
Abstract
Purpose
To investigate the expression profiles of Toll-like receptor 4 (TLR4), the effect of TLR4 on inflammation, and apoptosis of retinal ganglion cells (RGCs) cultured in high glucose and the underlying mechanism.
Methods
A high-glucose model was established in RGCs isolated from Sprague-Dawley (SD) rats (2–3 days old) and identified with Brn3a. Primary cultured RGCs were divided into control (0 mM), HG1 (10 mM glucose), HG2 (20 mM glucose), HG3 (30 mM glucose), HG (20 mM glucose) + TAK-242 (1.0 μM), and HG (20 mM glucose) + vehicle (1% DMSO) groups. The expression levels of TLR4, its downstream signalling molecules, and pro-inflammatory cytokines were measured by real-time PCR, Western blot or ELISA at 24 h and 48 h. The apoptosis rate of RGCs was measured by flow cytometry.
Results
The mRNA and protein expression levels of TLR4 were increased in high-glucose groups (10 mM, 20 mM, 30 mM). Consistent with these findings, four TLR4 downstream signalling molecules (MyD88, NF-κB, TRAF6, NLRP3) and pro-inflammatory cytokines (IL-1β, IL-18) were upregulated in the three high-glucose groups. Apoptosis of RGCs was clearly increased in the high-glucose group. The administration of TAK-242, an antagonist of TLR4, inhibited inflammation and apoptosis of RGCs in the high-glucose group.
Conclusion
Our results demonstrated that TLR4 plays a critical role in the inflammation and apoptosis of RGCs induced by high glucose. TLR4 might become a novel potential pharmacological target for preventing the progression of DR.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic retinopathy (DR) is one of the most common complications of diabetic mellitus (DM) [1,2,3]. DR is a major worldwide cause of sight-threatening disease among the working-age population [1, 4,5,6]. The mechanism of DR is complicated, and microvascular and neuron lesions are considered the most important factors [1, 7, 8]. It has been reported that retinal neuropathy is likely to occur in the early stages of DR; the damage of RGCs has been clinically demonstrable in DM without retinal microvascular abnormalities detectable on image analysis fluorescein angiography. This finding was further confirmed in human donor eyes and experimental animal models of diabetes [8,9,10,11,12]. Although the microvascular complications have been well documented, not much is known about the retinal neuropathies, in particular, the exact mechanism of RGC damage in DR [1, 12, 13].
Most recently, the role of inflammation and the immune system have become of greater interest in the understanding of DR [1, 14,15,16]. A number of metabolic and physiologic abnormalities characteristic of inflammation have been detected in the retinas of diabetic animals or patients [17, 18]. Toll-like receptors (TLRs) are essential components of innate immune responses; TLRs are a family of proteins primarily involved in recognizing pathogens and internal ligands [14, 19,20,21]. Among TLRs, TLR4 significantly participates in the induction of several immune-related diseases. The signalling transduction pathways activated by TLR4 are broadly divided into MyD88-dependent and MyD88-independent pathways. MyD88 is an immediate and common downstream adaptor molecule recruited by activated TLR4 [20]. It has been shown that the TLR4 signalling pathway affects the process of autoimmune myositis inflammation in mouse models with experimental autoimmune myositis [22, 23]. Previous studies have suggested that the TLR4 signalling pathway contributes to the development and progression of rheumatoid arthritis [24, 25]. Animals with TLR4 deficiency have been shown to higher protection against brain injuries, and the absence of TLR4 reduces neuroinflammation in brain trauma [26, 27]. Furthermore, studies in STZ (streptozotocin)-induced diabetic mouse models and in clinical patients have suggested that TLR4 signalling was activated and the inflammation in the retina was exaggerated [28, 29]. However, the effect of TLR4 on the inflammation and apoptosis of primary cultured RGCs induced by high glucose is unknown. Therefore, we hypothesised that TLR4 signalling would regulate the inflammation and apoptosis of RGCs induced by high glucose.
Materials and methods
Ethics statement
This study was carried out according to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. The protocol was approved by the ethics committee of Renmin Hospital of Wuhan University. Every effort was made to minimize animal discomfort and stress.
Animals and reagents
SPF SD rats (2–3 days old, either gender) were obtained from the Laboratory Animal Center of Wuhan University, quality certification: SCXK (Hubei Provence) 2014–0004.
DMDM/F12 culture medium and foetal bovine serum was purchased from Gibco (Gibco, Grand Island, NY, USA). 5-fluorouracil (5-Fu), uridine, and D-Glucose were obtained from Sigma-Aldrich (St. Louis, MO, USA). Papain was purchased from Worthington (Worthington Biochemical Corp, NJ, USA). Bovine, HEPES, left-handed L-glutamate (Lys), Penicillin (100 ku/L), streptomycin (100 mg/L), and dimethylsulphoxide (DMSO) were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). DAPI was purchased from Beyotime (Shanghai, China). Primary antibody against Brn3a was purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Inc., USA). Cy3-IgG donkey anti goat was purchased from Aspen Biotechnology (Aspen Biotechnology, Inc., USA). TKA-242 (MCE, USA) was prepared as a 10 mM stock solution in sterile 100% DMSO following the manufacturer’s recommended procedure, maintained at −4 °C, and diluted with 1% DMSO before use. A final concentration of 1% DMSO was used as vehicle for the treatment of the control cultures.
Cell culture
The primary cultures of rat RGCs were prepared according to the procedure as previously described with minor modifications [30]. Briefly, the six-well plates were coated with poly-L-lysine (100 μg/mL) and dried before use. Retinal nerve tissue was isolated from SD rats (2–3 days old) and neuronal cells isolated by incubation in papain (10.0 mg/mL) in sterile glass tubes at 37 °C for 20 min. The 10.0 mg/mL papain was deal with 0.22 μm pin type filter to keep it sterile and supplemented with bovine serum protein 0.5 mg/mL, left-handed cysteine (L-cys) 0.5 mg/mL, and HEPES 6.0 mg/mL in the solvent DMEM/F12. To yield a suspension of single cells, the tissues were titrated through a narrow-bore glass pipette in a DMEM/F12 solution containing 10% BSA. After centrifugation at 1000 rpm for 5 min, the cells were resuspended in another DMEM/F12 solution, and the cell density was adjusted to 15 × 106/mL. The cells were then seeded on poly-L-lysine-coated six-well plates incubated in DMEM/F12 containing 10% foetal bovine serum, penicillin G (100 U/mL), streptomycin sulphate (100 mg/mL), and KCl (final concentration are 15 mM). The viability of cells was assessed using a trypan blue exclusion assay before seeding in six-well plates. All procedures are performed under sterile conditions. Cells then were maintained at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h before 5-fluorouracil (5-Fu) and uridine (final concentrations of 20 μg/mL and 50 μg/mL, respectively) to inhibit the growth of non-neural cells. The culture medium was changed after 24 h, resulting in the establishment of a high-glucose model at that time. RGCs in these cultures were identified based on the cell morphology and the expression of cell markers.
Cell identification
For immunofluorescence staining, cells grown in six-well plates for 72 h were washed three times with PBS for 5 min each time and fixed with 4% buffered paraformaldehyde at 4 °C for 30 min. Then, the cells were washed again three times with PBS for 5 min each time and with the addition of permeabilization buffer at room temperature for 10 min. The cells were washed again with PBS 3 times (5 min each time) and incubated with a primary antibody against brain-specific homeobox/POU domain protein 3 A (Brn3a) [31] (1:50 dilution, anti-Goat, santa sc-31,984) overnight at 4 °C. After the incubation, the cells were washed with PBS three times (5 min each time) and incubated at room temperature for 1 h with secondary antibodies (Cy3-labelled donkey anti-goat IgG Aspen 1:50 dilution). The cells were again washed with PBS (5 min each time), fixed with DAPI dye 1 mL/well, incubation at room temperature for 5 min. Then, the cells were washed with PBS (5 min each time) and observed under a fluorescence microscope, and photographs of general and fluorescent images were taken of the same field. The double positive cells were considered as RGCs of SD rats.
Groups and treatment
After 24 h of purification with 5-Fu and uridine, the culture medium was changed, and the in vitro cultured RGCs were randomly divided into six groups as follows: control (0 mM), HG1 (10 mM), HG2 (20 mM), HG3 (30 mM), HG (20 mM) + TAK-242 (1.0 μM), and HG (20 mM) + vehicle (1% DMSO). Then, RGCs were cultivated in a 5% CO2 chamber at 37 °C for detection after 24 h and 48 h. RGCs with each treatment were cultured in triplicate.
Apoptosis analysis
The apoptosis of primary cultured RGCs induced by high glucose was measured by flow cytometry in a double labelling system. In short, cells grown on six-well plates for 48 h were washed two times with PBS for 30 s each time and digested with 0.25% trypsin 1 mL/well at room temperature for 5 min and blocked by culture medium. The cell suspension was collected and centrifuged twice at 300 g for 5 min, and finally re-suspended in 300 μL binding buffer with precooling at 4 °C. Then, the cell suspension was gently mixed with 10 μL of Annexin V-FITC solution and incubated for 10 min; then, 5 μL propidium iodide (1 μg/mL) (PI; Sigma, St. Louis, MO, USA) was added and incubated 5 min. Both incubations were at 37 °C in the dark. Then, the cells were analysed by fluorescence-activated cell sorting (FACS) within 1 h using flow cytometry (BD LSRFortessa™ Cell Analyser, USA).
Western blotting analysis
Western blotting was performed as described previously [32]. Total proteins were extracted from in vitro cultured RGCs using radio immunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China) including 1% protease inhibitor (Beyotime, Shanghai, China). The mixture was centrifuged at 12000 rpm at 4 °C for 10 min, and the supernatant was retained. The protein concentrations were determined with a BCA protein assay kit (Pierce). Each protein preparation (equal amounts 15–20 μg) in the supernatant were separated by sodium dodecyl sulphate polyacrylamide gel (SDS-PAGE) electrophoresis (10%). The proteins separated on the gel were then transferred to a polyvinylidene difluoride (PVDF) membrane (activated with methanol) using a semi-drying method. The membranes were washed with TBST before blocking with 5% non-fat milk in TBST (20 mM Tris, 150 mM NaCl, pH 7.5, and 0.1% Tween 20) for 1.5 h, non-specific binding sites were blocked by 5% non-fat milk powder. Then, the membranes were incubated with primary antibodies for TLR4 (1:500), nuclear factor-κB (NF-κB, 1:1000), myeloid differentiation factor 88 (MyD88, 1:1000), TNF receptor-associated factor 6 (TRAF6, 1:1500), nucleotide-binding oligomerization domain-like receptor with pyrin domain protein 3 (NLRP3, 1:500) and GAPDH (1:10,000) overnight at 4 °C. The membranes were then washed and incubated with secondary antibodies (HRP-goat anti-rabbit IgG, KPL074–1506 1:10,000) for 1 h at 37 °C. Excess antibody was washed off with TBST.
Enhanced chemiluminescence (ECL) chromogenic reagents were used to develop the membrane, which was then exposed to an X-ray. The bands were scanned to measure the densities using an automatic image analysis system (AlphaEaseFC software). The analysis was normalized against a housekeeping protein GAPDH. The measurements were repeated three times in each experiment. All studies were repeated three times (n = 3).
Real-time PCR analysis
Total RNA was extracted from RGCs with Trizol reagent (Invitrogen) according to the manufacturer protocols. Then, the total RNA was purified using gDNA Eraser (#RR047A Takara Japan). The RNA purity was verified by confirming that the OD260/OD280 ratio range from 1.8 to 2.0 with a nucleic acid protein analyser (Beckman BECHMAN DU 730). The total RNA (0.9 μg) was reverse transcribed using the Prime Script™ RT reagent Kit (#RR047A Takara Japan) to synthesize complementary DNA (cDNA). The target gene expression was quantified by real-time quantitative PCR using primers (summarized in Table 1) specific for SD rats. Rat TLR4, primer ID (NM 019178); rat MyD88, primer ID (NM 198130.1); rat p65, primer ID (NM 199267.2); rat NLRP3, primer ID (NM 001191642.1); rat TRAF6, primer ID (NM 001107754.2); rat IL-1β, primer ID (NM 031512.2); rat IL-18, primer ID (NM 019165) and GAPDH control primer (NM 017008.4) were used. Real-time PCR was performed with the following cycling parameters: 1 cycle of pre-denaturation at 95 °C for 30 s, 40 cycles of denaturation at 95 °C for 5 s, annealing at 60 °C for 40 s. The SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (#RR820A Takara, Japan) was used in all samples, and the reactions were carried out in a 20 μL final reaction volume using a 7500 Real-Time PCR System (Applied Biosystems, 7500 Fast). Fluorescence data were acquired at stage two in step two (60 °C for 40 s) to decrease the nonspecific signals, and the amplification of specific transcripts was confirmed by melting curve profiles at the end of each PCR. Measurements were masked to group assignment.
The relative amount of target mRNA was calculated from the obtained ΔCt values for the target and endogenous reference gene GAPDH, a housekeeping gene used as an endogenous control, using the 2-ΔΔCt cycle threshold method. The reactions were performed in triplicate in three separate experiments. The melting curve was also analysed after PCR amplification.
ELISA analysis
The levels of IL-1β and IL-18 in the cell culture supernatant were assessed using a standard mouse enzyme-linked immunosorbent assay (ELISA) kit (RLB00 RnD and KA0439 Novus, respectively) according to the manufacturer’s instructions. Briefly, after high glucose treatment, the serial diluted test samples (1:50–1:400) 100 μL/well were added to a 96-well plate in triplicate, along with equal volumes of working dilution standard samples. The samples were incubated with pre-coated capture antibody in the 96-well microplate at 37 °C. After the washing step, 100 μL of enzyme-linked reagents was added and incubated for 30 min at 37 °C. The colour of the samples was developed by incubating with the chromogenic substrate TMB 100 μL/well for 30 min under protecting from light. The reaction was stopped by the addition of the quenching buffer 50 μL/well. The optical density values of each well at 450 nm were obtained using a Microplate Reader (Winpact Scientific, Inc. USA). A standard curve was plotted to deduce the sample concentrations. All assays were performed in triplicate.
Statistical analysis
The quantitative data were expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. Statistical analysis was performed with GraphPad Prism software 6 (GraphPad Software, Inc. San Diego, CA, USA). Continuous variables of band intensity, optical density values and relative mRNA expression experiments between two groups were analysed with the unpaired Student’s t-test. One-way ANOVA followed by a Bonferroni correction were applied for multiple comparisons. A value of P < 0.05 was considered to indicate statistically significant differences.
Results
Retinal ganglion cell identification
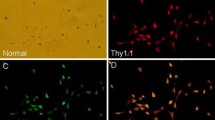
RGCs were isolated from SD rat retinas according to the protocol. The cells were cultured in six-well plates for 72 h and then double labelled with Brn3a and DAPI. The cells were observed under an inverted fluorescence electron microscope (10 × 20 × 0.55). The RGCs showed an uneven distribution and mutual confluence with obvious axons. Most stick wall cells were blue using the nonspecific nucleus dye DAPI. Brn3a is typically used as a phenotypic marker of RGCs. The red stick wall cells were specifically labelled with the immune fluorescent tag Brn3a. The same field of vision at low magnification showed most cells in red and blue double colour dye proving the cell culture extraction and purification for RGCs (Fig. 1 A-D). We also observed changes of morphology in RGCs under high glucose (Fig. 1 E-F).

A-D Imaging of the double-labelled RGCs by phase contrast microscopy at 200× magnification, (A) grey image, (B) blue image representing DAPI labelling, filter wavelength is 330–385 nm, (C) red fluorescence image representing Brn3a labelling, filter wavelength is 510–550 nm, (D) superimposed red and blue fluorescent images (Colour figure online). E-F Imaging of RGCs by phase contrast microscopy at 400 × magnification, (E) 20 mM for 24 h, (F) 20 mM for 48 h
TLR4 expression levels were increased in retinal ganglion cells under high glucose
The primary cultured RGCs were treated with three different high concentrations (10 mM, 20 mM, 30 mM) of glucose for 24 h and 48 h, then TLR4 protein and mRNA were measured by real-time PCR and Western blotting. Compared to the control group (0 mM glucose), the mRNA levels of TLR4 in the primary cultured RGCs were significantly increased in the three high-glucose groups for 24 h (all P < 0.05) and 48 h (all P < 0.05), respectively (Fig. 2 A and B).

Expression levels of TLR4 in RGCs under high glucose were detected by Western blotting and real-time PCR. TLR4 expression levels were increased in RGCs under high glucose (ranging from 0 mM to 30 mM) for 24 h (A) or 48 h (B). (C) TLR4 expression levels were increased in RGCs with the treatment of 20 mM glucose for 24 h and 48 h. The relative expression levels of mRNA and protein were normalized to GAPDH. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is one of the most commonly used housekeeping genes and serves as a common loading control for protein levels and a normalization control for gene expression analysis. Data are the mean ± SEM and are representative of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, # P < 0.05, # # P < 0.01
The results of Western blotting (Fig. 2 A and B) were consistent with those of real-time PCR; the primary RGCs cultured in the three high-glucose groups showed significantly elevated expression of TLR4 protein for 24 h and 48 h, respectively (P < 0.05). The level of TLR4 in 20 mM glucose was highest in the three high-glucose groups.
To investigate the effect of high-glucose incubation time on the TLR4 expression, the primary cultured RGCs were treated with 20 mM glucose at 0 h, 24 h and 48 h, then the expression levels of TLR4 protein and mRNA were detected. We found the levels of both TLR4 protein and mRNA were increased over time (0 h, 24 h, and 48 h) at 20 mM glucose (all P < 0.05). These results collectively suggested that TLR4 expression levels were increased in RGCs under high glucose, which was connected with the concentration of high glucose and the time of stimulation in some ranges.
The TLR4/NF-κB signalling pathway was activated in retinal ganglion cells under high glucose
Specific inflammatory cytokines are important biomarkers for reflecting the immune and inflammatory responses. The TLR4/NF-κB signalling pathway has been considered to be involved in an experimental mouse model of DR [29]. We also examined the expression of MyD88, NF-κB, TRAF6, and NLRP3 in RGCs under high glucose.
Our results showed that the expression levels of MyD88, NF-κB, TRAF6, and NLRP3 were significantly upregulated in RGCs under high glucose (20 mM glucose group, P < 0.05), according to the real-time PCR and Western blotting data (Fig. 3). These results suggested that the TLR4/NF-κB pathway-mediated immunomodulatory effect and neuroinflammatory response was involved in the damage of RGCs under high glucose.

Expression levels of NF-κB, MyD88, TRAF6 and NLRP3 in RGCs under high glucose were detected by Western blotting and real-time PCR. NF-κB (A), MyD88(B), TRAF6(C), and NLRP3(D) expression levels were upregulated in RGCs under high glucose. The relative expression levels of mRNA and protein were normalized to GAPDH, respectively. Data are the mean ± SEM and are representative of three independent experiments. **P < 0.01, ***P < 0.001
Pro-inflammatory cytokine expression levels were upregulated in retinal ganglion cells under high glucose
The expression levels of pro-inflammatory cytokines (IL-1β and IL-18) in the supernatant of primary cultured RGCs were investigated by ELISA and real-time PCR. The protein levels of IL-1β and IL-18 in the high-glucose group (368.13 ± 15.85 pg/mL, 220.80 ± 10.49 pg/mL, respectively) were significantly higher than those in the control group (94.24 ± 12.58 pg/mL, 70.61 ± 8.96 pg/mL). The data showed an obvious upregulation of IL-1β (P < 0.01) and IL-18 (P < 0.01) in RGCs under high glucose. In line with the results of ELISA, the real-time PCR results revealed that the mRNA levels of IL-1β and IL-18 increased in RGCs under high glucose (Fig. 4).

Expression levels of IL-1β and IL-18 in primary cultured RGCs were detected by ELISA and real-time PCR. IL-1β and IL-18 expression levels in primary cultured RGCs with the treatment of high glucose (20 mM) for 48 h. Both protein and mRNA levels of IL-1β (A) and IL-18 (B) were increased in RGCs under high glucose. *P < 0.05, **P < 0.01, ***P < 0.001
TAK-242 inhibited the inflammation of retinal ganglion cells under high glucose
Our earlier findings suggest that the activation of TLR4 signalling is involved in the inflammation of RGCs under high glucose. Therefore, we included a series of experiments with the pharmacological inhibitor TAK-242.
TAK-242, a selective inhibitor of TLR4, effectively decreased the expression levels of TLR4 downstream signalling molecules (MyD88, NF-κB, TRAF6, NLRP3) in primary cultured RGCs under a high-glucose environment for 48 h (P < 0.05). No significant difference (P > 0.05) was found between the 20 mM high-glucose group and the 20 mM glucose + vehicle (1% DMSO) group. These data showed that TAK-242 inhibited the expression levels of TLR4 downstream signalling molecules in primary cultured RGCs (P < 0.05) (Fig. 5).

The effect of TAK-242 on the expression levels of NF-κB, MyD88, TRAF6, NLRP3, IL-1β, IL-18 in RGCs under high glucose were detected by Western blotting, real-time PCR, and ELISA. NF-κB(A), MyD88(B), TRAF6(C), NLRP3(D), IL-1β (E), and IL-18 (F) expression levels were decreased in RGCs under high glucose (20 mM) + TAK-242(1.0 μM). *P < 0.05, **P < 0.01, ***P < 0.001
In line with these findings, the administration of TAK-242 led to a significant decrease in the mRNA levels of both IL-1β and IL-18. At the protein level, TAK-242 greatly suppressed the levels of IL-1β and IL-18 in the supernatant of primary cultured RGCs exposed to high glucose (Fig. 5).
TAK-242 inhibited the apoptosis of retinal ganglion cells under high glucose
The results of the FCM analysis showed a significantly increased apoptosis rate of RGCs under high glucose (P < 0.01). The administration of TAK-242 showed a significant attenuation of apoptosis in RGCs (P < 0.05) (Fig. 6). These results further indicated that the inhibition of TLR4 significantly alleviated the inflammation and apoptosis of RGCs induced by high glucose.

The apoptosis of RGCs under high glucose was detected by flow cytometry analysis. (A) The representative apoptosis rate of RGCs in control group. (B) The representative apoptosis rate of RGCs under high glucose (20 mM). (C) The representative apoptosis rate of RGCs under high glucose (20 mM) + TAK-242(1.0 μM). (D) TAK-242 inhibited the apoptosis of RGCs under high glucose. **P < 0.01, ***P < 0.001
Discussion
In the current study, we demonstrated that the expression levels of TLR4 and TLR4 downstream signalling molecules (MyD88, NF-κB, TRAF6, and NLRP3) were increased in primary cultured RGCs under high glucose. In addition, the levels of IL-1β and IL-18, two downstream pro-inflammatory cytokines of the TLR4 signalling pathway, were also significantly upregulated in primary cultured RGCs under high glucose. The apoptosis of RGCs under high glucose was clearly increased. Furthermore, the administration of TAK-242 attenuated the expression levels of TLR4 downstream signalling molecules and significantly reduced inflammatory cytokine levels in primary cultured RGCs, accompanied by a lower apoptosis rate of RGCs [33].
DR has long been defined as a sight-threatening retinal microvascular complication of diabetes mellitus [1, 3, 34]. It is becoming increasingly clear that diabetes induces a progressive dysfunction and degeneration of some neuronal cells in the inner retina, which contributes to vision deficits in the early stages of diabetic retinopathy [1, 11, 12, 35]. Neural retina abnormalities include neural apoptosis, reduction in the thickness of the inner retina, glial reactivity, slowing of optic nerve retrograde transport, and dysfunction in electrophysiological activity [11, 12, 36]. The available clinical evidence strongly suggests that the irreversible visual impairment in early DR is mostly a direct consequence of the dysfunction and/or loss of RGCs in the retina [10, 12, 31]. The loss of RGCs was also reflected by a reduction in the number of axons in the optic nerve [12]. These observations were complemented by mammal diabetic model studies, as a distinct reduction in the thickness of the retinal nerve fibre layer was revealed in an STZ-induced diabetic mouse model, accompanied by notable reduction in the number of RGCs [10, 11, 36]. Numerous reports using electroretinography have demonstrated that retinal neurodegeneration is compromised at the onset of DR [35]. Collectively, these data suggested that the neuroprotection, especially the protection of RGCs, should be emphasized as an approach to the treatment of early stage DR. However, few studies have reported the protective effect on the immune and inflammatory impairment of RGCs induced by high glucose in early stage DR. Therefore, we demonstrated that the expression levels of pro-inflammatory cytokines (IL-1β and IL-18) were significantly upregulated in primary cultured RGCs under high glucose. Furthermore, the apoptosis of RGCs under high glucose is increased.
DM is characterized by the dysregulation of innate immunity and is associated with an increased inflammatory response [28]. Recently, studies have shown that DR is a result of systemic neuroinflammation and is facilitated by innate immune responses [1, 14, 15, 17, 18, 35]. The immune system protects organisms against environmental and endogenous pathogens and regulates the homeostasis of inflammatory processes [14, 21]. The inflammation of the retina is a contributing factor in ocular diseases such as retinal microvascular abnormalities in DR, uveitis, glaucoma, and age-related macular degeneration [19, 32, 37, 38]. The Toll-like receptors (TLRs) are a class of highly preserved innate immunity receptors and function as pattern-recognition receptors. TLRs play an essential role in the initiation of innate immunity against invading microbial pathogens. TLRs have been shown to induce the transcriptional activation of genes encoding for pro-inflammatory cytokines, chemokines, and co-stimulatory molecules, which finally lead to a secondary cascade of inflammatory responses [14, 19,20,21].
TLR4, a cell surface TLR, is highly expressed on immune organs with pathogen surveillance activity and on non-immune organs such as the central nervous system, retina, or salivary glands [20, 21]. TLR4 has been considered a modulator of neuronal survival in many neurodegenerative diseases and neuronal injuries [26, 37,38,39,40]. It is responsible for sensing damage signals outside of the cell and intracellular non-infectious signals, which then initiate the innate immune response [20, 21]. The results of animal experiments showed that TLR4 mRNA is increased in the retina of DR rats [29, 41]. Furthermore, many studies have shown an upregulation of TLR4 accompanied by an increased inflammatory response in DR [28, 29]. Chronic neuroinflammation activated by the TLR4/NF-κB signalling pathway has been determined to be involved in the death of RGCs after optic nerve crush in an animal model of glaucoma [37]. Additionally, mice lacking TLR4 show reduced neuronal apoptosis and decreased pathology in the retina and brain [29, 42]. To date, very few studies have been performed to investigate the role of TLR4 in the inflammation and apoptosis of RGCs induced by high glucose. Zhao M et al. using human RGC-5 cells as an in vitro model of retinal ganglion cells, elucidated the expression profiles of TLR4 in RGC-5 in a high-glucose environment and its implications [43]. RGC-5 is a transformed cell line of mouse origin [44]. Research shows that the process of transformation invariably leads to dedifferentiation of the cells. Accordingly, transformed cell lines would be missing characteristic properties of their cells of origin, which is also true for RGC-5 [45, 46]. Primary RGCs cultured in vitro have been widely used in studies regarding retinal neuropathy diseases [30, 47, 48]. In the present study, we first isolated the RGCs from SD rats. Then, we investigated the expression profiles of TLR4 in RGCs primary cultured in high-glucose. Our data showed that the TLR4 expression levels were increased in RGCs under high glucose. The results were dependent on time and dose in a certain range. The level of TLR4 in 20 mM glucose was highest in the three high-glucose groups. This finding might indicate that TLR4 was involved in the immune inflammation of RGCs under high glucose. One interesting phenomenon is that the expression peak of TLR4 was in the 20 mM high-glucose group and was reduced in the 30 mM high-glucose group (Fig. 2). This finding might be related to the increasing damage of primary RGCs under 30 mM high glucose. This dose-dependent inconsistency displayed the complexity of activation and expression of TLR4 in primary RGCs under high glucose. Further studies are needed to explore the exact mechanisms.
The MyD88-dependent pathway, one of the signalling transduction pathways activated by TLR4, has been considered to be involved in the RGC damage in ischaemia reperfusion injury [49]. MyD88 is a common signalling molecule for most TLRs, leading to the downstream activation of NF-κB, a transcription factor, which is often used as a marker of TLR4 activity [21, 40]. TRAF6, a downstream adaptor, functions as a signal transducer in the NF-κB pathway that activates IkappaB kinase (IKK) in response to pro-inflammatory cytokines [50]. NLRP3 is a component of the inflammasome. The NLRP3 inflammasome is an upstream activator of NF-κB signalling and plays a role in the regulation of inflammation, the immune response, and apoptosis [51]. Our results demonstrated that the release of MyD88, NF-κB, TRAF6, and NLRP3 was upregulated in the inflammation of RGCs under high glucose. Meanwhile, the apoptosis of RGCs under high glucose was increased. These observations suggested that the TLR4 and TLR4/MyD88 signalling pathways played an essential role in the inflammation and apoptosis of primary RGCs under high glucose; we also revealed the potential therapeutic role of TAK-242 for the treatment of primary RGCs degeneration in DR. This is a meaningful supplement to the current body of knowledge of DR. Therefore, innovative strategies that target TLR4 against neuroinflammatory response cascades are regarded as promising therapeutic approaches for DR.
References
Cheung N, Mitchell P, Wong TY (2010) Diabetic retinopathy. Lancet 376:124–136. doi:10.1016/S0140-6736(09)62124-3
Group DER, Aiello LP, Sun W, Das A, Gangaputra S, Kiss S, Klein R, Cleary PA, Lachin JM, Nathan DM (2015) Intensive diabetes therapy and ocular surgery in type 1 diabetes. N Engl J Med 372:1722–1733. doi:10.1056/NEJMoa1409463
Leasher JL, Bourne RR, Flaxman SR, Jonas JB, Keeffe J, Naidoo K, Pesudovs K, Price H, White RA, Wong TY, Resnikoff S, Taylor HR, Vision Loss Expert Group of the Global Burden of Disease S (2016) Global estimates on the number of people blind or visually impaired by diabetic retinopathy: a meta-analysis from 1990 to 2010. Diabetes Care 39:1643–1649. doi:10.2337/dc15-2171
Ghassibi MP, Chien JL, Abumasmah RK, Liebmann JM, Ritch R, Park SC (2016) Optic nerve head Drusen prevalence and associated factors in clinically normal subjects measured using optical coherence tomography. Ophthalmology DOI. doi:10.1016/j.ophtha.2016.10.035
Grassi MA, Sun W, Gangaputra S, Cleary PA, Hubbard L, Lachin JM, Gao X, Kiss S, Barkmeier AJ, Almony A, Davis M, Klein R, Danis RP, Group DER (2013) Validity of self-report in type 1 diabetic subjects for laser treatment of retinopathy. Ophthalmology 120:2580–2586. doi:10.1016/j.ophtha.2013.06.002
Polack S, Yorston D, Lopez-Ramos A, Lepe-Orta S, Baia RM, Alves L, Grau-Alvidrez C, Gomez-Bastar P, Kuper H (2012) Rapid assessment of avoidable blindness and diabetic retinopathy in Chiapas, Mexico. Ophthalmology 119:1033–1040. doi:10.1016/j.ophtha.2011.11.002
Simo R, Hernandez C, European Consortium for the Early Treatment of Diabetic R (2014) Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab 25:23–33. doi:10.1016/j.tem.2013.09.005
Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB (2008) Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes 57:889–898. doi:10.2337/db07-1669
Rao RC, Dlouhy BJ (2012) Diabetic retinopathy. N Engl J Med 367:184. doi:10.1056/NEJMc1205011#SA2
Sohn EH, van Dijk HW, Jiao C, Kok PH, Jeong W, Demirkaya N, Garmager A, Wit F, Kucukevcilioglu M, van Velthoven ME, DeVries JH, Mullins RF, Kuehn MH, Schlingemann RO, Sonka M, Verbraak FD, Abramoff MD (2016) Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc Natl Acad Sci U S A 113:E2655–E2664. doi:10.1073/pnas.1522014113
Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW (1998) Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 102:783–791. doi:10.1172/JCI2425
Carrasco E, Hernandez C, Miralles A, Huguet P, Farres J, Simo R (2007) Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care 30:2902–2908. doi:10.2337/dc07-0332
Cheung N, Donaghue KC, Liew G, Rogers SL, Wang JJ, Lim SW, Jenkins AJ, Hsu W, Li Lee M, Wong TY (2009) Quantitative assessment of early diabetic retinopathy using fractal analysis. Diabetes Care 32:106–110. doi:10.2337/dc08-1233
Xu H, Chen M, Forrester JV (2009) Para-inflammation in the aging retina. Prog Retin Eye Res 28:348–368. doi:10.1016/j.preteyeres.2009.06.001
Adamis AP, Berman AJ (2008) Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol 30:65–84. doi:10.1007/s00281-008-0111-x
Powell ED, Field RA (1964) Diabetic retinopathy and rheumatoid arthritis. Lancet 2:17–18
Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS (2007) Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes 56:337–345. doi:10.2337/db06-0789
Adamis AP (2002) Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol 86:363–365
Tang J, Kern TS (2011) Inflammation in diabetic retinopathy. Prog Retin Eye Res 30:343–358. doi:10.1016/j.preteyeres.2011.05.002
Miyake K (2007) Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Semin Immunol 19:3–10. doi:10.1016/j.smim.2006.12.002
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680. doi:10.1038/90609
Reynolds JM, Martinez GJ, Chung Y, Dong C (2012) Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci U S A 109:13064–13069. doi:10.1073/pnas.1120585109
Racke MK, Hu W, Lovett-Racke AE (2005) PTX cruiser: driving autoimmunity via TLR4. Trends Immunol 26:289–291. doi:10.1016/j.it.2005.03.012
Babazada H, Yamashita F, Hashida M (2014) Suppression of experimental arthritis with self-assembling glycol-split heparin nanoparticles via inhibition of TLR4-NF-kappaB signaling. J Control Release 194:295–300. doi:10.1016/j.jconrel.2014.09.015
Sheedy FJ, Marinou I, O'Neill LA, Wilson AG (2008) The mal/TIRAP S180L and TLR4 G299D polymorphisms are not associated with susceptibility to, or severity of, rheumatoid arthritis. Ann Rheum Dis 67:1328–1331. doi:10.1136/ard.2007.083337
Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A (2012) Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med 18:911–917. doi:10.1038/nm.2749
Hakimizadeh E, Kazemi Arababadi M, Shamsizadeh A, Roohbakhsh A, Allahtavakoli M (2016) The possible role of toll-like receptor 4 in the pathology of stroke. Neuroimmunomodulation 23:131–136. doi:10.1159/000446481
Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I (2008) Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab 93:578–583. doi:10.1210/jc.2007-2185
Berger EA, Carion TW, Jiang Y, Liu L, Chahine A, Walker RJ, Steinle JJ (2016) Beta-adrenergic receptor agonist, compound 49b, inhibits TLR4 signaling pathway in diabetic retina. Immunol Cell Biol 94:656–661. doi:10.1038/icb.2016.21
Tezel G, Yang X (2004) Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci 45:4049–4059. doi:10.1167/iovs.04-0490
Mead B, Tomarev S (2016) Evaluating retinal ganglion cell loss and dysfunction. Exp Eye Res 151:96–106. doi:10.1016/j.exer.2016.08.006
Yang H, Zheng S, Qiu Y, Yang Y, Wang C, Yang P, Li Q, Lei B (2014) Activation of liver X receptor alleviates ocular inflammation in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci 55:2795–2804. doi:10.1167/iovs.13-13323
Xiao J, Fu C, Zhang X, Zhu D, Chen W, Lu Y, Ye Z (2015) Soluble monosodium urate, but not its crystal, induces toll like receptor 4-dependent immune activation in renal mesangial cells. Mol Immunol 66:310–318. doi:10.1016/j.molimm.2015.03.250
Murakami T, Nishijima K, Sakamoto A, Ota M, Horii T, Yoshimura N (2011) Foveal cystoid spaces are associated with enlarged foveal avascular zone and microaneurysms in diabetic macular edema. Ophthalmology 118:359–367. doi:10.1016/j.ophtha.2010.03.035
Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, KF LN, Norbury CC, Quinn PG, Sandirasegarane L, Simpson IA, Group JDRC (2006) Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 55:2401–2411. doi:10.2337/db05-1635
Kern TS, Barber AJ (2008) Retinal ganglion cells in diabetes. J Physiol 586:4401–4408. doi:10.1113/jphysiol.2008.156695
Xu Y, Yang B, Hu Y, Lu L, Lu X, Wang J, Xu F, Yu S, Huang J, Liang X (2016) Wogonin prevents TLR4-NF-kappaB-medicated neuro-inflammation and improves retinal ganglion cells survival in retina after optic nerve crush. Oncotarget DOI 10.18632/oncotarget.12700
Szatmari-Toth M, Kristof E, Vereb Z, Akhtar S, Facsko A, Fesus L, Kauppinen A, Kaarniranta K, Petrovski G (2016) Clearance of autophagy-associated dying retinal pigment epithelial cells - a possible source for inflammation in age-related macular degeneration. Cell Death Dis 7:e2367. doi:10.1038/cddis.2016.133
Zhao Y, Zhao Y, Zhang M, Zhao J, Ma X, Huang T, Pang H, Li J, Song J (2016) Inhibition of TLR4 Signalling-induced inflammation attenuates secondary injury after diffuse axonal injury in rats. Mediat Inflamm 2016:4706915. doi:10.1155/2016/4706915
Qi Y, Zhao M, Bai Y, Huang L, Yu W, Bian Z, Zhao M, Li X (2014) Retinal ischemia/reperfusion injury is mediated by toll-like receptor 4 activation of NLRP3 inflammasomes. Invest Ophthalmol Vis Sci 55:5466–5475. doi:10.1167/iovs.14-14380
Wang YL, Wang K, Yu SJ, Li Q, Li N, Lin PY, Li MM, Guo JY (2015) Association of the TLR4 signaling pathway in the retina of streptozotocin-induced diabetic rats. Graefes Arch Clin Exp Ophthalmol 253:389–398. doi:10.1007/s00417-014-2832-y
Badshah H, Ali T, Kim MO (2016) Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/NFkappaB signaling pathway. Sci Rep 6:24493. doi:10.1038/srep24493
Zhao M, Li CH, Liu YL (2016) Toll-like receptor (TLR)-2/4 expression in retinal ganglion cells in a high-glucose environment and its implications. Genet Mol Res 15. doi:10.4238/gmr.15026998
Van Bergen NJ, Wood JP, Chidlow G, Trounce IA, Casson RJ, Ju WK, Weinreb RN, Crowston JG (2009) Recharacterization of the RGC-5 retinal ganglion cell line. Invest Ophthalmol Vis Sci 50:4267–4272. doi:10.1167/iovs.09-3484
Krishnamoorthy RR, Clark AF, Daudt D, Vishwanatha JK, Yorio T (2013) A forensic path to RGC-5 cell line identification: lessons learned. Invest Ophthalmol Vis Sci 54:5712–5719. doi:10.1167/iovs.13-12085
Kritis AA, Stamoula EG, Paniskaki KA, Vavilis TD (2015) Researching glutamate - induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci 9:91. doi:10.3389/fncel.2015.00091
Li CP, Wang SH, Wang WQ, Song SG, Liu XM (2017) Long noncoding RNA-Sox2OT knockdown alleviates diabetes mellitus-induced retinal ganglion cell (RGC) injury. Cell Mol Neurobiol 37:361–369. doi:10.1007/s10571-016-0380-1
Tsutsumi T, Iwao K, Hayashi H, Kirihara T, Kawaji T, Inoue T, Hino S, Nakao M, Tanihara H (2016) Potential Neuroprotective effects of an LSD1 inhibitor in retinal ganglion cells via p38 MAPK activity. Invest Ophthalmol Vis Sci 57:6461–6473. doi:10.1167/iovs.16-19494
Chen S, Sorrentino R, Shimada K, Bulut Y, Doherty TM, Crother TR, Arditi M (2008) Chlamydia pneumoniae-induced foam cell formation requires MyD88-dependent and -independent signaling and is reciprocally modulated by liver X receptor activation. J Immunol 181:7186–7193
Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361
Kim Y, Wang W, Okla M, Kang I, Moreau R, Chung S (2016) Suppression of NLRP3 inflammasome by gamma-tocotrienol ameliorates type 2 diabetes. J Lipid Res 57:66–76. doi:10.1194/jlr.M062828
Author information
Authors and Affiliations
Contributions
Hongxia Yang and Lili Hu conceived and designed the project. Lili Hu performed most of the experiments and wrote the manuscript. Lili Hu and Shuanghong Jiang analysed the data. Ming Ai and Hongxia Yang reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Funding information
The funding organizations had no role in the design or conduct of this research.
Animal experiments
All procedures performed in the studies were in accordance with the ethical standards of the institution and national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
Hu, L., Yang, H., Ai, M. et al. Inhibition of TLR4 alleviates the inflammation and apoptosis of retinal ganglion cells in high glucose. Graefes Arch Clin Exp Ophthalmol 255, 2199–2210 (2017). https://doi.org/10.1007/s00417-017-3772-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-017-3772-0