugs against tumors involving Kv10.1 ectopic overexpression.
Moreover, although Kv10.1 inhibition hinders tumors development, it is also known that its tumorigenic role is not totally abolished upon inhibition of the K+ flow through the channel [12, 23thr]. This suggests that compounds capable of inhibiting K+ flux and altering the conformational changes of Kv10.1 channels, as we have shown here to be the case for amiodarone, might be of major interest in the quest to find drugs to treat cancer.
Regarding the above, it is interesting to mention that observations using relatively elevated [Ko+] and mutant channels suggest that setting the voltage sensors in pre-activated states could inhibit the cellular proliferation induced by Kv10.1 [6, 12, 23,24,25]. Hence, the capability of Ad to inhibit both the K+ conductance and the Cole-Moore shift of the channels makes this or related compounds particularly interesting to treat Kv10.1-expressing tumors (see below).
Amiodarone inhibition of the K+ conductance
The most conspicuous effect of Ad on Kv10.1 channels is the induction of a significant apparent inactivation, which inhibits K+ flow in a voltage-dependent manner. Frequently, an apparent inactivation is the result of a slow open-pore blockade of the channels [1]. However, the opposite is not necessarily true, as an apparent current inactivation does not by itself imply an open-pore block mechanism. The latter must be kept in mind particularly with regard to Kv10.1 channels, where communication between voltage sensor and pore modules follows a different mechanism than that in Shaker-related Kv channels, and in which the pore does not have the significant widening or central cavity of canonical K+ channels [20, 35, 40].
In the case of amiodarone, we found that the simplified model in Fig. 4 hinted that Ad reduction of the peak K+ current was too strong to be accounted for by an open-pore block mechanism. In agreement with the latter, the observations in Fig. 5 demonstrated that indeed Ad inhibits I K at potentials at which the K+ permeation pathway is closed. This demonstrated that, in contrast to an open-pore block mechanism, Ad inhibition is not strictly coupled to the opening of the Kv10.1 pore.
The question remained of whether a component of Ad inhibition, namely, the open-sate apparent inactivation observed at Vm > −50 mV, might be due to open-pore blockade. In this regard, we found that a tenfold increase of [K+]o (from 5 to 50 mM) did not affect at all the rate of recovery from apparent inactivation. Moreover, we observe that even in the presence of a highly external [K+], hyperpolarized HPs slow recovery from inactivation, instead of accelerating it as would be the case if inactivation were the result of an open-pore blockade mechanism. Therefore, the observations in Figs. 5 and 6 indicate that there is no evidence to support open-pore blockage as the main mechanism of Ad-promoted apparent inactivation of Kv10.1 and that pore opening and/or the fully activated position of the voltage sensor simply facilitates inactivation development whatever its actual mechanism (see below).
It has been reported that upon prolonged depolarizations, the K+ current through Kv10.1 channels may undergo a subtle decrement, and it is known that deletions of the protein N-terminal end yield channels that inactivate fast [9, 33, 34] (see below). These observations bring out the possibility that Ad-promoted inactivation might be the result of potentiation of the subtle wild-type Kv10.1 current decrement that has been reported.
Amiodarone reduction of the Cole-Moore shift of Kv10.1 channels
Kv10.1 channel activation acquires both an initial lag or time shift for current surge and a markedly sigmoidal time course, as the holding or resting potential becomes more negative (“Introduction”ntroduct).
The initial lag for current surge was first studied by Cole and Moore (1961) in their classical review of the Hodgkin and Huxley model of the squid K+ channel. Since then, this initial time shift, common to voltage-dependent channels, is referred to as the Cole-Moore shift, and it has been associated with the presence of multiple closed states. As the resting potential becomes more negative, the channels dwell in deeper closed states, hence the lag for current surge [4, 14, 43].
In the case of Kv10.1 channels, it is a widespread practice to use the term Cole-Moore shift in a relaxed manner including in it both the time shift of current surge (the Cole-Moore effect proper) and the sigmoidal kinetics of current activation that develops at negative holding potentials. In this work, we have followed this widespread practice.
The sigmoidal kinetics of the Kv10.1 current elicited from hyperpolarized potentials is attributed to the presence of a rate-limiting step connecting the passage from deep closed states to states near the final open conformation of the protein [e.g., 25g., .
Here, we reported that Ad inhibits the so-called Cole-Moore shift of Kv.10.1, in the sense mentioned before. This effect clearly demonstrates the involvement of the voltage sensor domain on Ad-induced modification of Kv10.1. Hence, based on this and on our formerly mentioned observations, we hypothesize that Ad binds to the voltage sensor module and/or to a region that interacts with this domain (e.g., the PAS domain), and thus modifies the voltage-dependent gating of the channels (see below).
Whatever the case, it is clear from Fig. 8 that Ad accelerates the rate-limiting transition that connects deep closed states with closed states near the open state. Operationally, this would be equivalent to set the voltage sensor in a pre-activated state, from which the K+ permeation pathway can open more easily.
It is interesting to note that an equivalent hypothesis was put forward in a study regarding the individual participation of S4 segments in the activation of Shaker channels [8]. Similar to the effects of Ad on Kv10.1, such study showed that charge-neutralizing mutations reduced the Cole-Moore shift, moving to the left the conductance-voltage relationship. The authors of that study hypothesized that the introduced mutations set the individual S4 voltage-sensing segments in a pre-activated state [8].
It is worth to mention that we recently published [10, 11] that mibefradil, a water-soluble compound up to a 30-mM concentration (in contrast to amiodarone which is water-insoluble), exerts similar effects on Kv10.1 channels than those reported here for Ad. In the case of mibefradil, we hypothesized that it modifies Kv10.1 gating by binding to the S1–S4 voltage sensor module [10, 11]. It is intriguing why two different compounds having so markedly different water solubility exert effects so qualitatively similar on Kv10.1.
Although the voltage sensor domain strictly comprises the S1–S4 module, in the case of Eag1 channels, evidence indicates the additional participation of the intracellular N-terminal PAS domain (Per-Arnt-Sim domain) on the voltage-dependent gating of these proteins e[.g., 15, 21, 33, 36, 40].
In this regard, it has been shown that deletion of the PAS domain, N-terminal amino acids, inhibits the Cole-Moore effect, shifts the conductance-voltage relationship to the left, and induces an inactivation gating on the channels. The mechanism by which these several effects are produced is not yet well understood, but it has been shown that it involves the interaction between the PAS N-terminal region and the S4 segment of the proteins [15, 33]. It turns out that the main difference among the effects produced by PAS domain deletions and Ad interaction with the Kv10.1 channels is that the reported deletions slow down the channel deactivation rate [33], whereas Ad does not.
Although, and as expected, the effects of PAS deletions are quantitatively different from those here reported for amiodarone, it seems reasonable to hypothesize that amiodarone may bind either to a hydrophobic region of Kv10.1 PAS domain or to the S1ble to hypo sensor module of the channels, in any case producing the effects on gating here reported.
Finally, we showed that, as a consequence of the reduction of the Cole-Moore shift, the current through Ad-modified Kv10.1 channels can indeed initially surpass I K of unmodified channels, depending on the HP. Therefore, the question arises of whether this transient current enhancement would affect the possible usefulness of amiodarone regarding the role of Kv10.1 in cancer pathology. Clearly, Ad reduction of the Cole-Moore shift does not undermine its possible utility against Kv10.1-expressing tumors, because the initial I K acceleration occurs only when channels activate from hyperpolarized potentials, whereas most if not all non-neural cells in which ectopic overexpression of Kv10.1 is associated with a cancerous phenotype are likely to have comparatively depolarized resting membrane potentials [42].
Change history
16 March 2018
The original publication of this article contained multiple technical errors that occurred during its production and printing. These errors included sentences and paragraphs with parts missing. The Publisher regrets these mistakes.
References
Armstrong CM (1971) Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol 58(4):413–437. https://doi.org/10.1085/jgp.58.4.413
Balser JR, Bennett PB, Hondeghem LM, Roden DM (1991) Suppression of time-dependent outward current in guinea pig ventricular myocytes. Actions of quinidine amiodarone. Circ Res, 69(2):519–529
Bauer CK, Schwarz JR (2001) Physiology of EAG K+ channels. J Membrane Biol 182:1–15
Cole KS, Moore JW (1960) Potassium ion current in the squid giant axon: dynamic characteristics. Biophys J 1(1):1–14. https://doi.org/10.1016/S0006-3495(60)86871-3
Cornish-Bowden A (2014) Fundamentals of enzyme kinetics. Fourth edition. Wiley-Blackwell
Downie BR, Sánchez A, Knötgen H, Contreras-Jurado C, Gymnopoulos M, Weber C, Stühmer W, Pardo LA (2008) Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem 283(52):36234–36240. https://doi.org/10.1074/jbc.M801830200
Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280(5360):69–77. https://doi.org/10.1126/science.280.5360.69
Gagnon DG, Bezanilla F (2009) A single charged voltage sensor is capable of gating the Shaker K+ channel. J Gen Physiol 133(5):467–483. https://doi.org/10.1085/jgp.200810082
Garg V, Sachse FB, Sanguinetti MC (2012) Tunning of EAG K+ channel inactivation: molecular determinants of amplification by mutations and small molecules. J Gen Physiol 140:307–324
Gómez-Lagunas F, Carrillo E, Pardo LA, Stühmer W (2017) Gating modulation of the tumor-related Kv10.1 channel by Mibefradil. J Cell Physiol 232(8):2019–2032. https://doi.org/10.1002/jcp.25448
Gómez-Lagunas F, Barriga-Montoya C (2017) Mibefradil inhibition of the Cole-Moore shift and K+-conductance of the tumor-related Kv10.1 channel. Channels (Austin) 26:1–4
Hegle AP, Marble DD, Wilson GF (2006) A voltage-driven switch for ion-independent signaling by ether-a-go-go K+ channels. Proc Natl Acad Sci U S A 103(8):2886–2891. https://doi.org/10.1073/pnas.0505909103
Holmgren M, Smith PL, Yellen G (1997) Trapping of organic blockers by closing of voltage-dependent K+ channels. Evidence for a trap door mechanism of activation kinetics. J Gen Physiol 109:527–535
Hoshi T, Armstrong CM (2015) The Cole-Moore shift still unexplained?. Biophys J 109:1312–1316, 7, DOI: https://doi.org/10.1016/j.bpj.2015.07.052
Ju M, Wray D (2006) Molecular regions responsible for differences in activation between heag channels. Biochem Biophys Res Commun 342(4):1088–1097. https://doi.org/10.1016/j.bbrc.2006.02.062
Kamiya K, Atsushi N, Yasui K, Hojo M, Sanguinetti MC, Kodama I (2001) Short- and long-term effects of amiodarone on the two components of cardiac delayed rectifier K+ current. Circulation 103(9):1317–1324. https://doi.org/10.1161/01.CIR.103.9.1317
Kodama I, Kamiya K, Toyama J (1997) Cellular electropharmacology of amiodarone. Cardiovasc Res 35:13–29, 1, DOI: https://doi.org/10.1016/S0008-6363(97)00114-4
Long SB, Campbell EB, MacKinnon R (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309:897–903, 5736, DOI: https://doi.org/10.1126/science.1116269
Loewe A, Lutz Y, Wilhelms M, Sinnecker D, Barthel P, Scholz EP, Dössel O, Schmidt G, Seemann G (2014) In silico assessment of the dynamic effects of amiodarone and dronedarone on human atrial patho-electrophysiology. Europace 16:iv30–iv38
Lörinczi É, Gómez-Posada JC, de la Peña P, Tomczak AP, Fernández-Trillo J, Leipscher U, Stühmer W, Barros F, Pardo LA (2015) Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat Commun 6:6672. https://doi.org/10.1038/ncomms7672
Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, MacKinnon R (1998) Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95(5):649–655. https://doi.org/10.1016/S0092-8674(00)81635-9
Occhiodoro T, Bernheim L, Liu JH, Bijlenga P, Sinnreich M, Bader CR, Fischer-Lougheed J (1998) Cloning of a human ether-à-go-go potassium channel expressed in myoblasts at the onset of fusion. FEBS Lett 434:177–182, 1-2, DOI: https://doi.org/10.1016/S0014-5793(98)00973-9
Ouadid-Ahidouch H, Ahidouch A, Pardo LA (2016) Kv10.1 K+ channel: from physiology to cancer. Pflugers Arch-Eur J Physiol 468:751–762
L A Pardo, D del Camino, A Sánchez, F Alves, A Brüggemann, S Beckh, and W Stühmer (1999) Oncogenic potential of EAG channels. EMBO J 18:5540–5547, 20, DOI: https://doi.org/10.1093/emboj/18.20.5540
Pardo L. A., Stühmer W. (2014) The role of K+ channels in cancer. Nat Rev Cancer 14:39–48, 1, DOI: https://doi.org/10.1038/nrc3635
Peña A, Calahorra C, Michel B, Ramírez J, Sánchez NS (2009) Effects of amiodarone on K+, inaternal pH and Ca2+ homeostasis in Saccharomyces cerevisiae. FEMS Yeast Res 9(6):832–848. https://doi.org/10.1111/j.1567-1364.2009.00538.x
Sato T, Takizawa T, Saito T, Kobayashi S, Hara Y, Nakaya H (2003) Amiodarone inhibits sarcolemmal but not mitochondrial KATP channels in guinea pig ventricular cells. J Pharmacol Exp Ther 307(3):955–960. https://doi.org/10.1124/jpet.103.055863
Schönherr R, Mannuzzu L, Isacoff EY, Heinemann S (2002) Conformational switch between slow and fast gating modes: allosteric regulation of voltage sensor movility in the eag K+ channel. Neuron 35(935–949):5. https://doi.org/10.1016/S0896-6273(02)00869-3
Silverman WR, Roux B, Papazian DM (2003) Structural basis of two-stage voltage-dependent activation in K+ channels. Proc Natl Acad Sci U S A 100(5):2935–2940. https://doi.org/10.1073/pnas.0636603100
Tamargo J, Caballero R, Gómez R, Valenzuela C, Delpón E (2004) Pharmacology of cardiac potassium channels. Cardiovasc Res 62:9–33, 1, DOI: https://doi.org/10.1016/j.cardiores.2003.12.026
Tang CY, Bezanilla F, Papazian DM (2000) Extracellular Mg2+ modulates slow gating transitions and the opening of ether-a-go-go potassium channels. J Gen Physiol 115(3):319–337. https://doi.org/10.1085/jgp.115.3.319
Terlau H, Ludwig J, Steffan R, Pongs O, Stngsan W, Heinemann SH (1996) Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflugers Arch 432(2):301–312. https://doi.org/10.1007/s004240050137
Terlau H, Heinemann S, Stuhmer W, Pongs O, Ludwig J (1997) Amino terminal-dependent gating of the potassium channel rat eag is compensated by a mutation in the S4-segment. J Physiol 502(3):537–543. https://doi.org/10.1111/j.1469-7793.1997.537bj.x
Ting-Feng L, Guey-Mei J, Hsin-Yu F, Ssu-Ju F, Hao-Han W, Mei-Miao C, Chung-Jiuan J (2014) The Eag domain regulates the voltage-dependent inactivation of rat Eag1 K+ channels. PLoS One 9:e110423
Tomczak A, Fernández-Trillo J, Bharill S, Papp F, Panyi G, Stühmer W, Isacoff EY, Pardo AL (2017) A new mechanism of voltage-dependent gating exposed by Kv10.1 channels interrupted between voltage sensor and pore. J Gen Physiol 30:577–593
Turker I, Yu C-C, Chang P-C, Chen Z, Sohma Y, Lin S-F, Chen PS, Ai T (2013) Amiodarone inhibits apamin-sensitive potassium currents. PLoS One 8(7):e70450. https://doi.org/10.1371/journal.pone.0070450
Waldhauser KM, Brecht K, Hebeisen S, Ha HR, Konrad D, Bur D, Krrradener S (2008) Interaction with the hERG channel and cytotoxicity of amiodarone and amiodarone analogs. Br J Pharmacol 155:585–595
Warmke JW, Ganetzky B (1994) A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A 91:3438–3442, 8, DOI: https://doi.org/10.1073/pnas.91.8.3438
Wei A, Jegla T, Salkoff L (1996) Eight potassium channel families revealed by the C. elegans genome proyect. Neuropharmacology 35(7):805–829. https://doi.org/10.1016/0028-3908(96)00126-8
Whicher J, MacKinnon R (2016) Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 353(6300):664–669. https://doi.org/10.1126/science.aaf8070
Wu L, Rajamani S, Shryock JC, Li H, Ruskin J, Antzelevitch C, Belardinelli L (2008) Augmentation of late sodium current unmasks the proarrythmic effects of amiodarone. Cardiovasc Res. 77:481–488
Yang M, Brackenbury WJ (2013) Membrane potential and cancer progression. Front Physiol 4,185, DOI: https://doi.org/10.3389/fphys.2013.00185
Zagotta WN, Hoshi T, Aldrich RW (1994) Shaker potassium channel gating III. Evaluation of kinetic models for activation. J Gen Physiol 103:321–362
Acknowledgements
The authors thank Dr. Daniel Balleza for his participation in the initial phase of this work and to Mrs. Josefina Bolado of the School of Medicine, UNAM, for reviewing the English language of the manuscript.
Funding
This research was supported by PAPIIT grants IN22461-RN22461 and IN219918 grants from CONACyT Laboratorios Nacionales Consolidaci461-RN224616, for reviewin05 and 153504, and PAPIIT IN211616 and IN220916. AHG is a DGAPA-UNAM postdoctoral fellow.
Author information
Authors and Affiliations
Contributions
CBM, AHG ARV, AHC, and AP made experiments and contributed to the analysis of results; FGL designed the work, made experiments, analyzed results and wrote the article.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
A correction to this article is available online at https://doi.org/10.1007/s00424-018-2131-2.
Electronic supplementary material
Supplementary Figure 1
A test for inactivation of control, unmodified, Kv10.1 channels. Non-inactivated current fraction vs. pre-pulse voltage (H616, for revi3 independent experiments carried out with non-modified, control, Kv10.1 channels. I K was evoked by a constant + 50 mV/0.1 s pulse following 1-s pre pulses from −140 to +50 mV, applied in 10 mV increments, as indicated. The non-inactivated fraction was obtained as I K(+50 mV)/I K,max, as a function of pre-pulse potential. See that at any voltage, I K drops less than 10% (for some voltages current drop is even less than 5%). Pair of pulses were applied every 20 sec. (GIF 124 kb)
Rights and permissions
About this article

Cite this article
Barriga-Montoya, C., Huanosta-Gutiérrez, A., Reyes-Vaca, A. et al. Inhibition of the K+ conductance and Cole-Moore shift of the oncogenic Kv10.1 channel by amiodarone. Pflugers Arch - Eur J Physiol 470, 491–503 (2018). https://doi.org/10.1007/s00424-017-2092-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-2092-x