
Abstract
Endothelial nitric oxide synthase (eNOS) plays an essential role in the regulation of endothelial function and acts as a master regulator of vascular tone and homeostasis through the generation of the gasotransmitter nitric oxide (NO). The complex network of events mediating efficient NO synthesis is regulated by post-translational modifications and protein-protein interactions. Dysregulation of these mechanisms induces endothelial dysfunction, a term often used to refer to reduced NO bioavailability and consequent alterations in endothelial function, that are a hallmark of many cardiovascular diseases. Endothelial dysfunction is linked to eNOS uncoupling, which consists of a switch from the generation of NO to the generation of superoxide anions and hydrogen peroxide. This review provides an overview of the eNOS signalosome, integrating past and recently described protein-protein interactions that have been shown to play a role in the modulation of eNOS activity with implications for cardiovascular pathophysiology. The mechanisms underlying eNOS uncoupling and clinically relevant strategies that were adopted to influence them are also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The lumen of all healthy blood vessels is lined by the vascular endothelium, a monolayer of cells that constitutes the interface between oxygen, nutrients, circulating cells and a variety of factors carried within the bloodstream and all tissues and organs of the body. As such, the endothelium is the first organ that is exposed to exogenous insults and altered endothelial cell function, or endothelial cell activation, is recognized as the initiating event of many cardiovascular diseases. Nitric oxide (NO) is a gasotransmitter generated by the “healthy” endothelium with well-documented effects on vascular tone as well as in the prevention of smooth muscle cell proliferation and migration, leukocyte adhesion and platelet aggregation [1]. Early studies demonstrated that a functional endothelial NO synthase (eNOS) enzyme is protective against pathological vascular remodelling [2], hypertension [3], atherosclerosis [4] and complications associated with diabetes [5, 6].
In endothelial cells, NO is synthesized by the eNOS, which is a multi-domain enzyme consisting of an N-terminal oxygenase domain containing binding sites for heme, the substrate l-arginine and the cofactor tetrahydrobiopterin (BH4) and a reductase domain where the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and calmodulin (CaM) bind. During the synthesis of NO, NADPH-derived electrons pass to flavins in the reductase domain and must be then transferred to the heme located in the oxygenase domain so that the heme iron can bind O2 and catalyse the stepwise synthesis of NO from l-arginine. The activity of eNOS is determined by intracellular calcium concentrations and CaM binding but can also be modulated at the transcriptional, post-transcriptional and post-translational levels (e.g. palmitoylation, phosphorylation, S-glutathionylation and S-nitrosylation). Phosphorylation of eNOS on serine (Ser), threonine (Thr) and tyrosine (Tyr) residues modulates its activity (Fig. 1). Many kinases including AKT [7, 8], adenosine monophosphate-activated kinase (AMPK) [9], CaM kinase II [10] and protein kinase (PK) A [11, 12] can phosphorylate eNOS on Ser1177, thus potentiating the enzyme’s catalytic activity in response to a variety of stimuli. Phosphorylation on Ser615 and Ser633 by AKT, PKA, AMPK or Pim1 (see below) has also been associated with increased NO production [13]. AMPK and PKC were shown to mediate the inhibitory phosphorylation on eNOS Thr495 which interferes with the binding of calcium-activated CaM [9, 10, 14]. Thr495 is basally phosphorylated in endothelial cells, and as the binding of CaM is required to initiate NO production, it follows that cell stimulation is usually linked with the rapid dephosphorylation of Thr495 [10, 15]. This is a general response elicited by calcium-elevating agonists and usually slightly precedes the phosphorylation of the serine residues. In fact, agonist-induced enzyme activity is associated with reciprocal changes in the phosphorylation of Ser1177 and Thr495 i.e. one transiently decreases while the other transiently increases. The role of eNOS tyrosine phosphorylation is less well studied, but Src activation has been linked with the phosphorylation of Tyr81 which indirectly increases eNOS activity and NO production [16–18], while proline-rich tyrosine kinase 2 (PYK2)-mediated phosphorylation of Tyr657 abrogates eNOS catalytic activity [19, 20]. Dysregulation of any of the post-translational modifications mentioned above can lead to the attenuated or altered enzymatic activity and decrease in bioavailable NO that characterizes the state referred to as “endothelial dysfunction”. The latter is linked with the switch of eNOS from an NO-generating enzyme to an enzyme that can also generate superoxide anions (O2 −) and hydrogen peroxide (H2O2), a phenomenon known as “eNOS uncoupling”. eNOS activity is also regulated by a series of protein-protein interactions with the ability to influence enzyme localisation, trafficking and catalytic activity. As the regulation of eNOS function by post-translational modifications has been reviewed elsewhere [21, 22], this review will focus on recent findings related to protein-protein interactions that modulate eNOS function, the context in which these interactions were identified and their link to endothelial dysfunction. Recent advances in understanding the mechanisms leading to eNOS uncoupling and the implications of these findings for cardiovascular medicine will also be discussed.

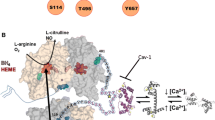
The eNOS signalosome. In its inactive state, human eNOS is phosphorylated on Ser114 and Thr495 and forms a complex with Cav-1. Hsp90 mediates the inhibitory interaction with CHIP and Cdc37. Direct binding of Pin1 to eNOS phosphorylated on Ser114 is considered inhibitory, though other reports suggest that Pin1 binding enables dephosphorylation of Ser114 and eNOS activation (see main text). Increases in intracellular Ca2+ concentrations lead to the displacement of Cav-1 by CaM, which initiates a burst in eNOS activity. A variety of protein kinases phosphorylate eNOS on Tyr81, Ser615, Ser633 and Ser1177, resulting in enhanced activation of the enzyme. SDF2 is required for the assembly of a functional eNOS complex including CaM and Hsp90. In endothelial cells, the interaction of ILK with Hsp90 and eNOS promotes NO production and prevents eNOS uncoupling. At least in sinusoidal endothelial cells, GIT1 binds directly to eNOS and facilitates AKT-mediated phosphorylation of eNOS on Ser1177. Small peptides and mutant proteins, such as Cavnoxin and Cav-1 Phe92Ala (F92A), can be effectively used to interfere with dynamic changes of the eNOS signalosome, thereby influencing eNOS activity
The eNOS signalosome
Core interactions
CaM and Cav-1: CaM was the first protein found to directly interact and regulate eNOS function [23], and eNOS activity is generally proportional to the level of intracellular calcium and the binding of calcium-activated CaM. There are however subtle differences in the relationship between calcium/CaM and NO output depending on the subcellular localisation of eNOS, with intracellular eNOS pools being less responsive to changes in calcium than the enzyme associated with membrane subdomains [24]. Under basal conditions, eNOS is anchored to plasma membrane caveolae via the N-myristoylation and palmitoylation of its N-terminus and is maintained in an inactive/basally active state through its interaction with the scaffolding domain of Cav-1 [25–27]. Upon stimulation with calcium-elevating agonists, Cav-1 is displaced by calcium-activated CaM, resulting in a conformational change that promotes NADPH-dependent electron flux to the heme moiety and the generation of NO [28–30]. The importance of this regulatory mechanism is demonstrated by the fact that peptides containing the Cav-1 scaffolding domain inhibit NO generation by eNOS [31]. Moreover, Cav-1−/− mice display enhanced basal and stimulated eNOS activity as well as subsequently enhanced vasorelaxation [32, 33], an effect that can be rescued by reintroduction of Cav-1 [34, 35]. Within the Cav-1 scaffolding domain, Thr90 and 91 and Phe92 are responsible for the interaction with and inhibition of eNOS [36], information that has been exploited to generate peptides based on the Cav-1 scaffolding domain that are able to increase NO bioavailability [37]. For example, the Cav-1 Phe92Ala mutant protein and Cavnoxin (a peptide containing the Thr90/91 and Phe92 substitutions) act as “dominant negative” scaffolding domains in that they release eNOS from the inhibitory interaction with endogenous Cav-1 to increase basal NO release from endothelial cells [37]. Moreover, Cavnoxin attenuates vessel tone ex vivo and lowers blood pressure in wild-type mice while leaving eNOS−/− and Cav-1−/− mice unaffected. The potential benefit of such peptides to increase eNOS activity was recently demonstrated in a mouse model of diabetes-associated atherosclerosis where it significantly attenuated atherosclerotic burden in vivo. The latter was accompanied by a decrease in oxidative stress markers, attenuated expression of pro-atherogenic mediators and reduced leukocyte-endothelial interactions [38].
Hsp90: The molecular chaperone heat shock protein 90 (Hsp90) is a major signalling hub in many cell types including endothelial cells. Hsp90 is involved in the folding of NOS enzymes and is likely to influence the insertion of heme into the immature protein and thus eNOS maturation and stability [39]. The inhibition of Hsp90 by prolonged exposure to geldanamycin results in the degradation of Hsp90 client proteins, including eNOS [40]. Domain mapping studies have shown that eNOS binds to the M domain of Hsp90 [41], and this interaction is increased in endothelial cells by several stimuli including vascular endothelial growth factor (VEGF), histamine, estrogen, and fluid shear stress [42, 43]. The binding of Hsp90 alone is able to induce a conformational change that promotes eNOS activity and increases NO production [29, 42, 44]. Moreover, Hsp90 functions as a molecular scaffold for the recruitment of many proteins that regulate the activity of eNOS. The best characterized of these proteins is the serine/threonine kinase AKT [41], but the list of proteins requiring the presence of Hsp90 to bind and modulate eNOS activity and/or localisation is increasing.
Interactions involving Hsp90
Cdc37: Cell division cycle 37 (Cdc37) is a co-chaperone of Hsp90 that was found to interact with and inhibit eNOS [45]. Despite studies that have assessed the consequences of inhibitors that disrupt the interaction between Cdc37 and Hsp90, especially in the context of cancer therapy (reviewed in [46]), there has been no detailed studies addressing the interaction between Cdc37 and eNOS. Although Cdc37 has potential to influence NO output, it is not an attractive therapeutic target given its central role in the maturation of the catalytic domains of many protein kinases.
CHIP: The subcellular localisation of eNOS is an important determinant of its activation state, as well as downstream NO-sensitive signal transduction pathways. One protein that affects localisation is the C-terminal hsp70-interacting protein (CHIP) which prevents eNOS from trafficking through the Golgi complex and the distribution of eNOS into an inactive detergent-insoluble compartment [47]. CHIP is also not an attractive target for potential intervention given its role as an Hsp90/Hsp70 co-chaperone and ubiquitin ligase.
ILK: Integrin-linked kinase (ILK) is a phosphoinositide 3-kinase-dependent serine/threonine kinase that binds to the cytoplasmic domain of β-integrin and lies upstream of many intracellular signalling pathways [48, 49]. ILK has been detected in a complex with eNOS and Hsp90 [50] and has been attributed roles in angiogenesis and vasculogenesis [51, 52] as well as endothelial cell survival and vascular development [53]. ILK has also been proposed as a regulator of endothelial function since ILK expression could not be detected in the endothelium of atherosclerotic arteries (human or mouse) [50]. Moreover, endothelial cells from conditional ILK knockout mice show signs of eNOS uncoupling i.e. reduced BH4 levels, increased 7,8-dihydro-l-biopterin (BH2) levels, decreased dihydrofolate reductase (DHFR) expression and increased eNOS-dependent generation of O2 − accompanied by extensive vascular protein nitration. The situation is however not entirely clear as the same conditional ILK knockout mice were used to generate essentially contradictory data in that ILK depletion in aortic vessels led to increased vascular expression and activity of the primary NO receptor i.e. soluble guanylate cyclase (sGC) and its downstream target protein kinase G (PKG), both of which promote relaxation [54]. To resolve these controversial findings, studies in conditional cell type-specific knockout mice may help dissecting the diverse actions of ILK in the vessel wall.
Pim1: A member of the Pim (proviral integration site for Moloney murine leukemia virus) family of serine/threonine kinases, Pim1, is a downstream effector of AKT that mediates cardiomyocyte survival in response to ischemic insults [55, 56]. Moreover, its downregulation contributes to the pathogenesis of diabetic cardiomyopathy [57]. Interestingly, a recent report identified a consensus Pim1 phosphorylation motif in eNOS that would target Ser633 to increase NO production [58]. Several kinases have been reported to phosphorylate eNOS on serine residues, and Pim1 may be more important in the longer-term phosphorylation and activation of the protein rather than its rapid and transient phosphorylation following agonist stimulation. Certainly, Pim1 has been implicated in the sustained activation of eNOS by VEGF, taking over from the transient phosphorylations attributed to AKT and PKA. These findings have also implications for vascular complications associated with diabetes and hyperglycaemia, as Pim1 expression as well as eNOS phosphorylation on Ser633 are reduced in endothelial cells and aortae from diabetic mice [58]. Also, Pim1 has a link to Hsp90 which protects Pim1 from proteasomal degradation [59, 60]. It is tempting to speculate that Hsp90 participates in the stabilisation of the eNOS/Pim1 complex, much in the same way that it does for AKT, thus facilitating eNOS phosphorylation on Ser633 and eNOS activation.
SDF2: Stromal cell-derived factor 2 (SDF2) was originally identified as a secreted protein using the signal sequence trap method in the mouse ST2 stromal cell line. Even though its function in mammals remains largely unknown, SDF2 is expressed in several mouse tissues where it localizes to the endoplasmic reticulum [61]. Its link to eNOS was revealed using a proteomic strategy of tandem affinity purification followed by mass spectrometry, and SDF2 was found preferentially in a complex with the active Hsp90-bound, Ser1177-phosphorylated form of eNOS [40]. SDF2 seems to directly affect eNOS activity as SDF2 knockdown decreases while overexpression enhances NO release. Moreover, the ability of SDF2 to stimulate NO production is markedly attenuated by the point mutation of Ser1177 to alanine. SDF2 was also identified as a novel Hsp90 client protein, and inhibition of Hsp90 triggered its degradation (similar to many client proteins including AKT and eNOS). Also, domain mapping studies revealed that SDF2 binds to the M domain of Hsp90, a common site for Hsp90-interacting proteins [62]. Stimulation of endothelial cells with VEGF triggers the formation of a complex comprising eNOS, SDF2, Hsp90 and CaM, an effect attenuated in cells lacking SDF2. The higher rates of NO synthesis accompanying increases in the expression of SDF2 suggest that the interaction between SDF2 and Hsp90 is mainly driven by the abundance of each protein. The interaction between Hsp90 and SDF2 is stable upon inhibition of PI3K or AKT activation or reduction of calcium concentrations [40]. However, it is possible that other post-translational modifications of Hsp90 may regulate the interaction. Future studies will be needed to address the regulatory aspects of the interaction as well as the direct effect of SDF2 on Hsp90 function as a signalling hub. Moreover, whether reduced amounts of SDF2 result in eNOS uncoupling and could therefore contribute to the development of cardiovascular diseases remains to be investigated.
Interactions indirectly affecting eNOS function
NOSIP and NOSTRIN: Yeast two hybrid studies identified eNOS interacting protein (NOSIP) and eNOS traffic inducer (NOSTRIN) as members of the eNOS signalosome that modulate NO generation by affecting the subcellular localisation of eNOS [63–65]. Initially, NOSIP or NOSTRIN overexpression was shown to promote the translocation of eNOS from the plasma membrane caveolae to intracellular compartments, such as the Golgi, thereby reducing overall NO output. While these initial reports seemed convincing, translocation is not always synonymous with inhibition and differentially distributed pools of eNOS exist within endothelial cells, which are all equally able to synthetize NO [66–68]. For example, the S-nitrosation of Golgi, mitochondrial and even nuclear proteins has been recently demonstrated, corroborating the existence of active NO synthesis also in cellular organelles [69, 70]. Meanwhile, NOSIP and NOSTRIN have been allocated alternative roles in the regulation of cell function. NOSIP belongs to the family of U-box ubiquitin E3 ligases, and global NOSIP deficiency in mice results in perinatal lethality due to holoprosencephaly and craniofacial malformations. The characterisation of NOSIP ubiquitination targets by interactomic studies revealed that NOSIP and protein phosphatase 2A (PP2A) interact and that loss of NOSIP results in reduced PP2A ubiquitination and increased PP2A catalytic activity [71]. NOSTRIN, on the other hand, has maintained its link to vascular function as its selective deletion from endothelial cells results in elevated blood pressure and diastolic dysfunction [72]. What has changed is the importance of direct actions on eNOS for its physiological actions as NOSTRIN interacts directly with the muscarinic acetylcholine receptor subtype M3 (M3R) and is required for its correct spatial localisation at the plasma membrane in aortic endothelial cells. In the absence of NOSTRIN, the function of the M3R is markedly impaired, resulting in abolition of the calcium response to acetylcholine, impaired activation of eNOS and inhibition of vascular relaxation, leaving responses to other eNOS activating endothelial cell agonists intact and fully functional [72]. Global NOSTRIN deletion was also found to impair post-natal retinal angiogenesis—an effect attributed to the fact that NOSTRIN assembles a signalling complex containing FGFR1, Rac1 and Sos1 thereby facilitating FGF‐2‐dependent activation of Rac1 in endothelial cells during developmental angiogenesis [73].
Interactions linked to endothelial dysfunction
PYK2: Interactions between kinases and their substrates can be difficult to capture as they are transient in nature, but one of the most recent kinases shown to contribute significantly to the modulation of eNOS activity is PYK2. The phosphorylation of eNOS by PYK2 (on Tyr657) has a direct inhibitory effect, and the fluid shear stress-induced association of eNOS with PYK2 was proposed as a mechanism to facilitate prolonged but low output eNOS activation (i.e. prevent uncoupling). In in vitro studies, the phosphorylation of eNOS Tyr657 within the FMN binding domain results in a complete loss of the ability of the enzyme to generate NO, O2 − or citrulline [19]. A clue as to why this particular tyrosine residue could have such dramatic effects can be found by considering the mechanisms known to regulate the activity of the neuronal NOS (nNOS), which was reported to be determined by a large-scale swinging motion of the FMN domain to deliver electrons to the catalytic module in the holoenzyme [74]. From the crystal structure of nNOS, the phosphorylation of a tyrosine residue (Tyr889, rat nNOS sequence), which is in the vicinity of the FMN domain, could prevent its movement, essentially locking the FMN domain into its electron-accepting position, thus inhibiting enzyme activity [74]. Since Tyr657 is the equivalent tyrosine residue in the human eNOS sequence, it is highly likely that its phosphorylation would inhibit NO production. A number of physiologically relevant stimuli can elicit the activation of PYK2 in endothelial cells, including insulin [19], angiotensin II (Ang II) [75] and oxidative stress [76].
The response to insulin is an interesting response to focus on as it highlights the need to study native endothelial cells in situ or low passaged endothelial cells in culture at the same time as demonstrating that eNOS phosphorylation on Ser1177 is not an absolute indicator of eNOS activation. Insulin has been attributed with vasodilator effects in vivo, but the role of NO and indeed endothelium in such responses has long been controversial. It is correct that the application of insulin to native or to cultured endothelial cells elicits the rapid phosphorylation of AKT as well as the phosphorylation of eNOS on Ser1177. However, in the same samples, there is neither a rapid NO-dependent relaxation nor an increase in cyclic GMP levels [77, 78]. The clue to the puzzle seems to be that insulin induces the simultaneous phosphorylation of Ser1177 and Tyr657, and as the latter event basically prevents electron transport through the FMN binding domain, the end result is a decrease in activity. In favour of this hypothesis is the report that the siRNA-mediated downregulation of PYK2 can couple endothelial cell stimulation with insulin with an increase in cyclic GMP [19]. Looking at the regulation of PYK2 expression in cultured endothelial cells also helps explain why there are so many reports in the literature of insulin-induced eNOS activation as PYK2 levels decrease relatively rapidly after cell isolation and the kinase—like the tyrosine phosphorylation of eNOS—can only be convincingly demonstrated in primary endothelial cell cultures [19].
The tyrosine phosphorylation of eNOS may be more important in pathophysiology than physiology as Ang II was also found to enhance the phosphorylation of eNOS Tyr657 in an angiotensin receptor 1-, H2O2- and PYK2-dependent manner [20]. In isolated mouse aortae, H2O2 induces phosphorylation of eNOS on Tyr657 and impairs acetylcholine-induced relaxation and endothelial overexpression of a dominant-negative PYK2 mutant protects against H2O2-induced endothelial dysfunction. Carotid arteries from eNOS−/− mice overexpressing the non-phosphorylatable eNOS Y657F mutant are also protected against H2O2. Chronic treatment with Ang II to elicit endothelial dysfunction and hypertension considerably increases levels of Tyr657-phosphorylated eNOS in aortae from wild-type but not Nox2y/− mice [20], suggesting that PYK2-mediated phosphorylation of eNOS on Tyr657 may contribute significantly to the impaired endothelial function characterizing many cardiovascular diseases (Fig. 2). Further studies are needed to clarify the relevance of the eNOS phosphorylation on Tyr657 in vivo as well as the molecular consequences of this phosphorylation on eNOS-dependent signalling and function in pathophysiological states. Interestingly, the post-translational modification of PYK2 by S-nitrosation [79, 80] has been linked to increased PYK2 activity [80]. If this event is confirmed in the endothelium, it may represent a novel negative feedback loop for the regulation of eNOS function and downstream signalling.

Mechanisms of endothelial dysfunction and eNOS uncoupling. In healthy endothelial cells, NADPH-donated electrons are transferred through the flavins FAD and FMN in the reductase domain to the heme located in the oxygenase domain so that the heme iron can bind O2 and catalyse the stepwise synthesis of NO from l-arginine. Trp447 is required for the binding of the cofactor BH4 to eNOS and is essential for efficient NO production. A high BH4/BH2 ratio is ensured by GTPCH, the rate-limiting enzyme for BH4 synthesis, and DHFR, which mediates BH2 recycling to BH4. Members of the intrinsic cellular redox machinery, such as thioredoxin (TRX1) and glutaredoxin (GRX1), maintain key cysteine residues in a reduced state, preventing S-glutathionylation. Many cardiovascular diseases are accompanied by increased inflammation and oxidative stress, reduced NO bioavailability and impaired endothelium-dependent vasorelaxation, a state referred to as endothelial dysfunction. In this situation, electron transfer from eNOS flavins becomes “uncoupled” from l-arginine (L-Arg) oxidation and O2 − is released from the oxygenase domain (eNOS uncoupling). Mechanisms responsible for reduced NO bioavailability/eNOS uncoupling include the following: (1) overexpression of arginase resulting in competition for l-Arg; (2) PYK2-mediated phosphorylation of Tyr657, resulting in complete abrogation of enzymatic activity; (3) downregulation of GTPCH and DHFR, leading to limited BH4 availability, and binding of catalytically incompetent BH2 to the BH4 binding site; and (4) S-glutathionylation of Cys689 and Cys908, facilitated by the oxidative environment generated by NOX2 in the reductase domain
GIT1: G-protein-coupled receptor (GPCR) kinase interactor-1 (GIT1) was recently described as a novel eNOS interactor and activator in sinusoidal endothelial cells [81]. Interestingly, GIT1 expression is reduced in sinusoidal endothelial cells after liver injury by liver duct ligation, consistent with previously described endothelial dysfunction in this disease. Re-expression of GIT1 after liver injury rescues eNOS phosphorylation on Ser1177 and NO synthesis [81]. A model for the fine regulation of the association between GIT1 and eNOS in sinusoidal endothelial cells has been recently proposed [82] as follows: upon endothelin-1 binding to the endothelin B receptor, Src is activated and phosphorylates GIT1 on Tyr293 and Tyr554. Phosphorylated GIT1 then associates with eNOS to facilitate its activating phosphorylation at Ser1177 by AKT. Thus, both Src and AKT kinases are crucial in the resultant phosphorylation-enhanced association of GIT1 and eNOS and in stimulating eNOS activity and NO production [82].
Pin1: The association between eNOS and prolyl isomerase (Pin) 1 is dependent on the constitutive phosphorylation of eNOS on Ser116. While the ability of Pin1 and eNOS to associate with each other is not controversial, the result of the interaction is. Initial studies indicated that Pin1 suppresses basal eNOS activity in a manner analogous to the tonic suppression of eNOS activity by its association with caveolin-1 [83], a mechanism that may be of particular relevance in endothelial cells exposed to high glucose concentrations. Certainly, pharmacological inhibition or genetic deletion of Pin1 in diabetic mice was shown to be protective against mitochondrial oxidative stress, endothelial dysfunction and vascular inflammation [84, 85]. Others have reported that the association of eNOS with Pin1 enables the dephosphorylation of Ser116 and stimulates NO production and demonstrated that a pharmacological inhibitor of Pin1 increased aortic eNOS Ser116 phosphorylation, endothelial dysfunction and hypertension, findings that were reproduced using Pin1-deficient mice [86]. To shed light in this controversial issue, a recent study demonstrated that Pin1 interacts directly with eNOS and the interaction increases when the phosphorylation of eNOS on Ser116 is mimicked. In bovine endothelial cells, TNFα induces ERK 1/2-mediated phosphorylation of eNOS on Ser116 (Ser114 in the human sequence), accompanied by Pin1 binding and a consequent reduction in NO release. This mechanism is however dependent on the presence of an adjacent proline residue (Pro117 in the bovine sequence, Pro115 in the human sequence); without a proline in this position, Pin1 binding and prolyl isomerisation, cannot occur. The importance of this residue is highlighted by the fact that in the mouse and rat eNOS sequences, Pro115 has been replaced by glutamine and can account for the fact that eNOS phosphorylation of Ser116 seems not to be detectable in mouse tissues [87]. The stability of Pim1 is thought to be determined by its ability to complex with Pin1, which promotes Pim1 degradation [88]. It is therefore tempting to speculate that in diabetes, the increased binding of Pin1 to eNOS may explain also the reported reduction of Pim1 protein levels and subsequent decreased Ser633 phosphorylation.
Cx37/Cx40: First evidence of the interaction between eNOS and connexin 37 (Cx37) came from a high-throughput phage display screening in search for peptide sequences that bind to Cx37 C-terminus, as a polymorphism in this region is associated with arterial stenosis and myocardial infarction in humans [89]. Experiments in vitro confirmed that a Cx37/eNOS complex also exists in native murine and human endothelial cells and that Cx37 exerts an inhibitory action on NO synthesis [89]. However, these results were not supported by in vivo or ex vivo evidence (see below). Another endothelial-specific connexin, connexin 40 (Cx40), has been shown to play a pivotal role in the regulation of blood pressure and vasorelaxation [90, 91]. This effect has been linked to reduced eNOS expression in Cx40-deficient mice [92]. Restoration of Cx40 expression in endothelial cells from Cx40-deficient mice normalizes eNOS levels [93], suggesting a link between these proteins. Volume-dependent hypertension (one kidney, one clip model) promotes the interaction of both Cx40 and Cx37 with eNOS resulting in increased release of NO [93]. Contrary to what was previously shown for Cx37’s effect on eNOS function in vitro, vascular reactivity studies demonstrated that basal NO release and the sensitivity to acetylcholine are decreased in aortae from Cx37−/− and Cx40−/− mice but not in Cx40+/− mice [94]. A more detailed analysis of the mechanisms regulating the interaction between these two connexins and eNOS and the generation of endothelial-specific knockout mice will deepen our understanding of the complex role of these connexins in eNOS signalling and function.
Hbα: Haemoglobin (Hb) is a well-characterised NO scavenger, so the first report that Hbα is expressed in endothelial cells exclusively at the sites of myo-endothelial junctions [95] was rather surprising. Mechanistically, Hbα was proposed to regulate NO diffusion to vascular smooth muscle during α1-adrenergic-dependent vasoconstriction and prevent NO flooding the microcirculation. The somewhat paradox association of Hbα with eNOS at myo-endothelial junctions has been substantiated by different lines of evidence including immunofluorescence, proximity ligation assays and co-immunoprecipitations from both cell lysates and purified proteins [95]. However, although an interaction between Hbα and eNOS has been proposed by in silico modelling [96], rigorous domain mapping and mutation experiments remain to be performed. Nevertheless, the modelling studies allowed the generation of a 10 amino acid-long Hbα mimetic peptide (called Hbα X) able to interfere with the association between Hbα and eNOS. Incubation of thoracodorsal arteries with Hbα X enhanced phenylephrine-induced cyclic GMP production and decreased vasoconstriction. In addition, when injected into normotensive and hypertensive mice, Hbα X induced a significant decrease in blood pressure, whereas injection of Hbα X into eNOS−/− mice had no effect [96]. Future studies are required to dissect the molecular mechanism behind the assembly of the Hbα/eNOS complex in endothelial cells and whether endothelial Hbα expression may participate in arteriogenesis at the myo-endothelial junction as well as anti-inflammatory signalling and what role it may potentially play in eNOS coupling.
NOX2: The S-glutathionylation of eNOS reversibly decreases its activity, resulting in an increase in endothelial cell O2 − generation [97]. A role for NOX2 in this particular post-translational modification has recently been reported [98]. NOX2-specific inhibition prevents lipopolysaccharide-induced eNOS S-glutathionylation and reduces O2 − production and permeability in lung microvascular endothelial cells. Lipopolysaccharide exposure induces the formation of a complex between eNOS and NOX2 that in turn enables low-level NOX2-catalysed O2 − and/or reactive metabolites of O2 − to oxidize cysteine residues in the eNOS reductase domain, making them susceptible to S-glutathionylation [98]. Though BH2 and BH4 were not measureable in lung microvascular endothelial cells in vitro, NOX2-derived O2 − could also promote the oxidation of BH4 to BH2, resulting in further eNOS uncoupling. Future studies will need to explore how this interaction is regulated and whether targeting the interaction may prove successful in preventing eNOS uncoupling.
The eNOS uncoupling phenomenon
The efficient conversion of the substrate l-arginine into NO and l-citrulline by an active eNOS homodimer requires a very specific and controlled balance between availability of substrate and cofactors, regulated post-translational modifications and protein-protein interactions. There are however some situations in which the enzyme can be switched from a purely NO-generating enzyme to one that generates NO as well as O2 − (Fig. 2). The switch to O2 − production is referred to as “eNOS uncoupling”, which basically means that the transport of electrons to ferrous-heme-O2 species generated during the stepwise activation of O2 by NOS does not occur fast enough to prevent their oxidative decay, the result being the generation of reactive oxygen species. The enhanced generation of O2 − is likely to result in the formation of the highly potent oxidant peroxynitrite (ONOO−), which may further enhance O2 − production by oxidation of the zinc cluster within eNOS and dissociation of the functional dimer [99].
l-arginine and arginase
Under physiological conditions, the intracellular concentration of l-arginine is generally high enough to be in excess of the Km for eNOS. However, cardiovascular disease and the associated oxidative stress have been linked with reduced l-arginine transport and/or competition with other arginine-utilizing enzymes such as arginase, leading ultimately to eNOS uncoupling. Several studies have convincingly demonstrated that increased arginase activity is associated with endothelial dysfunction. This is shown in various experimental models of hypertension [100], atherosclerosis [101], diabetes [102] and aging [103] (reviewed in [104]). In all of these studies, the disease state induces elevated expression of arginase and oxidative stress, thus resulting in decreased NO bioavailability and impaired endothelial-dependent vasorelaxation and function. Accordingly, inhibition of arginase decreases oxidative stress and restores NO bioavailability and normal endothelial function. Certainly, the results obtained in animal models have encouraged clinical studies to test the use of arginase inhibitors to treat cardiovascular diseases. In patients with coronary artery disease and type 2 diabetes, the intra-arterial infusion of the arginase inhibitor Nω-hydroxy-nor-l-arginine (nor-NOHA) was shown to improve forearm endothelial function [105]. Coronary arterioles obtained from patients with type 1 and type 2 diabetes displayed reduced endothelium-dependent relaxation in vitro and increased expression of arginase I in endothelial cells, and treatment with nor-NOHA improved coronary arteriolar endothelial function [106]. Plasma levels of arginase in patients with heart failure were higher than in control subjects and proportional to the severity of the disease. Again, local administration of nor-NOHA resulted in improved sublingual microcirculation by an NO-dependent mechanism [107]. Lastly, reflex cutaneous vasodilatation in patients with hypertension was increased following administration of S-(2-boronoethyl)-l-cysteine and nor-NOHA via skin microdialysis catheters [108]. Thus, arginase presents an attractive and promising pharmacological target against cardiovascular disease.
BH4/BH2 ratio
Suboptimal concentrations of the essential cofactor BH4 result in eNOS uncoupling. Replenishment of BH4 levels with sepiapterin or the overexpression of the guanosine triphosphate cyclohydrolase I (GTPCH), the rate-limiting enzyme in BH4 biosynthesis, effectively augments BH4 levels in cultured endothelial cells and improves NO output. It is also known that BH4 levels decline quite rapidly in cultured endothelial cells, requiring exogenous BH4, for example by supplementation of sepiapterin to the culture media in order to maintain NO production. Although fully reduced BH4 supports catalysis by eNOS, oxidized species such as BH2 and biopterin are catalytically incompetent, having the same allosteric effects without the ability to catalyse NO production (for review, see [109]). Uncoupled eNOS in dysfunctional endothelium, therefore, generates O2 − and ONOO– causing oxidation of BH4, creating a vicious feedback loop sustaining further eNOS uncoupling, increased oxidative stress and reduced NO bioavailability. Another mechanism leading to reduced BH4 bioavailability is by downregulation of GTPCH expression [109]. Regardless of the cause for BH4 deficiency, BH4 supplementation has been shown to enhance NO-mediated effects in cell culture (sepiapterin), in animal models and also in patients with cardiovascular disease [110]. For example, gene transfer of human GTPCH in hypertensive rats restores BH4 levels, improves endothelial function [111] and constitutive endothelial-specific overexpression of GTPCH in diabetic mice as well as in ApoE−/− mice and prevents the loss of endothelial BH4, eNOS uncoupling and endothelial dysfunction [112, 113]. Conversely, other reports show that in atherosclerosis, BH4 levels may also be depleted due to its oxidative degradation to BH2 by ONOO– and O2 − in the vascular wall [114]. Numerous clinical studies have tested whether the pharmacological supplementation of BH4 can sufficiently improve endothelium-dependent relaxation and endothelial function. In these studies, acute intra-arterial infusions of BH4 led to short-term improvements in endothelial function. This has been demonstrated in patients with risk factors for cardiovascular disease, such as chronic cigarette consumption [115] and hypercholesterolemia [116], in patients with established coronary artery disease [117] and disease states such as diabetes and hypertension [118, 119]. Despite the general enthusiasm generated by these clinical studies, these findings are, however, difficult to interpret in light of the high doses of BH4 that were used as well as due to the lack of long-term studies. A caveat of such approaches, especially for their application to advanced cardiovascular diseases, is that in the presence of a highly oxidizing environment, exogenous BH4 is oxidized to BH2, which lacks eNOS cofactor activity. This was, in fact, the case in a recent study that reported no net modification of the ratio of reduced to oxidized biopterins in patients with coronary artery disease receiving oral administration of BH4, despite elevated BH4 levels in the blood after administration. As a consequence, beneficial effects on eNOS coupling, endothelial function, or vascular O2 − production could not be demonstrated [120]. Until now, the regulation of the binding of BH4 to eNOS received little attention, the major focus being the relative availability of the cofactor in endothelial cells. A recent study, however, revealed a novel layer of complexity in the relationship between BH4 and eNOS uncoupling as a tryptophan residue at position 447 within the BH4 binding site of eNOS is required for efficient NO production by the enzyme, by preserving eNOS coupling and dimerisation [121]. However, while mutation of Trp447 switched eNOS to an O2 −-generating enzyme and highlights the role of BH4 in the uncoupling phenomenon, there is no information available regarding modification of Trp447 in pathophysiological situations.
Another important enzyme that contributes to the balance between BH4 and BH2 is DHFR, which is able to reduce BH2 thus regenerating BH4. DHFR expression is reduced by Ang II, leading to reduced BH4 levels and eNOS uncoupling [122]. While targeting BH4 remains a rational and attractive therapeutic strategy in cardiovascular disease, future studies should aim to manipulate the BH4/BH2 ratio in favour of BH4, to enhance the binding of BH4 to eNOS while preventing its oxidation or to boost BH4 recycling pathways.
S-glutathionylation
Manipulation of l-arginine or BH4 metabolism is not sufficient to completely restore eNOS activity and NO-dependent vasodilatation, indicating the existence of additional mechanisms contributing to eNOS uncoupling and dysfunction. S-glutathionylation is the reversible binding of a glutathione tripeptide (glycine, cysteine and glutamic acid) to a protein via the formation of disulphide bond with a protein thiol, and the S-glutathionylation of eNOS has been recently identified as a main cause of eNOS uncoupling. In an oxidative environment, S-glutathionylation can be mediated by thiol-disulfide exchange with oxidized glutathione, reaction with oxidant-induced protein thiyl radicals with reduced glutathione or reaction of a nitrosothiol with another thiol [97]. S-glutathionylation of two conserved cysteines (Cys), Cys689 and Cys908, in the reductase domain of eNOS leads to increased O2 − generation, and this form of eNOS uncoupling is (unlike uncoupling induced by BH4 depletion) insensitive to NOS inhibitors and calcium chelators [97]. These findings have clear pathophysiological significance, and vessels from spontaneously hypertensive rats display higher levels of eNOS S-glutathionylation and impaired endothelium-dependent vasorelaxation, which can be rescued by treatment with thiol-specific reducing agents [97]. Also, Ang II-mediated endothelial dysfunction was shown to involve eNOS S-glutathionylation in cultured endothelial cells and in intact vessels. Also, the attenuation of Ang II signalling in vivo by administration of an angiotensin-converting enzyme inhibitor reduces eNOS S-glutathionylation and eNOS uncoupling, improves endothelium-dependent vasorelaxation and reduces blood pressure [123]. Noticeably, S-glutathionylation and loss of eNOS activity were shown to depend on NOX2 activity in two independent studies [98, 123]. When considering the causes of S-glutathionylation, it is clear that any approach that restores a reducing environment within endothelial cells, including, for example, the potentiation of the intrinsic cellular thioredoxin and glutaredoxin anti-oxidant systems, would inhibit S-glutathionylation and eNOS uncoupling.
Outlook
The last 20 years have seen an enormous advance in knowledge regarding the role of endothelium-derived NO in the regulation of vascular tone and cardiovascular homeostasis in general. The fine regulation of eNOS activity has however turned out to be exceedingly complicated and regulated by subcellular location, associated proteins and post-translational modifications. It is clear that the eNOS signalosome changes rapidly in response to endogenous and exogenous stimuli, and it is crucial to investigate the temporal dynamics of these changes in a qualitative and, more importantly, quantitative manner to develop effective strategies to optimize NO output. Our increasing understanding of the molecular mechanisms underlying the phenomenon of eNOS uncoupling has facilitated the implementation of strategies to restore NO bioavailability and inhibit the eNOS-dependent generation of oxygen and nitrogen radicals. Strategies that target the enzymes involved in the metabolism of the substrate l-arginine or the cofactor BH4 have demonstrated some degree of benefit in clinical studies, but results remain below expectation. Further studies targeting the cellular redox state, i.e. reducing overall oxidative stress, seem to be a sensible and promising approach to limit the causes that bring about eNOS uncoupling and endothelial dysfunction.
References
Forstermann U, Munzel T (2006) Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113:1708–1714. doi:10.1161/CIRCULATIONAHA.105.602532
Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC (1998) Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 101:731–736. doi:10.1172/JCI1699
Shesely G, Nobuyo M, Kim S, Desai M, Krege H, Laubach E, Sherman A, Sessa C, Oliver S (1996) Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci 93:13176–13181. doi:10.1073/pnas.93.23.13176
Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL (2001) Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 104:448–454. doi:10.1161/hc2901.091399
Cook S, Hugli O, Egli M, Menard B, Thalmann S, Sartori C, Perrin C, Nicod P, Thorens B, Vollenweider P, Scherrer U, Burcelin R (2004) Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes 53:2067–2072. doi:10.2337/diabetes.53.8.2067
Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD (2000) Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 49:684–687. doi:10.2337/diabetes.49.5.684
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399:601–605. doi:10.1038/21224
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399:597–601. doi:10.1038/21218
Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE (1999) AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443:285–289. doi:10.1016/S0014-5793(98)01705-0
Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R (2001) Phosphorylation of Thr (495) regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res 88:E68–E75. doi:10.1161/hh1101.092677
Butt E, Bernhardt M, Smolenski A, Kotsonis P, Frohlich LG, Sickmann A, Meyer HE, Lohmann SM, Schmidt HH (2000) Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem 275:5179–5187. doi:10.1074/jbc.275.7.5179
Gangopahyay A, Oran M, Bauer EM, Wertz JW, Comhair SA, Erzurum SC, Bauer PM (2011) Bone morphogenetic protein receptor II is a novel mediator of endothelial nitric-oxide synthase activation. J Biol Chem 286:33134–33140. doi:10.1074/jbc.M111.274100
Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE (2002) Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem 277:42344–42351. doi:10.1074/jbc.M205144200
Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE (2001) Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem 276:17625–17628. doi:10.1074/jbc.C100122200
Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC (2001) Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem 276:16587–16591. doi:10.1074/jbc.M100229200
Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, Jennings IG, Venema RC (2005) Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J Biol Chem 280:35943–35952. doi:10.1074/jbc.M504606200
Fulton D, Ruan L, Sood SG, Li C, Zhang Q, Venema RC (2008) Agonist-stimulated endothelial nitric oxide synthase activation and vascular relaxation. Role of eNOS phosphorylation at Tyr83. Circ Res 102:497–504. doi:10.1161/CIRCRESAHA.107.162933
Venema RC (2002) Post-translational mechanisms of endothelial nitric oxide synthase regulation by bradykinin. Int Immunopharmacol 2:1755–1762. doi:10.1016/S1567-5769(02)00185-6
Fisslthaler B, Loot AE, Mohamed A, Busse R, Fleming I (2008) Inhibition of endothelial nitric oxide synthase activity by proline-rich tyrosine kinase 2 in response to fluid shear stress and insulin. Circ Res 102:1520–1528. doi:10.1161/CIRCRESAHA.108.172072
Loot AE, Schreiber JG, Fisslthaler B, Fleming I (2009) Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J Exp Med 206:2889–2896. doi:10.1084/jem.20090449
Dudzinski DM, Michel T (2007) Life history of eNOS: partners and pathways. Cardiovasc Res 75:247–260. doi:10.1016/j.cardiores.2007.03.023
Fleming I (2010) Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 459:793–806. doi:10.1007/s00424-009-0767-7
Busse R, Mulsch A (1990) Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett 265:133–136. doi:10.1016/0014-5793(90)80902-U
Church JE, Fulton D (2006) Differences in eNOS activity because of subcellular localization are dictated by phosphorylation state rather than the local calcium environment. J Biol Chem 281:1477–1488. doi:10.1074/jbc.M505968200
Garcia-Cardena G, Fan R, Stern DF, Liu J, Sessa WC (1996) Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J Biol Chem 271:27237–27240. doi:10.1074/jbc.271.44.27237
Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC (1997) Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem 272:25437–25440. doi:10.1074/jbc.272.41.25437
Ju H, Zou R, Venema VJ, Venema RC (1997) Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem 272:18522–18525. doi:10.1074/jbc.272.30.18522
Fulton D, Gratton JP, Sessa WC (2001) Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 299:818–824
Gratton JP, Fontana J, O’Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC (2000) Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem 275:22268–22272. doi:10.1074/jbc.M001644200
Michel JB, Feron O, Sacks D, Michel T (1997) Reciprocal regulation of endothelial nitric-oxide synthase by Ca2 + -calmodulin and caveolin. J Biol Chem 272:15583–15586. doi:10.1074/jbc.272.25.15583
Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G, Sessa WC (2000) In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med 6:1362–1367. doi:10.1038/82176
Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293:2449–2452. doi:10.1126/science.1062688
Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di VD, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138. doi:10.1074/jbc.M105408200
Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC (2007) Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med 204:2373–2382. doi:10.1084/jem.20062340
Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC (2006) Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116:1284–1291. doi:10.1172/JCI27100
Bernatchez PN, Bauer PM, Yu J, Prendergast JS, He P, Sessa WC (2005) Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Natl Acad Sci U S A 102:761–766. doi:10.1073/pnas.0407224102
Bernatchez P, Sharma A, Bauer PM, Marin E, Sessa WC (2011) A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J Clin Invest 121:3747–3755. doi:10.1172/JCI44778
Sharma A, Sellers S, Stefanovic N, Leung C, Tan SM, Huet O, Granville DJ, Cooper ME, de Haan JB, Bernatchez P (2015) Direct endothelial nitric oxide synthase activation provides atheroprotection in diabetes-accelerated atherosclerosis. Diabetes 64:3937–3950. doi:10.2337/db15-0472
Billecke SS, Bender AT, Kanelakis KC, Murphy PJ, Lowe ER, Kamada Y, Pratt WB, Osawa Y (2002) Hsp90 is required for heme binding and activation of apo-neuronal nitric-oxide synthase: geldanamycin-mediated oxidant generation is unrelated to any action of hsp90. J Biol Chem 277:20504–20509. doi:10.1074/jbc.M201940200
Siragusa M, Frohlich F, Park EJ, Schleicher M, Walther TC and Sessa WC (2015) Stromal cell-derived factor 2 is critical for Hsp90-dependent eNOS activation. Sci Signal 8:ra81. doi: 10.1126/scisignal.aaa2819
Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N, Tsuruo T, Sessa WC (2002) Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res 90:866–873. doi:10.1161/01.RES.0000016837.26733.BE
Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC (1998) Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392:821–824. doi:10.1038/33934
Russell KS, Haynes MP, Caulin-Glaser T, Rosneck J, Sessa WC, Bender JR (2000) Estrogen stimulates heat shock protein 90 binding to endothelial nitric oxide synthase in human vascular endothelial cells. Effects on calcium sensitivity and NO release. J Biol Chem 275:5026–5030. doi:10.1074/jbc.275.7.5026
Takahashi S, Mendelsohn ME (2003) Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J Biol Chem 278:30821–30827. doi:10.1074/jbc.M304471200
Harris MB, Bartoli M, Sood SG, Matts RL, Venema RC (2006) Direct interaction of the cell division cycle 37 homolog inhibits endothelial nitric oxide synthase activity. Circ Res 98:335–341. doi:10.1161/01.RES.0000203564.54250.0b
Gray PJ Jr, Prince T, Cheng J, Stevenson MA, Calderwood SK (2008) Targeting the oncogene and kinome chaperone CDC37. Nat Rev Cancer 8:491–495. doi:10.1038/nrc2420
Jiang J, Cyr D, Babbitt RW, Sessa WC, Patterson C (2003) Chaperone-dependent regulation of endothelial nitric-oxide synthase intracellular trafficking by the co-chaperone/ubiquitin ligase CHIP. J Biol Chem 278:49332–49341. doi:10.1074/jbc.M304738200
Chiswell BP, Zhang R, Murphy JW, Boggon TJ, Calderwood DA (2008) The structural basis of integrin-linked kinase-PINCH interactions. Proc Natl Acad Sci U S A 105:20677–20682. doi:10.1073/pnas.0811415106
Lal H, Verma SK, Foster DM, Golden HB, Reneau JC, Watson LE, Singh H, Dostal DE (2009) Integrins and proximal signaling mechanisms in cardiovascular disease. Front Biosci (Landmark Ed) 14:2307–2334. doi:10.2741/3381
Herranz B, Marquez S, Guijarro B, Aracil E, Aicart-Ramos C, Rodriguez-Crespo I, Serrano I, Rodríguez-Puyol M, Zaragoza C, Saura M (2012) Integrin-linked kinase regulates vasomotor function by preventing endothelial nitric oxide synthase uncoupling: role in atherosclerosis. Circ Res 110:439–449. doi:10.1161/CIRCRESAHA.111.253948
Legate KR, Montanez E, Kudlacek O, Fassler R (2006) ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol 7:20–31. doi:10.1038/nrm1789
Werner C, Bohm M, Friedrich EB (2008) Role of integrin-linked kinase for functional capacity of endothelial progenitor cells in patients with stable coronary artery disease. Biochem Biophys Res Commun 377:331–336. doi:10.1016/j.bbrc.2008.09.081
Friedrich EB, Liu E, Sinha S, Cook S, Milstone DS, MacRae CA, Mariotti M, Kuhlencordt PJ, Force T, Rosenzweig A, St-Arnaud R, Dedhar S, Gerszten RE (2004) Integrin-linked kinase regulates endothelial cell survival and vascular development. Mol Cell Biol 24:8134–8144. doi:10.1128/MCB.24.18.8134-8144.2004
Serrano I, De FS, Griera M, Medrano D, Rodriguez-Puyol M, Dedhar S, Ruiz-Torres MP, Rodriguez-Puyol D (2013) Ilk conditional deletion in adult animals increases cyclic GMP-dependent vasorelaxation. Cardiovasc Res 99:535–544. doi:10.1093/cvr/cvt131
Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, Fischer K, Martindale JJ, Glembotski CC, Leri A, Kajstura J, Magnuson N, Berns A, Beretta RM, Houser SR, Schaefer EM, Anversa P, Sussman MA (2007) Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med 13:1467–1475. doi:10.1038/nm1671
Siragusa M, Katare R, Meloni M, Damilano F, Hirsch E, Emanueli C, Madeddu P (2010) Involvement of phosphoinositide 3-kinase gamma in angiogenesis and healing of experimental myocardial infarction in mice. Circ Res 106:757–768. doi:10.1161/CIRCRESAHA.109.207449
Katare R, Caporali A, Zentilin L, Avolio E, Sala-Newby G, Oikawa A, Cesselli D, Beltrami AP, Giacca M, Emanueli C, Madeddu P (2011) Intravenous gene therapy with PIM-1 via a cardiotropic viral vector halts the progression of diabetic cardiomyopathy through promotion of prosurvival signaling. Circ Res 108:1238–1251. doi:10.1161/CIRCRESAHA.110.239111
Chen M, Yi B, Zhu N, Wei X, Zhang GX, Huang S, Sun J (2016) Pim1 kinase promotes angiogenesis through phosphorylation of endothelial nitric oxide synthase at Ser-633. Cardiovasc Res 109:141-150. doi:10.1093/cvr/cvv250
Mizuno K, Shirogane T, Shinohara A, Iwamatsu A, Hibi M and Hirano T (2001) Regulation of Pim-1 by Hsp90. Biochem Biophys Res Commun 281:663-669. doi: 10.1006/bbrc.2001.4405
Shay KP, Wang Z, Xing PX, McKenzie IF, Magnuson NS (2005) Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol Cancer Res 3:170–181. doi:10.1158/1541-7786.MCR-04-0192
Lorenzon-Ojea AR, Caldeira W, Ribeiro AF, Fisher SJ, Guzzo CR, Bevilacqua E (2014) Stromal cell derived factor-2 (Sdf2): a novel protein expressed in mouse. Int J Biochem Cell Biol 53:262–270. doi:10.1016/j.biocel.2014.05.024
Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S (2012) Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150:987–1001. doi:10.1016/j.cell.2012.06.047
Dedio J, König P, Wohlfart P, Schroeder C, Kummer W, Müller-Esterl W (2001) NOSIP, a novel modulator of endothelial nitric oxide synthase activity. FASEB J 15:79-89. doi:10.1096/fj.00-0078com
Schleicher M, Brundin F, Gross S, Muller-Esterl W, Oess S (2005) Cell cycle-regulated inactivation of endothelial NO synthase through NOSIP-dependent targeting to the cytoskeleton. Mol Cell Biol 25:8251–8258. doi:10.1128/MCB.25.18.8251-8258.2005
Zimmermann K, Opitz N, Dedio J, Renne C, Müller-Esterl W, Oess S (2002) NOSTRIN: a protein modulating nitric oxide release and subcellular distribution of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 99:17167–17172. doi:10.1073/pnas.252345399
Fulton D, Fontana J, Sowa G, Gratton JP, Lin M, Li KX, Michell B, Kemp BE, Rodman D, Sessa WC (2002) Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J Biol Chem 277:4277–4284. doi:10.1074/jbc.M106302200
Jagnandan D, Sessa WC, Fulton D (2005) Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol 289:C1024–C1033. doi:10.1152/ajpcell.00162.2005
Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D (2006) Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler Thromb Vasc Biol 26:1015–1021. doi:10.1161/01.ATV.0000216044.49494.c4
Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC (2006) Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A 103:19777–19782. doi:10.1073/pnas.0605907103
Sangwung P, Greco TM, Wang Y, Ischiropoulos H, Sessa WC, Iwakiri Y (2012) Proteomic identification of S-nitrosylated Golgi proteins: new insights into endothelial cell regulation by eNOS-derived NO. PLoS One 7, e31564. doi:10.1371/journal.pone.0031564
Hoffmeister M, Prelle C, Kuchler P, Kovacevic I, Moser M, Muller-Esterl W, Oess S (2014) The ubiquitin E3 ligase NOSIP modulates protein phosphatase 2A activity in craniofacial development. PLoS One 9, e116150. doi:10.1371/journal.pone.0116150
Kovacevic I, Muller M, Kojonazarov B, Ehrke A, Randriamboavonjy V, Kohlstedt K, Hindemith T, Schermuly RT, Fleming I, Hoffmeister M, Oess S (2015) The F-BAR protein NOSTRIN dictates the localization of the muscarinic M3 receptor and regulates cardiovascular function. Circ Res 117:460–469. doi:10.1161/CIRCRESAHA.115.306187
Kovacevic I, Hu J, Siehoff-Icking A, Opitz N, Griffin A, Perkins AC, Munn AL, Muller-Esterl W, Popp R, Fleming I, Jungblut B, Hoffmeister M, Oess S (2012) The F-BAR protein NOSTRIN participates in FGF signal transduction and vascular development. EMBO J 31:3309–3322. doi:10.1038/emboj.2012.176
Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED (2004) Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem 279:37918–37927. doi:10.1074/jbc.M406204200
Yin G, Yan C, Berk BC (2003) Angiotensin II signaling pathways mediated by tyrosine kinases. Int J Biochem Cell Biol 35:780–783. doi:10.1016/S1357-2725(02)00300-X
Tai LK, Okuda M, Abe J, Yan C, Berk BC (2002) Fluid shear stress activates proline-rich tyrosine kinase via reactive oxygen species-dependent pathway. Arterioscler Thromb Vasc Biol 22:1790–1796. doi:10.1161/01.ATV.0000034475.40227.40
Fleming I, Schulz C, Fichtlscherer B, Kemp BE, Fisslthaler B, Busse R (2003) AMP-activated protein kinase (AMPK) regulates the insulin-induced activation of the nitric oxide synthase in human platelets. Thromb Haemost 90:863–871. doi:10.1160/TH03-04-0228
Randriamboavonjy V, Schrader J, Busse R, Fleming I (2004) Insulin induces the release of vasodilator compounds from platelets by a nitric oxide-G kinase-VAMP-3-dependent pathway. J Exp Med 199:347–356. doi:10.1084/jem.20030694
Lee YI, Giovinazzo D, Kang HC, Lee Y, Jeong JS, Doulias PT, Xie Z, Hu J, Ghasemi M, Ischiropoulos H, Qian J, Zhu H, Blackshaw S, Dawson VL, Dawson TM (2014) Protein microarray characterization of the S-nitrosoproteome. Mol Cell Proteomics 13:63–72. doi:10.1074/mcp.M113.032235
Yan XL, Liu DH, Zhang GL, Hu SQ, Chen YG, Xu T (2015) S-Nitrosylation of proline-rich tyrosine kinase 2 involves its activation induced by oxygen-glucose deprivation. Neurosci Lett 597:90–96. doi:10.1016/j.neulet.2015.04.043
Liu S, Premont RT, Rockey DC (2012) G-protein-coupled receptor kinase interactor-1 (GIT1) is a new endothelial nitric-oxide synthase (eNOS) interactor with functional effects on vascular homeostasis. J Biol Chem 287:12309–12320. doi:10.1074/jbc.M111.320465
Liu S, Premont RT, Rockey DC (2014) Endothelial nitric-oxide synthase (eNOS) is activated through G-protein-coupled receptor kinase-interacting protein 1 (GIT1) tyrosine phosphorylation and Src protein. J Biol Chem 289:18163–18174. doi:10.1074/jbc.M113.521203
Ruan L, Torres CM, Qian J, Chen F, Mintz JD, Stepp DW, Fulton D, Venema RC (2011) Pin1 prolyl isomerase regulates endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol 31:392-398. doi:10.1161/ATVBAHA.110.213181
Costantino S, Paneni F, Luscher TF, Cosentino F (2016) Pin1 inhibitor Juglone prevents diabetic vascular dysfunction. Int J Cardiol 203:702–707. doi:10.1016/j.ijcard.2015.10.221
Paneni F, Costantino S, Castello L, Battista R, Capretti G, Chiandotto S, D’A D, Scavone G, Villano A, Rustighi A, Crea F, Pitocco D, Lanza G, Volpe M, Sal GD, Luscher TF, Cosentino F (2014) Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: insights in patients with diabetes. Eur Heart J 36:817–828. doi:10.1093/eurheartj/ehu179
Chiasson VL, Munshi N, Chatterjee P, Young KJ, Mitchell BM (2011) Pin1 deficiency causes endothelial dysfunction and hypertension. Hypertension 58:431–438. doi:10.1161/HYPERTENSIONAHA.111.172338
Kennard S, Ruan L, Buffett RJ, Fulton D and Venema RC (2016) TNFα reduces eNOS activity in endothelial cells through Serine 116 phosphorylation and Pin1 binding: confirmation of a direct, inhibitory interaction of Pin1 with eNOS. Vasc Pharmacol. doi:10.1016/j.vph.2016.04.003
Ma J, Arnold HK, Lilly MB, Sears RC, Kraft AS (2007) Negative regulation of Pim-1 protein kinase levels by the B56β subunit of PP2A. Oncogene 26:5145–5153. doi:10.1038/sj.onc.1210323
Pfenniger A, Derouette JP, Verma V, Lin X, Foglia B, Coombs W, Roth I, Satta N, Dunoyer-Geindre S, Sorgen P, Taffet S, Kwak BR, Delmar M (2010) Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler Thromb Vasc Biol 30:827–834. doi:10.1161/ATVBAHA.109.200816
de Wit C, Roos F, Bolz SS, Kirchhoff S, Kruger O, Willecke K, Pohl U (2000) Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res 86:649–655. doi:10.1161/01.RES.86.6.649
Wagner C, de WC, Kurtz L, Grunberger C, Kurtz A and Schweda F (2007) Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res 100:556-563. doi: 10.1161/01.RES.0000258856.19922.45
Alonso F, Boittin FX, Beny JL, Haefliger JA (2010) Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am J Physiol Heart Circ Physiol 299:H1365–H1373. doi:10.1152/ajpheart.00029.2010
Le GL, Alonso F, Mazzolai L, Meda P, Haefliger JA (2015) Interplay between connexin40 and nitric oxide signaling during hypertension. Hypertension 65:910–915. doi:10.1161/HYPERTENSIONAHA.114.04775
Meens MJ, Alonso F, Le GL, Kwak BR, Haefliger JA (2015) Endothelial Connexin37 and Connexin40 participate in basal but not agonist-induced NO release. Cell Commun Signal 13:34. doi:10.1186/s12964-015-0110-1
Adam CS, Alexander WL, Marie B, Scott RJ, Scott TD, Monica YL, Pamela SB, Angela KB, Linda C, Benjamin G, Isakson BE (2012) Endothelial cell expression of haemoglobin α regulates nitric oxide signalling. Nature 491:473–477. doi:10.1038/nature11626
Straub AC, Butcher JT, Billaud M, Mutchler SM, Artamonov MV, Nguyen AT, Johnson T, Best AK, Miller MP, Palmer LA, Columbus L, Somlyo AV, Le TH, Isakson BE (2014) Hemoglobin α/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler Thromb Vasc Biol 34:2594–2600. doi:10.1161/ATVBAHA.114.303974
Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL (2010) S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468:1115–1118. doi:10.1038/nature09599
Feng W, William S.Szczepaniak, Sruti S, Huanbo L, Yinna W, Ling W, Ying W, Eric E.Kelley, Alex FC, Mark TG and Bryan JM (2014) Nox2-dependent glutathionylation of endothelial NOS leads to uncoupled superoxide production and endothelial barrier dysfunction in acute lung injury. Am J Physiol Lung Cell Mol Physiol 307:L987–L997. doi: 10.1152/ajplung.00063.2014
Zou MH, Shi C, Cohen RA (2002) Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 109:817–826. doi:10.1172/JCI14442
Michell DL, Andrews KL, Chin-Dusting JP (2011) Endothelial dysfunction in hypertension: the role of arginase. Front Biosci (Schol Ed) 3:946–960. doi:10.2741/199
Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, Lim HK, Sohi J, Santhanam L, Soucy K, Tuday E, Baraban E, Ilies M, Gerstenblith G, Nyhan D, Shoukas A, Christianson DW, Alp NJ, Champion HC, Huso D, Berkowitz DE (2008) Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res 102:923–932. doi:10.1161/CIRCRESAHA.107.169573
Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, Caldwell RB, Caldwell RW (2008) Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res 102:95–102. doi:10.1161/CIRCRESAHA.107.155028
Santhanam L, Christianson DW, Nyhan D, Berkowitz DE (2008) Arginase and vascular aging. J Appl Physiol 105:1632–1642. doi:10.1152/japplphysiol.90627.2008
Pernow J, Jung C (2013) Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res 98:334–343. doi:10.1093/cvr/cvt036
Shemyakin A, Kovamees O, Rafnsson A, Bohm F, Svenarud P, Settergren M, Jung C, Pernow J (2012) Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 126:2943–2950. doi:10.1161/CIRCULATIONAHA.112.140335
Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z (2011) Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol 300:H777–H783. doi:10.1152/ajpheart.00831.2010
Quitter F, Figulla HR, Ferrari M, Pernow J, Jung C (2013) Increased arginase levels in heart failure represent a therapeutic target to rescue microvascular perfusion. Clin Hemorheol Microcirc 54:75–85. doi:10.3233/CH-2012-1617
Holowatz LA, Kenney WL (2007) Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J Physiol 581:863–872. doi:10.1113/jphysiol.2007.128959
Channon KM (2004) Tetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med 14:323–327. doi:10.1016/j.tcm.2004.10.003
Alp NJ, Channon KM (2004) Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 24:413–420. doi:10.1161/01.ATV.0000110785.96039.f6
Zheng JS, Yang XQ, Lookingland KJ, Fink GD, Hesslinger C, Kapatos G, Kovesdi I, Chen AF (2003) Gene transfer of human guanosine 5′-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension. Circulation 108:1238–1245. doi:10.1161/01.CIR.0000089082.40285.C3
Alp NJ, McAteer MA, Khoo J, Choudhury RP, Channon KM (2004) Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler Thromb Vasc Biol 24:445–450. doi:10.1161/01.ATV.0000115637.48689.77
Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, Goh N, Rockett KA, Channon KM (2003) Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest 112:725–735. doi:10.1172/JCI17786
Antoniades C, Shirodaria C, Crabtree M, Rinze R, Alp N, Cunnington C, Diesch J, Tousoulis D, Stefanadis C, Leeson P, Ratnatunga C, Pillai R, Channon KM (2007) Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 116:2851–2859. doi:10.1161/CIRCULATIONAHA.107.704155
Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, Munzel T (2000) Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res 86:E36–E41. doi:10.1161/01.RES.86.2.e36
Fukuda Y, Teragawa H, Matsuda K, Yamagata T, Matsuura H, Chayama K (2002) Tetrahydrobiopterin restores endothelial function of coronary arteries in patients with hypercholesterolaemia. Heart 87:264–269. doi:10.1136/heart.87.3.264
Maier W, Cosentino F, Lutolf RB, Fleisch M, Seiler C, Hess OM, Meier B, Luscher TF (2000) Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J Cardiovasc Pharmacol 35:173–178. doi:10.1155/2014/850312
Heitzer T, Krohn K, Albers S, Meinertz T (2000) Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 43:1435–1438. doi:10.1007/s001250051551
Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, Chayama K (2002) Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens 15:326–332. doi:10.1016/S0895-7061(01)02317-2
Cunnington C, Van AT, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, Antoniades C, Margaritis M, Lee R, Cerrato R, Crabtree MJ, Francis JM, Sayeed R, Ratnatunga C, Pillai R, Choudhury RP, Neubauer S, Channon KM (2012) Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation 125:1356–1366. doi:10.1161/CIRCULATIONAHA.111.038919
Benson MA, Batchelor H, Chuaiphichai S, Bailey J, Zhu H, Stuehr DJ, Bhattacharya S, Channon KM, Crabtree MJ (2013) A pivotal role for tryptophan 447 in enzymatic coupling of human endothelial nitric oxide synthase (eNOS): effects on tetrahydrobiopterin-dependent catalysis and eNOS dimerization. J Biol Chem 288:29836–29845. doi:10.1074/jbc.M113.493023
Chalupsky K, Cai H (2005) Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 102:9056–9061. doi:10.1073/pnas.0409594102
Galougahi KK, Liu CC, Gentile C, Kok C, Nunez A, Garcia A, Fry NA, Davies MJ, Hawkins CL, Rasmussen HH, Figtree GA (2014) Glutathionylation mediates angiotensin II-induced eNOS uncoupling, amplifying NADPH oxidase-dependent endothelial dysfunction. J Am Heart Assoc 3, e000731. doi:10.1161/JAHA.113.000731
Acknowledgments
We are grateful to Dr. Claudia Koch for her assistance in the preparation of the figures. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 834/A9 and Exzellenzcluster 147 “Cardio-Pulmonary Systems”).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Siragusa, M., Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflugers Arch - Eur J Physiol 468, 1125–1137 (2016). https://doi.org/10.1007/s00424-016-1839-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1839-0