
Abstract
The carotid body (CB) is the principal arterial chemoreceptor that mediates the hyperventilatory response to hypoxia. Our understanding of CB function and its role in disease mechanisms has progressed considerably in the last decades, particularly in recent years. The sensory elements of the CB are the neuron-like glomus cells, which contain numerous transmitters and form synapses with afferent sensory fibers. The activation of glomus cells under hypoxia mainly depends on the modulation of O2-sensitive K+ channels which leads to cell depolarization and the opening of Ca2+ channels. This model of sensory transduction operates in all mammalian species studied thus far, including man. However, the molecular mechanisms underlying the modulation of ion channel function by changes in the O2 level are as yet unknown. The CB plays a fundamental role in acclimatization to sustained hypoxia. Mice with CB atrophy or patients who have undergone CB resection due to surgical treatments show a marked intolerance to even mild hypoxia. CB growth under hypoxia is supported by the existence of a resident population of neural crest-derived stem cells of glia-like phenotype. These stem cells are not highly affected by exposure to low O2 tension; however, there are abundant synapse-like contacts between the glomus cells and stem cells (chemoproliferative synapses), which may be needed to trigger progenitor cell proliferation and differentiation under hypoxia. CB hypo- or hyper-activation may also contribute to the pathogenesis of several prevalent human diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oxygen (O2) is essential for cells to generate ATP through oxidative phosphorylation. Adaptive responses to the lack of O2 (hypoxia) have therefore evolved, particularly in mammals, to ensure O2 supply to the most O2-dependent organs such as the brain or the heart. Responses to hypoxia can be acute or chronic [41]. In mammalian cells, sustained hypoxia (hours or days) causes inhibition of O2-sensitive prolyl hydroxylases, which results in the stabilization of hypoxia-inducible transcription factors, in particular HIF1α and HIF2α. Increased levels of HIFs modulate the expression of a broad array of genes that lead to an increased number of O2-transporting red blood cells, non-aerobic metabolism, and the formation of new blood vessels [76]. However, survival in hypoxia requires the activation of fast, cardiorespiratory, and circulatory systemic reflexes (e.g., hyperventilation or sympathetic activation), with a time course of seconds, to increase the uptake of O2 and its distribution to the tissues. These acute reflexes are mediated by specialized O2-sensitive cells in the arterial chemoreceptors and other organs, which constitute the homeostatic oxygen sensing system [86]. The main arterial chemoreceptor is the carotid body (CB), a small, paired organ, located at the bifurcation of the carotid arteries, which is necessary for the adaptation of mammals (including man) to environments with low O2 tension (PO2) [86]. In addition to its critical minute-to-minute regulatory actions on the respiratory and cardiovascular systems, the CB has a fundamental role in acclimatization to hypoxia, growing in size in high altitude residents or in patients with cardiorespiratory diseases, and thereby increasing the activation of sensory fibers impinging upon the central respiratory neurons to evoke hyperventilation [1, 46]. CB hypertrophy under hypoxia occurs due to the existence within the organ of a resident population of neural crest-derived stem cells, which remain active in adult life [60]. CB changes during adaptation to hypoxia have become an area of increasing medical interest as a result of their potential involvement in tumorigenesis or in the pathophysiology of highly prevalent diseases such as obstructive sleep apnea (OSA) syndrome. In this paper, we present a summary of the cellular physiology of CB chemoreceptor cells, followed by a description of the role of CB in adaptation to chronic hypoxia and the properties of CB adult stem cells. Finally, we discuss the participation of the CB in mechanisms of disease.
Cellular physiology of O2-sensitive carotid body glomus cells
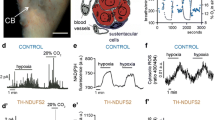
The CB is composed of clusters of cells, called glomeruli, in close contact with a profuse network of capillaries (Fig. 1a). Each glomerulus contains several neuron-like glomus, or type I, cells which possess numerous secretory vesicles filled with neurotransmitters, in particular dopamine, ATP, acetylcholine, and several neuropeptides. These cells normally express catecholaminergic markers, such as tyrosine hydroxylase (TH). Glomus cells are O2-sensitive presynaptic-like elements that form synaptic contacts (“chemosensory synapses”) with afferent excitatory nerve fibers of the nodose and petrosal ganglia, which terminate at the brain stem respiratory centers. Glomus cells are enveloped by the processes of glia-like, sustentacular or type II, cells which can be identified by immunostaining against glial fibrillary acidic protein (GFAP) [41, 60].

Structure and function of carotid body (CB) cells. a Schematic representation of a CB glomerulus with indication of glomus (type I, red) and sustentacular (type II, purple) cells, blood vessels and afferent sensory nerve fibers. b Top. Ionic currents recorded from a patch-clamped rabbit glomus cell during a depolarizing pulse from −80 to +20 mV. Inward calcium (ICa) and outward potassium (IK) currents are indicated. Voltage-gated sodium channels were blocked with tetrodotoxin. Note the selective reversible inhibition of the K+ current by exposure to hypoxia (switching from an O2 tension of 150 to 15 mmHg). b Bottom. Inhibition of single K+ channel activity recorded in an excised membrane patch exposed to hypoxia. c Catecholamine secretion from rodent glomus cells as a function of O2 tension. The inset illustrates the secretory response of a cell to hypoxia as monitored by amperometry in CB slices. Modified from [36]. d, e Number of tyroxine hydroxylase (TH) and dopamine decarboxylase (DDC) positive cells in human CB slices. Scale bar 50 μm. Quantification in e was obtained from 23 (DDC) and 28 (TH) donors from 10 to 67 years. f, g Secretory activity of cells in human CB slices in response to hypoxia (~10 mmHg) and 40 mM K+. Modified from [48]
Although the chemosensory nature of the CB body has been known for almost a century, the physiology of glomus cells only begun to be explored in detail after the development of in vitro preparations such as enzymatically dispersed cells [19, 21, 39] or CB thin slices [56, 59], which facilitated experiments using patch-clamp techniques as well as the recording of single cell parameters by microfluorimetry, amperometry, and other methodologies. Glomus cells are excitable, contain voltage-gated Na+, Ca2+, and K+ channels, and can therefore generate action potentials. They have both voltage-dependent [39, 42, 63, 80] and background [7, 14] membrane K+ channels, which are inhibited during exposure to hypoxia. Cationic channels activated by hypoxia have also been described in glomus cells, although their role in sensory transduction needs to be confirmed [34]. Modulation of O2-sensitive ion channels by hypoxia leads to membrane depolarization, Ca2+ influx through voltage-gated Ca2+ channels, and transmitter release (Fig. 1b, c) [8, 84]. Among the rich variety of neurotransmitters stored in glomus cell synaptic vesicles, ATP and acetylcholine are the best candidates for the activation of the afferent sensory fibers at the chemosensory synapse [22, 85, 90]. Dopamine, which is highly abundant in glomus cells, is believed to have an inhibitory autocrine/paracrine role [4] and some neuropeptides, such as endothelin-1 (ET1), appear to be involved in the regulation of CB plasticity during exposure to sustained hypoxia [10, 59]. Interestingly, ET-1 has also been involved in the chemosensory plasticity induced by chronic intermittent hypoxia (62, 65, 71). The “membrane model” of chemotransduction, based on depolarization by hypoxia and transmitter secretion dependent on extracellular Ca2+ influx, is now broadly accepted and also explains the effect of other chemostimulants, such as acidosis or hypercapnia, which induce cell depolarization. CB glomus cells have also been shown to be sensitive to hypoglycemia by a mechanism independent of O2-sensing, which involves activation of a background cationic inward current [24]. However, whether CB cells actually function as physiologically relevant glucose sensors that mediate counter-regulatory responses to hypoglycemia is still under debate [23].
Recently, human CB glomus cells have been shown to have similar electrophysiological and chemosensory properties to those in lower mammalian species [31, 54]. Interestingly, the number of TH+ cells in the human CB is much smaller than that in lower mammals, although this seems to be due to down-regulation of TH expression rather than an actual decrease in the number of glomus cells. Staining with antibodies against dopamine decarboxylase (DDC), another enzyme characteristic of glomus cells, demonstrates the abundance of glomeruli with DDC+ glomus cells in human CB tissue (Fig. 1d, e). The responsiveness of human CB cells to hypoxia, even at advance age, is similar to that reported in other species (Fig. 1f, g) [54]. These technical advances should facilitate investigations on the potential alterations of CB function in man and their contribution to human diseases.
Despite the advances in our understanding of CB cell physiology summarized above, the mechanism whereby glomus cells actually detect changes in O2 tension to signal membrane K+ channels is still unknown. This is probably a consequence of the small size of the CB, which prevents studies based on large-scale biochemical and molecular biology techniques. In addition, sensitivity to hypoxia is a labile process that is frequently altered by the vigorous enzymatic and mechanical treatment necessary to obtain dispersed glomus cells. The CB slice preparation can overcome some of these limitations, thereby facilitating the ability to conduct highly reproducible studies in animals and man [54, 59]. Although several appealing putative O2-sensing mechanisms have been postulated to function in the CB and other peripheral chemoreceptors [20, 38, 70], none of the existing proposals has gathered solid experimental support. In some cases, it has been reported that the responsiveness to hypoxia is unaffected in genetically modified animals lacking putative O2-sensor molecules (i.e., NADPH oxidase or hemeoxygenase 2) [27, 55]. In other cases, pharmacological analyses have cast doubts on the need for the proposed O2-sensing molecules or messengers, such as NO or H2S, in acute responsiveness to hypoxia. O2-sensitive prolyl hydroxylases are known to have a fundamental role in adaptation to chronic hypoxia. However, although this system might be necessary for the maintenance of CB homeostasis, it does not seem to directly participate in acute O2 sensing [56]. Nonetheless, prolyl hydroxylation can occur in some ion channel classes that are expressed in chemoreceptor cells [81]. Therefore, it is possible that the function, expression and/or membrane distribution of these channels are modulated by O2-dependent hydroxylation, thereby allowing them to participate in the adaptation to hypoxia.
A classical view is that mitochondria are involved in acute O2 sensing due to the exquisite sensitivity of CB cells to cyanide and other electron transport chain (ETC) inhibitors [9, 18, 53, 88]. The term “metabolic hypothesis”, coined to stress the role played by mitochondria in O2-sensing [49, 51], is not in contradiction with the membrane model of chemotransduction discussed above as mitochondria utilize O2 and therefore could detect changes in O2 availability and signal the membrane channels in response to hypoxia. Indeed, modulation by hypoxia of mitochondrial reactive oxygen species (ROS) production has been proposed to be an essential component of O2 sensing by cells, particularly in pulmonary arterial myocytes [26, 48, 86]. However, the potential mechanisms whereby these organelles sense O2 and signal membrane K+ channels are unknown. As mitochondrial parameters (e.g., mitochondrial membrane potential or NAD(P)H autofluorescence) in chemoreceptor cells are highly sensitive to changes in PO2, it was proposed that Ca2+ release from these organelles is the signal that triggers secretion during exposure to hypoxia [18]. Although this model lost support once it was established that the secretory response of chemoreceptor cells to a decrease in PO2 depends on extracellular Ca2+ influx [8, 84], it still remains possible that hypoxic mitochondria could provide a modulatory signal that affects ion channels and thus alters the membrane potential of glomus cells. In accord with this concept, activation of CB cells by ETC inhibitors depends on extracellular Ca2+ influx [53, 88]. A new perspective on the role of mitochondria as O2 sensors resulted from experiments showing that rotenone, an inhibitor of mitochondrial complex I (MCI), selectively occludes responsiveness to hypoxia in CB glomus [24, 53] and adrenomedullary (AM) chromaffin [82] cells without altering the response to hypoglycemia. These pharmacological data led us to postulate that a rotenone-binding molecule is involved in CB O2 sensing [53], although definitive experimental proof is still to be obtained.
Carotid body and acclimatization to hypoxia
The CB is thought to play an essential role in acclimatization to chronic hypoxia, a frequent condition in the human population that affects millions of people who live at or travel to high altitudes and are therefore exposed to low atmospheric air pressure and decreased O2 diffusion into the blood. Hypoxemia is also highly prevalent in medical disorders, such as chronic obstructive pulmonary disease (COPD), that cause a reduction of the O2 exchange capacity between the alveolar gas and the pulmonary capillaries [30, 79]. Adaptation to hypoxic environments depends on the hypoxic ventilatory response (HVR) [30, 69], a respiratory reflex that disappears in patients who have undergone surgical CB resections (most commonly due to tumors or asthma treatment) [83]. These individuals appear to live unaffected in normoxic environments, although disturbances during sleep and cases of sudden death have been attributed to a lack of functional chemoreceptors. In addition, it is known that the CB grows in individuals who are exposed to sustained hypoxemia for several days. This adaptive response increases the excitatory signals reaching the brain stem respiratory center to maintain hyperventilation [1, 30, 46].
Intolerance to hypoxia after disruption of the carotid body-adrenal medulla axis
Investigations of the role played by the CB in adaptation to hypoxia have proven difficult due to a lack of appropriate experimental models. The CB is generated during embryogenesis by the migration of sympathetic precursor cells from the superior cervical ganglion to the primordial carotid artery [28], and mutations of genes that prevent either carotid artery formation or sympathetic development are known to result in CB defects. However, most of the animals affected by these mutations die during embryonic life or shortly after birth and therefore cannot be studied in adulthood [13, 33]. Recently, we generated a new animal model (denoted as TH-VHLKO mice) in which ablation of the gene encoding the von Hippel-Lindau (Vhl) protein is restricted to TH+ catecholaminergic cells [43]. Vhl has a well-established role as a substrate recognition subunit of an ubiquitin ligase complex that targets HIFα for proteasomal degradation [44]. However, other functions of Vhl are not fully understood, and mutations of the Vhl gene are associated with the appearance of tumors. In normoxic conditions, our Vhl-deficient animals show full development of the brain and other organs and normal physiological functions although they exhibit hypotrophy of the CB, AM, and peripheral sympathetic system. TH-VHLKO mice do not have an HVR (Fig. 2a) and exhibit a striking intolerance to sustained hypoxia (Fig. 2b). Exposure of the animals to even mild hypoxia (14 % O2) causes strong erythropoietin induction and erythropoiesis, which within a few days is followed by splenomegaly, severe pulmonary hypertension, and right cardiac hypertrophy leading to death (Fig. 2c–e) [43]. These observations demonstrate the absolute necessity of peripheral chemoreceptor cells in the CB body and the AM for acclimatization and survival in hypoxic conditions. They also indicate that the TH-VHLKO mice provide an optimal model in which to study the consequences of functional inhibition of peripheral chemoreceptors throughout life. In this way, investigations of the early signs of hypoxia intolerance or the identification of biomarkers sensitive to maladaptation to hypoxia can be conducted. This could help to prevent hypoxia-associated morbidities affecting the brain or cardiorespiratory system, which are highly prevalent in susceptible individuals [74, 79, 87].

Responses to hypoxia in mice with atrophy of the carotid body/adrenal medulla axis. a Plethysmograph recordings showing changes in respiration rate during exposure to normoxia (Nx, 21 % O2) and hypoxic (Hx, 10 % O2) environments in control (VHLWT) and Vhl-deficient (TH-VHLKO) mice. b Kaplan–Meier survival curves comparing VHLWT and TH-VHLKO mice after exposure to chronic hypoxia (10 % O2). c Hematocrit and plasma erythropoietin (Epo) levels in animals maintained in chronic hypoxia (14 % O2). d Enlargement of the spleen (top) and heart (bottom) of TH-VHLKO mice after exposure to chronic hypoxia (10 % O2). e Hematoxylin/eosin staining of thin sections of the heart, lung, and spleen of VHLWT and TH-VHLKO mice maintained for 7 days in chronic hypoxia. Note the marked increase in size of the right ventricle, as well as pulmonary edema and changes in the histological architecture of the spleen. Scale bars: heart, 2 mm; spleen and lung, 0.2 mm. Modified from [39]
Carotid body stem cell niche
An intriguing property of the CB is its ability to grow in response to chronic hypoxia (Fig. 3a). Although this response was first described many years ago [1, 30, 46, 69], the underlying mechanisms have remained largely unstudied [10]. It has recently been shown that CB plasticity during sustained hypoxia depends on the existence in this organ of a population of multipotent adult neural crest-derived stem cells, which are the sustentacular or type II cells [60]. These cells are quiescent in normoxia but during exposure to hypoxia, lose their characteristic GFAP+ phenotype in parallel with the appearance of a progressive increase in the number of BrdU+ proliferating cells within the CB. Cell fate mapping experiments in vivo have shown that type II cells change their phenotype in response to hypoxia and are converted into proliferating (nestin+) progenitors, which in turn differentiate into new TH+ glomus cells [60]. Nestin + progenitors can also give rise to smooth muscle and endothelial cells [43, 60]. These changes in the CB stem cell niche can also be observed in vitro using neurosphere assays. CB neurospheres are cell colonies that normally derive from a single stem cell and are composed of a core of nestin + proliferating progenitors surrounded by differentiating TH+ cells budding out of the core. The newly generated TH+ cells form blebs attached to the neurosphere core, which grow in size with time in culture (Fig. 3b). Mature glomus cells derived from CB stem cells have similar functional properties to normal glomus cells in situ. They have voltage-gated ion channels, can release dopamine and other transmitters in response to hypoxia or hypoglycemia, and synthesize glial cell line-derived neurotrophic factor, which is highly expressed in adult CB cells [60]. In many aspects, differentiation of neuron-like glomus cells from GFAP+ stem cells in the CB resembles the organization of the central neurogenic centers located in the subventricular zone (SVZ) or the hippocampus. In both cases, there are neural stem cells with a quiescent astrocytic (GFAP+) phenotype that are converted to nestin + transient amplifying progenitors which in turn differentiate into neuroblasts giving rise to mature neurons [60]. Support for this model has also came from recent studies on TH-VHLKO mice, which have a strong reduction in the number of TH+ cells in the carotid bifurcation (Fig. 3c, top) but contain a large number of GFAP+ progenitor cells in apparently good condition (Fig. 3c, bottom). Indeed, neurosphere assays have confirmed the increased number of CB stem cells in these animals (Fig. 3d, e) and the loss of TH+ differentiated glomus cells (Fig. 3d, e). Newly generated TH+ cells in Vhl-deficient neurospheres appear to die as soon as TH-dependent Cre recombinase is induced and the Vhl gene is ablated (Fig. 3f) [43]. Multipotent neural stem cells have also been found in other parts of the peripheral nervous system, such as the enteric nervous system or the dorsal root ganglion. However, whether these cells participate in the maintenance of homeostasis of the organs in which they reside has not been established [29]. In addition to the growth of the CB parenchyma, sustained hypoxia also leads to an increase in CB glomus cell excitability due to, among other factors, an increase in Ca2+ current amplitude and a decrease in K+ channel expression [10, 68].

Carotid body (CB) growth in chronic hypoxia and CB stem cells. a Increase in size of CB parenchyma in mice exposed to 10 % O2 for 21 days. b Sequential photographs of a colony illustrating the formation of a typical CB neurosphere from a single CB stem cell (top) and time course of rat CB neurosphere formation in vitro (bottom). Appearance of blebs of (TH+) glomus cells is preceded by the formation of the neurosphere core containing nestin + progenitors. Scale bars, 50 μm. Modified from [54]. c Top. Marked decrease in the number of TH+ cells (superior cervical ganglion and carotid body) in the carotid bifurcation in TH-VHLKO mouse. c Bottom. Increase in the number of GFAP+ (green) type II cells in the carotid bifurcation in TH-VHLKO mice. The areas inside the rectangles are shown at a higher magnification in the insets. ca carotid artery. d, e Comparison of neurosphere composition and size (e), and number of neurospheres (f) in CB from control (VHLWT) and TH-VHLKO mice. f Differentiation of neuron-like glomus cells in neurospheres from control and TH-VHLKO mice. Note the appearance of pyknotic TH+ cells in TH-VHLKO preparations. Scales bars 100 μm (c, e), 2 μm (d), 10 μm (g). Modified from [39]
O2-sensitive chemoproliferative synapses
In contrast with the exquisite sensitivity of the CB to hypoxia, CB stem cell proliferation in vitro is virtually unaffected by lowering O2 tension. However, hypoxia strongly induces TH expression and promotes differentiation to glomus cells (Fig. 4a–d). These data are in accord with recent reports suggesting that hypoxia is a critical condition of the adult and embryonic stem cell niches. Quiescent somatic stem cells seem to maintain a predominantly anaerobic metabolism, which helps to preserve them from excessive production of ROS and other stressors [50, 78]. Resistance to hypoxia is a general property of neural stem cells as it has been found that the survival and proliferation of stem cells in the SVZ is not affected by a reduction in O2 tension from 21 to 1 % [12]. In line with these in vitro observations, CB growth is not induced by systemic administration of dimethyloxalylglycine (DMOG), an inhibitor of prolyl hydroxylases that mimics hypoxia and stabilizes HIFs in all tissues [5, 67]. Whereas mice treated with DMOG exhibit induction of classical HIF-dependent genes, such as erythropoietin and vascular endothelial growth factor, DMOG fails to induce CB cell proliferation (Fig. 4e–g). Taken together, these findings suggest that hypoxia does not directly activate the proliferation of CB stem cells. Indeed, morphological and functional data have indicated the existence of synaptic contacts between the O2-sensitive glomus cells and the GFAP+ stem cells, thus providing the structural basis for O2-sensitive chemoproliferative synapses which trigger CB growth in sustained hypoxia (Fig. 4h). Among the putative chemical transmitters in the glomus cell-stem cell synapse, we have identified ET1, a vasoactive and pro-proliferative agent involved in neural crest progenitor specification and migration [6, 77] that is synthesized in glomus cells [10, 47, 58, 67, 68]. ET1 receptors, in particular subtype B, are also highly expressed in type II cells [67].

Resistance of carotid body stem cells to hypoxia and O2-sensitive glomus cell-stem cell synapse. a CB neurospheres (NS) cultured under normoxia (21 % O2) or hypoxia (3 % O2). Differentiating blebs are colored in red in the insets. b Representative NS with a core of nestin + progenitors and blebs of TH+ cells. c NS core diameter in cultures grown at different levels of hypoxia. d Measurement of nestin + or TH+ staining area in normoxic and hypoxic NS sections. Note that the TH+ area clearly increased under hypoxia, whereas the nestin + area did not change. e–g Quantification of hematocrit (e), brain mRNA levels of vascular endothelial growth factor (VEGF) (f) and proliferating (Ki67+) cells in the carotid body (g) in rats maintained in normoxia (21 % O2), hypoxia (10 % O2) or in normoxia under DMOG treatment. Scale bars a 100 μm (50 μm in insets) b 50 μm. Modified from [59]
The picture that emerges from these studies is that the CB glomerulus is a highly sophisticated organoid-like structure with complex autocrine and paracrine interactions among its different cellular components. The O2-sensitive glomus cells are the central elements that, in response to hypoxia, mediate the activation of afferent sensory fibers (chemosensory synapse), thereby evoking an acute hyperventilatory response. In parallel, glomus cells trigger the proliferation of quiescent stem (type II) cells (“chemoproliferative synapses”) that initiates CB growth (see Fig. 4h). In addition, to these synapses, recent data have suggested even more elaborate interactions among cells within the CB glomerulus. Glomus cells are known to release ATP in response to hypoxia, which activates metabotropic receptors in type II cells [89]. In turn, activated type II cells appear to release ATP through large pannexin channels, increasing the excitatory response. All these responses seem to be further modulated by the interaction between the different chemical signals present in the CB and the afferent autonomic innervation [52].
Carotid body and human disease
Although the main biomedical interest in CB physiology derives from its role in the control of respiration, there is mounting evidence to indicate that the CB could be involved in the pathogenesis of several human diseases, some of which are highly prevalent. CB sensitivity to hypoxia develops during the early postnatal period and correlates with enhanced chemosensitivity in glomus cells. This process is important in the newborn given that, in addition to increasing ventilation and sympathetic tone, activation of the CB facilitates arousal from sleep and the switch from nasal to oral breathing. Defects in CB development or maturation have been associated with several neonatal respiratory deficiencies, such as sudden infant death or congenital central hypoventilation syndrome [40, 86]. CB denervation around the time of birth produces respiratory disturbances, exposing the newborn to respiratory instability and unexpected death. However, in several adult animal species, the hyperventilatory response to hypoxia, which is abolished by CB removal, is totally or partially re-established several months after the surgery (possibly due to the activation of other chemoreceptors). In man, CB resection due to tumor surgery or treatment for asthma results in complete and sustained lack of hypoxia responsiveness, particularly during sleep [83]. In recent years, alterations of CB function in adult life have been gaining increasing interest due to their participation in the pathogenesis of human pathologies, such as neural crest-derived tumors and the development of chronic neurogenic hypertension.
Carotid body tumorigenesis
Although relatively uncommon, CB tumors (chemodectomas or paragangliomas) are normally associated with medical syndromes presenting tumorigenesis in organs of the peripheral nervous system. These are mostly benign tumors that, besides the symptoms due to local compression, can also produce systemic hypertension [73]. Chemodectomas present with CB hypertrophy and cellular hyperplasia/anaplasia mainly affecting TH+ glomus cells although type-II cells are also affected. The grade of malignancy of CB paragangliomas appears to be inversely associated with the number of GFAP+ type-II cells, a fact that could indicate that deregulation of CB progenitors (with a type-II cell phenotype) participates in tumorigenesis. The most frequent cause of chemodectoma is hereditary CB paraganglioma due to loss of function mutations in SDHD, a gene that encodes the small membrane anchoring subunit of succinate dehydrogenase in mitochondrial complex II [3]. In affected individuals (heterozygous for mutated SDHD), the accidental loss of the normal allele (loss of heterozygosity, LOH) triggers tumor initiation. Indeed, tumorigenesis is favored in conditions that stimulate CB cell proliferation, such as living at high altitudes [2]. The pathogenesis of hereditary paragangliomas is unknown, although it has been proposed that the accumulation of succinate in the LOH-affected cells inhibits prolyl hydroxylases and stabilizes HIFs, thus causing a “pseudohypoxic status” leading to abnormal cell proliferation. Other molecular processes, such as oxidative stress or chromatin remodeling have also been proposed to play a causative role. Although bi-allelic ablation of SdhD causes embryonic lethality, heterozygous SdhD knockout mice are apparently normal and simply show a mild CB hypertrophy with normal sensitivity to hypoxia and no histological signs of tumorization [66]. Moreover, conditional knockout animals with SdhD ablation restricted to catecholaminergic (TH+) cells do not develop tumors but rather display a marked cell loss in the CB and other peripheral sympathetic organs [17]. It seems, therefore, that although SDHD is considered a “tumor suppressor gene” in humans, its role in tumor generation is not as direct as previously thought. It is possible that in addition to the SDHD mutation, additional “hits” are necessary to trigger tumor initiation. The possibility that these unknown hits are favored by chronic hypoxia (lasting months or years) is currently under study in heterozygous SdhD knockout mice.
Tumors of neural crest lineage, such as CB paragangliomas and AM pheochromocytomas, are also frequent in some forms of VHL disease, a hereditary syndrome caused by mutations in the VHL gene and characterized by the occurrence of tumors in multiple tissues [25, 32]. It has been suggested that VHL might participate in the molecular cascade leading to physiological apoptosis of sympathetic progenitor cells and that impairment of this protein could predispose to tumorigenesis in adulthood [36]. In contrast with this proposal, we have found that Vhl deletion in mice results in sympathoadrenal cell loss and impairment of survival of stem cell-derived catecholaminergic glomus cells [43] (see Fig. 3). The data indicate that Vhl is not only necessary for the survival of sympathoadrenal cells during development but is also required for the maintenance (full differentiation and survival) of these cells in adulthood. Interestingly, bi-allelic loss of VHL does not seem to produce pheochromocytomas or paragangliomas, and most of these tumors appear in patients with missense mutations of the VHL gene [36]. Hence, it is possible that pheochromocytomas/paragangliomas are generated by gain-of-function VHL mutations that, in the AM/CB setting, lead to abnormal cell proliferation [43]. It is therefore clear that Vhl does not act as a classical tumor suppressor gene in CB cells.
Carotid body over-activation
In addition to its critical role in adaptation to acute and chronic hypoxia, substantial evidence suggests that the CB can also have maladaptive effects. An exaggerated CB chemoreflex is known to contribute to the autonomic imbalance (tonic sympathetic over-activation) characteristic of several highly prevalent disorders, such as OSA, chronic heart failure (CHF), hypertension, obesity, metabolic syndrome, and diabetes [16, 61, 72, 75]. Recently, ablation of the CB was proposed for the treatment of refractory neurogenic hypertension [45]. The mechanisms underlying the changes in the CB-mediated chemoreflex in these pathologies seem to be intrinsic to the CB although their exact nature is as yet poorly known. In OSA, repeated episodes of intermittent hypoxia alter CB glomus cell excitability and sensitivity to lowered PO2 [11, 57, 64]. In addition, several groups have shown that the enhancement of CB activity in animals subjected to chronic intermittent hypoxia depends on oxidative stress-mediated alteration of ET-1 and angiotensin II signaling [15, 35, 62, 65, 71]. In CHF, a progressive sustained reduction of blood flow (even in the presence of a normal arterial PO2) seems to be the cause of CB activation, as this organ normally receives an extraordinarily high arterial blood supply. Among other factors, alterations of angiotensin II-dependent modulation of O2-sensitive membrane K+ channels in glomus cells or dysfunction of redox-mediated pathways have been proposed to have an important pathogenic role in CB over-activation in CHF models [75]. Insulin has been proposed to be an intermittent hypoxia-like factor that stimulates CB activity [37, 72]. Thus, increased CB-dependent sympathetic outflow is considered to play a key role in metabolic syndrome and in the development of hypertension, diabetes, and their associated morbidities.
Conclusions and perspectives
The CB is the principal arterial chemoreceptor that mediates the fast reflex cardiorespiratory response to hypoxia. Glomus cells, the O2-sensing elements in the CB, contain O2-sensitive K+ channels, which are inhibited by hypoxia, thereby triggering cell depolarization, Ca2+ influx, and activation of the chemosensory synapse connecting the CB with the brainstem respiratory center. Other O2-sensitive ion channel classes (e.g., non-selective cation channels) may also contribute to this response in particular during long hypoxic exposures. This membrane model of sensory transduction, generated from data obtained in lower mammalian species, has also been shown to be applicable to human CB cells. However, despite considerable research effort, the mechanisms whereby changes in O2 tension are detected by membrane channels are still unknown. Currently, there are several appealing proposals but, as yet, none is supported by sufficient experimental evidence. As the cellular processes underlying acute O2-sensing are fundamental to our understanding of hypoxia and O2 homeostasis, we expect that they will soon be elucidated. CB-dependent hyperventilation is necessary for acclimatization to sustained hypoxia, and CB hypertrophy is an adaptive response that is characteristic of high altitude residents or patients with diseases that compromise alveolar gas exchange. Recent work has proven that animals with atrophy of the CB-AM axis survive well in normoxia but show an absolute intolerance to even mild hypoxia, presenting exaggerated erythrocytosis, pulmonary edema, and right heart dilation leading to impending death. It will be important to investigate whether CB dysfunction can explain cases of respiratory depression (e.g., during anesthesia or opioid treatment) or disacclimatization to hypoxia in man. CB growth, a classic response to chronic hypoxia, is an intriguing property that is unusual for an organ of the peripheral nervous system. The mechanisms of CB plasticity are still not known in detail, but some of them are based on the activation of a resident population of adult neural crest-derived multipotent stem cells, which have a type II (glia-like) cell phenotype. Under hypoxia, CB stem cells are converted into nestin + progenitor cells, which in turn can give rise to glomus cells with normal chemosensitive properties as well as smooth muscle and endothelial cells. CB neural stem cells are the only ones in the adult peripheral nervous system that have a recognized physiological role. Interestingly, CB stem cells are insensitive to hypoxia, and their activation upon exposure to low PO2 seems to depend on the stimulation of glomus cells, which form abundant synaptic-like contacts with type II cells. Among the neurotransmitters involved in these O2-sensitive glomus cell-stem cell chemoproliferative synapses, ET1 appears to have a prominent role. Therefore, under hypoxia O2-sensitive glomus cells activate the chemosensory synapse to produce an acute hyperventilatory response and the chemoproliferative synapse to trigger stem cell proliferation and differentiation. The fact that these two synapses have different neurochemical properties makes pharmacological modulation of CB expansion in chronic hypoxia possible without affecting the acute chemosensory response. The chemical synapses existing between the glomus cell and the sensory fibers or the stem cells are examples of the complex reciprocal paracrine interactions that exist among the various elements of the CB glomerulus. Unraveling the functional topology within the CB glomeruli may help us to understand organizational principles in the peripheral and central nervous systems. Advances in CB cell physiology can now provide valuable information on the pathogenesis of diseases presenting hypo- or hyper-function of the CB chemoreceptors. CB tumors, although relatively rare, provide an excellent model for investigating the molecular basis of tumorigenesis in peripheral neural tissues. CB over-activation, secondary to chronic intermittent hypoxia, chronic cardiac failure, or diabetes, is an essential component of the exaggerated sympathetic outflow that occurs in these pathologies. Selective functional CB inhibition could therefore help to improve the prognosis in these diseases and associated comorbidities, while avoiding the need for surgical interventions.
References
Arias-Stella J, Valcarcel J (1976) Chief cell hyperplasia in the human carotid body at high altitudes; physiologic and pathologic significance. Hum Pathol 7:361–373
Astrom K, Cohen JE, Willett-Brozick JE, Aston CE, Baysal BE (2003) Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet 113:228–237
Baysal BE (2003) On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab 14:453–459
Benot AR, Lopez-Barneo J (1990) Feedback inhibition of Ca2+ currents by dopamine in glomus cells of the carotid body. Eur J Neurosci 2:809–812
Bishop T, Talbot NP, Turner PJ, Nicholls LG, Pascual A, Hodson EJ, Douglas G, Fielding JW, Smith TG, Demetriades M, Schofield CJ, Robbins PA, Pugh CW, Buckler KJ, Ratcliffe PJ (2013) Carotid body hyperplasia and enhanced ventilatory responses to hypoxia in mice with heterozygous deficiency of PHD2. J Physiol 591:3565–3577
Bonano M, Tribulo C, De Calisto J, Marchant L, Sanchez SS, Mayor R, Aybar MJ (2008) A new role for the endothelin-1/endothelin-A receptor signaling during early neural crest specification. Dev Biol 323:114–129
Buckler KJ (1997) A novel oxygen-sensitive potassium current in rat carotid body type I cells. J Physiol 498:649–662
Buckler KJ, Vaughan-Jones RD (1994) Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol 476:423–428
Buckler KJ, Turner PJ (2013) Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J Physiol 591:3549–3563
Chen J, He L, Dinger B, Stensaas L, Fidone S (2002) Role of endothelin and endothelin A-type receptor in adaptation of the carotid body to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 282:L1314–1323
Cutler MJ, Swift NM, Keller DM, Wasmund WL, Smith ML (2004) Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol 96:754–761
d’Anglemont de Tassigny X, Sirerol-Piquer MS, Gómez-Pinedo U, Pardal R, Bonilla S, Capilla-Gonzalez V, López-López I, De la Torre-Laviana FJ, García-Verdugo JM, López-Barneo J (2105) Resistance of subventricular neural stem cells to chronic hypoxemia despite structural disorganization of the germinal center and impairment of neuronal and oligodendrocyte survival. Hypoxia 3:15–33
Dauger S, Pattyn A, Lofaso F, Gaultier C, Goridis C, Gallego J, Brunet J-F (2003) Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development 130:6635–6642
Delpiano MA, Hescheler J (1989) Evidence for a PO2-sensitive K+ channel in the type-I cell of the rabbit carotid body. FEBS Lett 249:195–198
del Rio R, Moya EA, Iturriaga R (2010) Carotid body and cardiorespiratory alterations in intermittent hypoxia: the oxidative link. Eur Resp J 36:143–150
del Rio R, Moya EA, Parga MJ, Madrid C, Iturriaga R (2012) Carotid body inflammation and cardiorespiratory alterations in intermittent hypoxia. Eur Resp J 39:1492–1500
Diaz-Castro B, Pintado CO, Garcia-Flores P, Lopez-Barneo J, Piruat JI (2012) Differential impairment of catecholaminergic cell maturation and survival by genetic mitochondrial complex II dysfunction. Mol Cell Biol 32:3347–3357
Duchen MR, Biscoe TJ (1992) Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J Physiol 450:13–31
Duchen MR, Caddy KW, Kirby GC, Patterson DL, Ponte J, Biscoe TJ (1988) Biophysical studies of the cellular elements of the rabbit carotid body. Neuroscience 26:291–311
Evans AM, Hardie DG, Peers C, Wyatt CN, Viollet B, Kumar P, Dallas ML, Ross F, Ikematsu N, Jordan HL et al (2009) Ion channel regulation by AMPK: the route of hypoxia-response coupling in the carotid body and pulmonary artery. Ann N Y Acad Sci 1177:89–100
Fishman MC, Greene WL, Platika D (1985) Oxygen chemoreception by carotid body cells in culture. Proc Natl Acad Sci USA 82:1448–1450
Fitzgerald RS, Shirahata M, Ide T (1997) Further colinergic aspects of carotid body chemotransduction of hypoxia in rats. J Appl Physiol 82:819–827
Gao L, Ortega-Saenz P, Garcia-Fernandez M, Gonzalez-Rodriguez P, Caballero-Eraso C, Lopez-Barneo J (2014) Glucose sensing by carotid body glomus cells: potential implications in disease. Front Physiol 5:398
Garcia-Fernandez M, Ortega-Saenz P, Castellano A, Lopez-Barneo J (2007) Mechanisms of low-glucose sensitivity in carotid body glomus cells. Diabetes 56:2893–2900
Haase VH (2005) The VHL tumor suppressor in development and disease: functional studies in mice by conditional gene targeting. Semin Cell Dev Biol 16:564–574
Hamanaka RB, Chandel NS (2009) Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol 21:894–899
He L, Chen J, Dinger B, Sanders K, Sundar K, Hoidal J, Fidone S (2002) Characteristics of carotid body chemosensitivity in NADPH oxidase-deficient mice. Am J Physiol Cell Physiol 282:C27–C33
Hempleman SC, Warburton SJ (2013) Comparative embryology of the carotid body. Respir Physiol Neurobiol 185:3–8
Joseph NM, He S, Quintana E, Kim YG, Nunez G, Morrison SJ (2011) Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest 121:3398–3411
Joseph V, Pequignot J-M (2009) Breathing at high altitude. Cell Mol Life Sci 66:3565–3573
Kahlin J, Mkrtchian S, Ebberyd A, Hammarstedt-Nordenvall L, Nordlander B, Yoshitake T, Kehr J, Prabhakar N, Poellinger L, Fagerlund MJ, Eriksson LI (2014) The human carotid body releases acetylcholine, ATP and cytokines during hypoxia. Exp Physiol 99:1089–1098
Kaelin WG (2008) The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 8:865–873
Kameda Y, Ito M, Nishimaki T, Gotoh N (2008) FRS2 alpha 2F/2F mice lack carotid body and exhibit abnormalities of the superior cervical sympathetic ganglion and carotid sinus nerve. Dev Biol 314:236–247
Kang D, Wang J, Hogan JO, Vennekens R, Freichel M, White C, Kim D (2014) Increase in cytosolic Ca2+ produced by hypoxia and other depolarizing stimuli activates a non-selective cation channel in chemoreceptor cells of the rat carotid body. J Physiol 592:1975–1992
Lam SY, Liu Y, Ng KM, Liong EC, Yipoe GL, Leung PS, Fung ML (2014) Upregulation of a local renin-angiotensin system in the rat carotid body during chronic intermittent hypoxia. Exp Physiol 99:220–231
Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Schlisio S (2005) Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8:155–167
Limberg JK, Curry TB, Prabhakar NR, Joyner MJ (2014) Is insulin the new intermittent hypoxia? Med Hypotheses 82:730–735
Lopez-Barneo J (2003) Oxygen and glucose sensing by carotid body glomus cells. Curr Opin Neurobiol 13:493–499
Lopez-Barneo J, Lopez-Lopez JR, Urena J, Gonzalez C (1988) Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science 241:580–582
Lopez-Barneo J, Ortega-Saenz P, Pardal R, Pascual A, Piruat JI (2008) Carotid body oxygen sensing. Eur Respir J 32:1386–1398
Lopez-Barneo J, Pardal R, Ortega-Saenz P (2001) Cellular mechanism of oxygen sensing. Annu Rev Physiol 63:259–287
López-López J, González C, Ureña J, López-Barneo J (1989) Low pO2 selectively inhibits K channel activity in chemoreceptor cells of the mammalian carotid body. J Gen Physiol 93:1001–1015
Macias D, Fernandez-Aguera MC, Bonilla-Henao V, Lopez-Barneo J (2014) Deletion of the von Hippel-Lindau gene causes sympathoadrenal cell death and impairs chemoreceptor-mediated adaptation to hypoxia. EMBO Mol Med 6:1577–1592
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399:271–275
McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJ, Sobotka PA, Paton JF (2013) The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4:2395
McGregor KH, Gil J, Lahiri S (1984) A morphometric study of the carotid body in chronically hypoxic rats. J Appl Physiol 57:1430–1438
McQueen DS, Dashwood MR, Cobb VJ, Bond SM, Marr CG, Spyer KM (1995) Endothelins and rat carotid body: autoradiographic and functional pharmacological studies. J Auton Nerv Syst 53:115–125
Michelakis ED, Thebaud B, Weir EK, Archer SL (2004) Hypoxic pulmonary vasoconstriction: redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J Mol Cell Cardiol 37:1119–1136
Mills E, Jobsis FF (1972) Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J Neurophysiol 35:405–428
Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A (2010) Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7:150–161
Mulligan E, Lahiri S (1982) Separation of carotid body chemoreceptor responses to O2 and CO2 by oligomycin and antimycin A. Am J Physiol 242:C200–C206
Nurse C (2014) Synaptic and paracrine mechanisms at carotid body arterial chemoreceptors. J Physiol 592:3419–3426
Ortega-Saenz P, Pardal R, Garcia-Fernandez M, Lopez-Barneo J (2003) Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J Physiol 548:789–800
Ortega-Saenz P, Pardal R, Levitsky K, Villadiego J, Munoz-Manchado AB, Duran R, Bonilla-Henao V, Arias-Mayenco I, Sobrino V, Ordonez A, Oliver M, Toledo-Aral JJ, Lopez-Barneo J (2013) Cellular properties and chemosensory responses of the human carotid body. J Physiol 591:6157–6173
Ortega-Saenz P, Pascual A, Gomez-Diaz R, Lopez-Barneo J (2006) Acute oxygen sensing in heme oxygenase-2 null mice. J Gen Physiol 128:405–411
Ortega-Saenz P, Pascual A, Piruat JI, Lopez-Barneo J (2007) Mechanisms of O2-sensing by the carotid body: lessons from genetically modified animals. Respir Physiol Neurobiol 157:140–147
Ortiz FC, Del Rio R, Ebensperger G, Reyes VR, Alcayaga J, Varas R, Iturriaga R (2013) Inhibition of rat carotid body glomus cells TASK-like channels by acute hypoxia is enhanced by chronic intermittent hypoxia. Respir Physiol Neurobiol 185:600–607
Paciga M, Vollmer C, Nurse C (1999) Role of ET-1 in hypoxia-induced mitosis of cultured rat carotid body chemoreceptors. Neuroreport 10:3739–3744
Pardal R, Ludewig U, Garcia-Hirschfeld J, Lopez-Barneo J (2000) Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci USA 97:2361–2366
Pardal R, Ortega-Saenz P, Duran R, Lopez-Barneo J (2007) Glia-like stem cells sustain physiologic neurogenesis in the adult mammalian carotid body. Cell 131:364–377
Paton FR, Sobotka A, Fudim M, Engelman J, Hart CJ, McBryde D, Abdala P, Marina N, Gourine AV, Lobo M, Patel N, Burchell A, Ratcliffe L, Nightingale A (2013) The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 61:5–13
Pawar A, Nanduri J, Yuan G, Khan SA, Wang N, Kumar GK, Prabhakar NR (2009) Reactive oxygen species-dependent endothelin signaling is required for augmented hypoxic sensory response of the neonatal carotid body by intermittent hypoxia. Am J Physiol 296:R735–R742
Peers C (1990) Hypoxic suppression of K+ currents in type I carotid body cells: selective effect on the Ca2+-activated K+ current. Neurosci Lett 119:253–256
Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR (2003) Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A 100:10073–10078
Peng YJ, Nanduri J, Raghuramen G, Wang N, Kumar GK, Prabhakar NR (2013) Role of oxidative stress-induced endothelin-converting enzyme activity in the alteration of carotid body function by chronic intermittent hypoxia. Exp Physiol 98:1620–1630
Piruat JI, Pintado CO, Ortega-Saenz P, Roche M, Lopez-Barneo J (2004) The mitochondrial SDHD gene is required for early embryogenesis, and its partial deficiency results in persistent carotid body glomus cell activation with full responsiveness to hypoxia. Mol Cell Biol 24:10933–10940
Platero-Luengo A, Gonzalez-Granero S, Duran R, Diaz-Castro B, Piruat JI, Garcia-Verdugo JM, Pardal R, Lopez-Barneo J (2014) An O2-sensitive glomus cell-stem cell synapse induces carotid body growth in chronic hypoxia. Cell 156:291–303
Powell FL (2007) The influence of chronic hypoxia upon chemoreception. Respir Physiol Neurobiol 157:154–161
Powell F, Milsom W, Mitchell G (1998) Time domains of the hypoxic ventilatory response. Respir Physiol 112:123–134
Prabhakar NR, Peers C (2014) Gasotransmitter regulation of ion channels: a key step in O2 sensing by the carotid body. Physiology 29:49–57
Rey S, del Rio R, Iturriaga R (2006) Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res 1086:152–159
Ribeiro MJ, Sacramento JF, Gonzalez C, Guarino MP, Monteiro EC, Conde SV (2013) Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes 62:2905–2916
Rustin P, Munnich A, Rötig A (2002) Succinate dehydrogenase and human diseases: new insights into a well-known enzyme. Eur J Hum Genet 10:289–291
Schou L, Østergaard B, Rasmussen LS, Rydahl-Hansen S, Phanareth K (2012) Cognitive dysfunction in patients with chronic obstructive pulmonary disease—a systematic review. Respir Med 106:1071–1081
Schultz HD, Marcus NJ, Del Rio R (2013) Role of the carotid body in the pathophysiology of heart failure. Curr Hypertens Rep 15:356–362
Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148:399–408
Shin MK, Levorse JM, Ingram RS, Tilghman SM (1999) The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature 402:496–501
Suda T, Takubo K, Semenza GL (2011) Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9:298–310
Sutherland ER, Cherniack RM (2004) Management of chronic obstructive pulmonary disease. N Engl J Med 350:2689–2697
Stea A, Nurse CA (1991) Whole-cell and perforated-patch recordings from O2-sensitive rat carotid body cells grown in short- and long-term culture. Pflügers Arch 418:93–101
Takahashi N, Kuwaki T, Kiyonaka S, Numata T, Kozai D, Mizuno Y, Yamamoto S, Naito S, Knevels E, Carmeliet P, Oga T, Kaneko S, Suga S, Nokami T, Yoshida J, Mori Y (2011) TRPA underlies a sensing mechanism for O2. Nat Chem Biol 7:701–711
Thompson RJ, Buttigieg J, Zhang M, Nurse CA (2007) A rotenone-sensitive site and H2O2 are key components of hypoxia-sensing in neonatal rat adrenomedullary chromaffin cells. Neuroscience 145:130–141
Timmers HJLM, Wieling W, Karemaker JM, Lenders JWM (2003) Denervation of carotid baro- and chemoreceptors in humans. J Physiol 553:3–11
Urena J, Fernandez-Chacon R, Benot AR, Alvarez de Toledo GA, Lopez-Barneo J (1994) Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci USA 91:10208–10211
Varas R, Alcayaga J, Iturriaga R (2003) ACh and ATP mediate excitatory transmission in cat carotid identified chemoreceptor units in vitro. Brain res 988:154–163
Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL (2005) Acute oxygen-sensing mechanisms. N Engl J Med 353:2042–2055
Wilson MH, Newman S, Imray CH (2009) The cerebral effects of ascent to high altitudes. Lancet Neurol 8:175–191
Wyatt CN, Buckler KJ (2004) The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J Physiol 556:175–191
Xu J, Tse FW, Tse A (2003) ATP triggers intracellular Ca2+ release in type II cells of the rat carotid body. J Physiol 549:739–747
Zhang M, Zhong H, Vollmer C, Nurse CA (2000) Co-release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J Physiol 525:143–158
Acknowledgements
This work has been supported by grants from the Botín Foundation, the Spanish Ministries of Health and Science, and the European Research Council (European Union).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
López-Barneo, J., Macías, D., Platero-Luengo, A. et al. Carotid body oxygen sensing and adaptation to hypoxia. Pflugers Arch - Eur J Physiol 468, 59–70 (2016). https://doi.org/10.1007/s00424-015-1734-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-015-1734-0