
Abstract
Purpose
The purpose of our study was to investigate the effects of pleiotrophin (PTN) in proliferative vitreoretinopathy (PVR) both in vitro and in vivo.
Methods
Immunofluorescence was used to observe the PTN expression in periretinal membrane samples from patients with PVR and controls. ARPE-19 cells were exposed to TGF-β1. The epithelial-to-mesenchymal transition (EMT) of the ARPE-19 cells was confirmed by observed morphological changes and the increased expression of α-SMA and fibronectin at both the mRNA and protein levels. We used specific small interfering (si)RNA to knock down the expression of PTN. The subsequent effects of PTN inhibition were assessed with regard to the EMT, migration, proliferation, cytoskeletal arrangement, TGF-β signaling, PTN signaling, integral tight junction protein expression (e.g., claudin-1 and occludin), and p38 MAPK and p-p38 MAPK levels. Additionally, a PVR rat model was established by the intravitreal injection of ARPE-19 cells transfected with PTN-siRNA and was evaluated accordingly.
Results
PTN was highly expressed in PVR membranes compared to controls. PTN knockdown attenuated the TGF-β1-induced migration, proliferation, cytoskeletal rearrangement, and expression of EMT markers such as α-SMA and fibronectin in the ARPE-19 cells, and these effects may have been mediated through p38 MAPK signaling pathway activation. PTN silencing inhibited the up-regulation of claudin-1 and occludin stimulated by TGF-β1, and PTN knockdown inhibited the proliferative aspects of severe PVR in vivo.
Conclusions
PTN is involved in the process of EMT induced by TGF-β1 in human ARPE-19 cells in vitro, and PTN knockdown attenuated the progression of experimental PVR in vivo. These findings provide new insights into the pathogenesis of PVR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proliferative vitreoretinopathy (PVR) is a serious complication of retinal detachment (RD), and is the primary cause of RD surgical failure [1]. PVR is characterized by the migration and proliferation of cells following a retinal break or trauma, leading to the formation of membranes in the periretinal area, followed by the contraction of these cell membranes and traction on the retina, which causes RD. Vitreous surgery is currently the standard treatment; however, recurrent vitreoretinal traction can lead to re-detachment, extensive membrane reformation, and significant vision loss [1]. The pathogenesis of PVR involves several cell types, including retinal pigment epithelial (RPE) cells, fibroblasts, glial cells, and inflammatory cells [2]. The RPE layer represents a critical group of cells that participate in the progression of the proliferation in PVR [3]. The vitreous humor is rich in cytokines and growth factors, and in the process of PVR, RPE cells are detached from Bruch’s membrane and migrate through the retinal tear into the vitreous humor [4, 5]. RPE cells are then transformed from fully differentiated epithelial cells into mesenchymal cells in a process called epithelial-to-mesenchymal transition (EMT). Via the EMT, RPE cells can become contractile myofibroblasts, and these cells may contribute to the formation of fibrotic membranes around the retina [6]. Under stimulation of growth factors/cytokines in the vitreous humor, these membranes contract, leading to tractional RD [6]. Therefore, identifying potential therapeutic targets for the treatment of PVR and understanding the mechanisms of the EMT in RPE cells is crucial [7].
Transforming growth factor β (TGF-β) is known as a potent fibrogenesis factor. TGF-β is over-expressed in the vitreous of patients with proliferative vitreoretinal diseases [8, 9]. TGF-β has been suggested to play an important role in the EMT process in RPE cells, and studies have reported that the development of PVR membranes was accompanied by a pronounced up-regulation of TGF-β1 rather than TGF-β2 [10].
Pleiotrophin (PTN), an 18-kDa secreted growth factor, is also called heparin-binding growth-associated molecule (HB-GAM) or heparin affin regulatory peptide. It is activated by the binding of three cell surface receptors: syndecan-3, anaplastic lymphoma kinase (ALK), and receptor protein tyrosine phosphatase (RPTPβ/ζ) [11]. PTN is a multifunctional cytokine that is involved in cell growth, transformation, carcinogenesis, and metastasis; in addition, it is up-regulated in many pathological processes [12]. The oncogenic function of PTN has been identified in a variety of human tumors. PTN is not only enhanced in angiogenesis and tumor cell proliferation, thereby promoting tumor formation, but it also affects the invasion and metastasis of malignant cells into peripheral nerves by remodeling of the microenvironment [13]. In addition, PTN is involved in inflammation and fibrosis in various conditions and diseases [13, 14], including fibrous tissues [15], hypertrophic scars [16], and rheumatoid arthritis [17]. Studies have shown that PTN plays a role as a hepatocyte growth factor and contributes to liver cirrhosis and hepatocarcinogenesis [18]. Additionally, in a previous study, we showed that PTN plays an important role in proliferative diabetic retinopathy [13]. Therefore, we questioned whether PTN contributes to the progression of fibrotic diseases, such as to the EMT in PVR, and whether PTN interacts with TGF-β1 in the process of EMT in RPE cells. In the present study, we found that the expression of PTN was increased in the proliferative membranes obtained from patients with PVR, and the knockdown of PTN inhibited TGF-β1-induced EMT, a pathogenic marker of PVR. We also demonstrated that PTN knockdown inhibited the development of PVR in vivo.
Methods
Subjects and sample collection
The study protocol with regard to human patients was approved by the Ethics Committee and Institutional Review Board of Peking University (Beijing, China), and written informed consent was obtained from each study subject in accordance with the Declaration of Helsinki. All subjects underwent a standard eye examination by a retinal specialist (Dr. Mingwei Zhao). Patients diagnosed with PVR in the ophthalmology department of Peking University People’s Hospital (Beijing, China) were retrospectively reviewed from August 2014 to September 2015. PVR membranes were surgically removed from six patients with RD and PVR via pars plana vitrectomy. The controls consisted of idiopathic epiretinal macular membranes (EMMs) from six eyes. These epiretinal membranes were surgically removed via pars plana vitrectomy and frozen at −80°C. After membrane tissues were fixed in optimal cutting temperature medium (Merck, Darmstadt, Germany), 8-μm sections were prepared. The retinal specialist, Dr. Mingwei Zhao, performed all the surgeries at Peking University People’s Hospital.
Immunofluorescence staining of membranes
The membranes were dried and fixed in 4% paraformaldehyde. After blocking the samples with 5% bovine serum albumin for 1 h, 1:80 anti-PTN polyclonal antibody (Catalog No. ab95391; Abcam, Cambridge, MA, USA) and 1:100 anti-pan-cytokeratin (Chemicon, CA, USA) were applied to the tissue sections at 4°C overnight and incubated with 1:100 goat anti-rabbit fluorescein isothiocyanate (FITC)-conjugated and goat anti-mouse tetramethylrhodamine isothiocyanate (TRITC) secondary antibody (ZSGB-BIO, Beijing, China) for 1 h. The cell nuclei were then stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000 dilution, D9542; Sigma-Aldrich, St. Louis, MO, USA). Images were acquired using a fluorescence microscope equipped with a digital camera (Axiophot, Zeiss, Thornwood, NY, USA). In all immunostaining procedures, negative controls were prepared by omitting the primary antibody and applying an irrelevant polyclonal or isotype-matched monoclonal primary antibody; in all cases, the negative controls showed weak, negligible staining of PTN.
Cell culture and transfection assays
The human ARPE-19 cells (American Type Culture Collection, Manassas, VA, USA) used in this study have been previously described [16]. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO/Invitrogen, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, GIBCO/Invitrogen). Small interfering RNA (siRNA) specific to PTN (PTN-siRNA) was used in this study (Catalog No. SC-39713; Santa Cruz Biotechnology, Dallas, TX, USA).
RPE cells were transfected with siRNA using a transfection reagent according to the manufacturer’s instructions (Qiagen GmbH, Hilden, Germany). On the day of transfection, cells were plated at an appropriate density. The siRNA and transfection reagent were mixed to a final concentration of 10 nM, which was then applied to the cells. Non-silencing RNA (NS-siRNA) (Catalog No. 301799, HiPerFect; Qiagen GmbH) was used as a control. The cells were starved for 6 h before treatment with TGF-β1 (10 ng/ml; R&D Systems, Minneapolis, MN, USA). Lipofectamine 2000 (Invitrogen/Life Technologies, Carlsbad, CA, USA) was used to transfect RPE cells with siRNA. The cells were then collected for the following experiments.
Cell proliferation assay
We used a Cell Counting Kit-8 (CCK-8, Dojindo, Tokyo, Japan) assay to assess cell proliferation. ARPE-19 cells transfected with PTN-siRNA were incubated with 10 ng/ml TGF-β1 for 24, 48, or 72 h. As previously described [19], the number of cells was measured using a CCK-8 assay; the absorbance at 450 and 600 nm was measured using a plate reader (Finstruments Multiskan Model no. 347; MTX Lab Systems, Inc., Vienna, VA, USA). Each experiment included at least five replicates and was carried out three times.
Immunofluorescence microscopy and phalloidin staining
After the siRNA transfection and TGF-β1 treatment, the cells, which had been seeded on coverslips, were fixed with 4% paraformaldehyde (PFA) and then treated with 0.1% Triton X-100 for permeabilization. For the phalloidin staining, the cells were incubated for 1 h with rhodamine-labeled phalloidin diluted 1:100. After DAPI staining, the slides were visualized using a fluorescence microscope (Zeiss Axiophot).
Migration assay
Transwell systems (Catalog No. 3422, Corning Life Sciences, Tewksbury, MA, USA) were used to study RPE cell migration. After transfection with PTN-siRNA, ARPE-19 cells were cultured with TGF-β1 for 48 h. The cells were then collected, and 2 × 104 cells in 200 μl of serum-free medium were seeded on the upper chamber of the Transwell. DMEM containing 10% FBS was added to the lower chamber at a final volume of 600 μl. The cells were allowed to migrate for 6 h at 37°C and were then fixed and stained with DAPI (Roche Diagnostics, Indianapolis, IN, USA). Non-migrating RPE cells were gently removed from the front surface of the membrane, and the migrated cells were visualized by fluorescence imaging. The number of cells in five random fields of view were counted. Every experiment was performed in triplicate.
RNA isolation and real-time PCR
Total RNA was isolated from ARPE-19 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Reverse transcriptase reactions were performed using a RevertAid First Strand cDNA Synthesis Kit with oligo-dT primers (Fermentas, Pittsburgh, PA, USA). The mixture was incubated for 60 min at 42°C, and the reaction was terminated by incubation at 95°C for 5 min [20]. Real-time polymerase chain reaction (RT-PCR) was performed using an ABI7300 real-time PCR system (Applied Biosystems/Life Technologies, Foster City, CA, USA), SYBR Green PCR mix (Thermo Scientific, Pittsburgh, PA, USA), and sequence-specific primers (Table 1). Data were normalized to GAPDH, a housekeeping gene. The results are expressed as fold amplification, and each experiment was carried out five times.
Western blot analysis
The RPE cells were first treated as described above. The cells were then lysed on ice for 20 min in RIPA buffer (Beyotime Institute of Biotechnology) supplemented with protease inhibitors (Pierce/Thermo Scientific, Rockford, IL, USA), and centrifuged at 12,000 g and 4 °C to clear the lysates. The supernatant of each lysate was then collected, and the protein content was measured using a BCA protein assay kit (Pierce/Thermo Scientific) according to the manufacturer’s instructions. Equal amounts of protein (40 ng) were loaded on a 10% sodium dodecyl sulfate polyacrylamide gel and analyzed by immunoblotting. The primary antibodies used are listed in Table 2. The membrane was washed and then incubated with a secondary horseradish peroxidase (HRP)-conjugated antibody (goat anti-rabbit IgG, 1:4000; ZSGB-BIO, Beijing, China). The proteins were visualized using enhanced chemiluminescence Western blot detection reagents (Pierce/Thermo Scientific). The density of the protein bands was measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and normalized to that of GAPDH. All immunoblot analyses were performed three times and showed qualitatively similar results.
Immunocytochemistry assay
RPE cells were seeded in a 24-well plate with inlaid glass coverslips. After the siRNA transfection and TGF-β treatment, the cells were washed, fixed with 4% PFA, permeabilized with 0.1% Triton X-100, and then thoroughly blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. The slides were incubated at 4°C overnight with anti-PTN antibody (1:80, Catalog No. ab95391; Abcam, Cambridge, MA, USA) and then with secondary anti-rabbit FITC-conjugated antibody for 1 h. After DAPI staining, the slides were mounted with coverslips and visualized by fluorescence microscopy (Zeiss Axiophot).
Rat PVR model
Eight rats, each weighing 200–250 g, were used for the in vivo studies. All experiments adhered to the Association for Research in Vision and Ophthalmology statements for the use of animals in ophthalmology and vision research. All procedures were approved by the Animal Care and Use Committee of Peking University. The animals were housed with free access to food and water and kept in the laboratory on a 12-h light–dark cycle.
RPE cells were used for intravitreal injection (Ivit). However, before the Ivit, the rats were anesthetized with chloral hydrate, and their pupils were dilated with tropicamide (Santen, Osaka, Japan). Their eyes were topically anesthetized with 0.4% oxybuprocaine hydrochloride eye drops (Eisai Co., Ltd., Tokyo, Japan) to reduce the level of discomfort. For the RPE cell injection within the vitreous, a 29-gauge needle on a microsyringe (Hamilton Company, Franklin, MA, USA) was loaded with 5 μl of an RPE cell suspension with PBS and 10 ng/ml TGF-β1. PVR rats received Ivit in the right eye; at 1 mm posterior to the limbus, the needle was inserted through the sclera and the cells injected into the vitreous.
Ocular images were captured for analysis on day 14. PVR classification in our study was performed as in previous studies, as follows: stage 0: no lesions; stage 1: vitreous membrane; stage 2: local vascular changes, focal traction, and hyperemia; stage 3: local detachment of medullary rays; stage 4: extensive peripapillary RD; stage 5: total RD, retinal folds and macular holes [3, 21].
Statistical analysis
Prism 6 (GraphPad, San Diego, CA, USA) and SPSS (version 16.0, SPSS Inc, Chicago, IL, USA) statistical software packages were used for data analysis. All data are presented as the mean ± standard deviation, and the normality of the distributions was assessed. Differences were evaluated with analysis of variance (ANOVA). P values less than 0.05 were considered significant.
Results
PTN was intensely expressed in the proliferative membranes of PVR patients
PTN expression was detected by immunofluorescence in the proliferation membranes of six PVR and six EMM patients. Immunofluorescence revealed that PTN coexisted with cytokeratin, a marker of RPE cells. In addition, it was positively and strongly expressed in the proliferative membranes of all patients with PVR and was only weakly expressed in the EMMs (Fig. 1).

Immunofluorescence staining of the pre-retinal membranes. Images of epiretinal PVR membrane sections (left column) and EMM sections (right column) stained for PTN (green) (a, b). Cytokeratin (red) (c, d) was co-localized with PTN. Nuclei were stained with DAPI (e, f), and the merged images were presented (g, h). PTN was strongly expressed in the PVR membranes. PTN: green; nuclei: blue. PVR proliferative vitreoretinopathy, EMM epiretinal macular membrane, PTN pleiotrophin; DAPI: 4′,6-diamidino-2-phenylindole. Scale bars, 100 μm
PTN knockdown attenuated TGF-β1-induced EMT and cytoskeletal rearrangement in ARPE-19 cells
To assess the potential involvement of PTN in the process of TGF-β1-induced EMT in ARPE-19 cells, we first cultured RPE cells with TGF-β1, as previously described [22–24]. After treatment with TGF-β1 for 48 h, ARPE-19 cells underwent EMT, and showed increased expression of mesenchymal markers, including fibronectin and α-smooth muscle actin (α-SMA) (Fig. 2f). We then examined the expression of PTN in APRE-19 cells after stimulation with TGF-β1, and found that PTN expression gradually increased in the TGF-β-treated cells (Figs. 2a and 3a). We next examined PTN signaling after TGF-β1 stimulation by determining the expression levels of the ALK [25] and β-catenin signaling proteins, which are known to be involved in PTN signaling, using Western blot and RT-PCR (Fig. 2b and c). The observed up-regulation of PTN, ALK and β-catenin indicates that PTN possibly contributes to the EMT in RPE cells (Fig. 2).

Western blot and RT-PCR results of PTN signaling and epithelial and mesenchymal marker expression of ARPE-19 cells. After the knockdown of PTN (a), the relative mRNA and protein expression levels of ALK and β-catenin (b, c) were down-regulated. The elevation of the epithelial and mesenchymal markers α-SMA and fibronectin were inhibited after PTN knockdown in RPE cells (e, f). The relative protein expression levels were quantified via normalization to GAPDH expression (d, g). RT-PCR and Western blot analysis was carried out at least three times, and qualitatively similar results were found. Data are presented as the mean ± SEM; ** P < 0.01

Immunocytochemical assays for PTN and phalloidin staining in RPE cells. RPE cells were transfected with PTN-siRNA or negative control, non-silencing siRNA (NS-siRNA), and were treated with or without TGF-β1 (10 ng/ml) for 48 h. The expression of PTN was detected by immunofluorescence (a). The organization of the actin cytoskeleton was examined using fluorescein-conjugated phalloidin to stain F-actin (b). PTN knockdown abrogated TGF-β1-induced cytoskeletal rearrangement in RPE cells. Scale bars, 20 μm
PTN knockdown was used to explore its role in EMT induced by TGF-β1 in RPE cells. RT-PCR and immunofluorescence were used to examine the effectiveness of RPE cell transfection with PTN-siRNA. RT-PCR showed that compared to the NS-siRNA, the PTN-siRNA reduced the mRNA levels of PTN in RPE cells (P < 0.01; Fig. 2a). The immunocytochemistry results also confirmed the knockdown of PTN. We then examined the expression of EMT-related genes, including fibronectin and α-SMA, by RT-PCR and Western blot. PTN knockdown was found to significantly attenuate the TGF-β1-induced up-regulation of α-SMA and fibronectin (Fig. 2e and f). In addition, we investigated the effect of TGF-β1 on cytoskeletal structures. Actin cytoskeletal organization was examined via the fluorescein-conjugated phalloidin staining of F-actin. Fibers in the negative control cells were randomly oriented in the cytoplasm (Fig. 3b), and the TGF-β1-treated cells had much thicker stress fibers that were largely parallel to the longitudinal axis of the cell (Fig. 3b). Furthermore, PTN knockdown reversed the TGF-β1-induced cytoskeletal arrangements in RPE cells (Fig. 3b). These results indicate that in RPE cells, PTN participates in the EMT induced by TGF-β1.
PTN down-regulation inhibited RPE cell migration and proliferation
A CCK-8 assay was used to assess the effects of PTN on cell proliferation in vitro, as described in the “Methods” section. The results showed that the knockdown of PTN significantly inhibited the promoted ARPE-19 cell proliferation stimulated by TGF-β1 (Fig. 4c). In addition, the Transwell chamber assay showed that the knockdown of PTN significantly hampered RPE cell migration (Fig. 4a and b). Compared with the control group, the mean number of cells that migrated through the membrane in the PTN knockdown group was significantly lower.

Effects of PTN on RPE proliferation and migration. Nuclei were stained with DAPI and appeared as a blue dot. Five random field cells were counted, and the average was used for the statistical analysis. TGF-β1 stimulated the migration of RPE cells. After the knockdown of PTN, the migration of RPE cells treated by TGF-β1 was inhibited (a, b). Cell proliferation was measured with a CCK-8 assay at 0, 24, 48, and 72 h. PTN knockdown inhibited the RPE proliferation stimulated by TGF-β1 (c). The data are shown as the mean ± SD; *P < 0.05; **P < 0.01
Phosphorylated p38 MAPK levels were down-regulated by PTN knockdown
Next, we examined the impact of PTN modulation on TGF-β signaling. The phosphorylation of p38 [7] is known to be involved in TGF-β signaling. In our study, PTN knockdown inhibited the phosphorylation of p38 with TGF-β1 treatment compared to TGF-β1 treatment without PTN knockdown. These results show that PTN mediated the progression of the EMT induced by TGF-β1 in RPE cells, possibly through activation of the p38 signal transduction pathway (Fig. 5a).

Phosphorylated p38 MAPK, occludin, and claudin-1 were down-regulated by PTN knockdown. The level of p-p38 MAPK was down-regulated by PTN knockdown (a, b). The relative mRNA and protein expression levels of occludin and claudin-1 were down-regulated after PTN knockdown (c, d, e). The relative protein expression levels were quantified via normalization to GAPDH expression (b, e). RT-PCR and Western blot analysis were performed at least three times, and yielded qualitatively similar results. Data are presented as the mean ± SEM; *P < 0.05, **P < 0.01
Occludin and claudin-1 were down-regulated by PTN knockdown
We then examined the role of TGF-β1 in the expression of the integral tight junction proteins claudin-1 and occludin. In our study, PTN knockdown significantly attenuated the TGF-β1-induced up-regulation of mRNA and protein of claudin-1 and occludin (Fig. 5c–e).
PTN inhibited membrane formation in an experimental PVR rat model
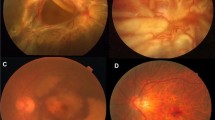
The fundus examination findings were normal in the control group (Fig. 6). Our in vitro data suggest that PTN plays an important role in the proliferation and EMT of cultured ARPE-19 cells. Therefore, we employed an experimental rat model of PVR to further explore the role of PTN. On day 14, three rats (100%) in the TGF-β1 treatment group developed stage 2 PVR, and two rats (66.7%) in the negative control group and the PTN-siRNA treatment group displayed stage 2 PVR, whereas the pre-retinal membranes were less extensive in the TGF-β1-treated PTN-siRNA group than in the TGF-β1-treated group (Fig. 6).

Effects of PTN on retinal morphological changes in experimental PVR. Color fundus images of BN rats after 14 days. Images of a healthy vitreous body in vivo (a), an eye injected with RPE cells (b), an eye injected with TGF-β1-treated RPE cells (c), and an eye injected with TGF-β1-treated RPE cells after PTN knockdown (d). The arrows point to the proliferative membranes. Injection of TGF-β1-treated RPE cells after PTN knockdown led to reduced PVR
Discussion
There are four main findings in the present study. First, we observed that PTN was strongly expressed in the proliferative membranes of PVR patients. Second, we found that the knockdown of PTN inhibited TGF-β1-induced EMT, attenuated TGF-β1-mediated cell activity, reduced cytoskeletal rearrangement, and impaired RPE integral tight junctions. Third, PTN knockdown down-regulated p38 MAPK phosphorylation in RPE cells treated with TGF-β1. Finally, we demonstrated that PTN knockdown inhibited the development of PVR in vivo.
The ocular fibrotic disease PVR is a process of fibrocellular proliferation in the vitreous cavity that can form contractile epiretinal membranes on both sides of the retina. PVR is still the leading cause of rhegmatogenous RD surgery failure, and the anatomical and functional results are still unsatisfactory [26]. The EMT plays a vital role in the formation of the body plan and the differentiation of many tissues and organs. This process is re-engaged in adult systems during wound healing, fibrosis, and tumor progression [27, 28]. Recent studies show that the EMT in RPE cells is a major contributor to the pathogenesis of PVR [1, 2, 6], and PTN has been reported to stimulate the EMT in glioblastoma cells as a secreted growth factor [29]. However, the basic mechanisms of the EMT in RPE cells in PVR are poorly understood. To our knowledge, this study shows for the first time that PTN plays a crucial role in the process of the EMT induced by TGF-β1 in human RPE cells and that PTN expression is activated during PVR progression.
PTN has been widely studied in several fibrotic, angiogenic, and other diseases. PTN expression in most normal tissues is low or non-existent, but it has been found to be expressed during development, after ischemic brain injury, during wound healing in skin and bone models, and during tumor cell growth [30–33]. Perez-Pinera et al. reported that PTN induced the EMT in glioblastoma cells [29]. Previous results from our research group have also demonstrated that PTN plays an important role in proliferative diabetic retinopathy [13]. In the present study, we intended to address the role of PTN in PVR. As shown in Fig. 1, PTN was highly expressed in the PVR membranes, but was not expressed in the EMMs. The difference between PVR and EMM is the presence of cell migration and the progression of the EMT. PTN has a variety of functions, including roles in proliferation, mitogenic activity, apoptosis, and oncogenic and angiogenic activity. PTN expression was found to be up-regulated in a mouse model of peritoneal fibrosis, and it was shown to trigger inflammation and increased permeability of the peritoneum, resulting in human peritoneal fibrosis [17]. Additionally, PTN expression has been observed in the epiretinal membranes of patients with proliferative diabetic retinopathy [13]. The intense expression of PTN in PVR membranes is an indicator and the basic rationale for this research.
As demonstrated in several studies, TGF-β1 is the most established mediator of the EMT in RPE cells [26, 34]. Previous studies have reported that TGF-β1 was detected in vitreous samples, subretinal fluid, and epiretinal membranes surgically obtained from patients diagnosed with PVR [35–37]. PTN stimulates the EMT and disrupts cell–cell adhesion in human glioblastoma cells [32]. The up-regulation of α-SMA and fibronectin is the main marker of the EMT [26]. However, we did not know whether TGF-β1 correlated with the high expression of PTN in PVR membranes that we detected. Thus, in the present study, we used TGF-β1 to stimulate the EMT in RPE cells, and we aimed to analyze the relationship of TGF-β1, PTN, and the EMT with the proliferative effects of PVR. As expected, we found that the EMT was induced in RPE cells with increasing expression of the mesenchymal markers α-SMA and fibronectin after treatment with TGF-β1 (Fig. 2), which was consistent with the findings of previous studies [26, 38]. Additionally, we verified that PTN expression was increased in RPE cells after stimulation by TGF-β1 (Fig. 2), indicating that PTN may play a vital role in the TGF-β1-induced EMT in RPE cells, which is a sign of the development of PVR. Therefore, we then knocked down the expression of PTN using siRNA to further explore the role of PTN in RPE cells and a PVR animal model. SiRNA can be used to inhibit virtually any target gene and is profoundly useful in therapeutics [39]. Several RNAi-based therapeutics have been or are currently being investigated in human clinical trials [39]. We found that the knockdown of PTN achieved in RPE cells, i.e., 99%, was sufficient (Fig. 2a). The application of PTN-siRNA significantly inhibited the EMT induced by TGF-β1, including the observed cytoskeletal rearrangement (Fig. 3b) and the up-regulation of α-SMA and fibronectin (Fig. 2f). These findings indicate that PTN could be downstream of TGF-β1. In a previous study, Weng et al. reported that TGF-β1 influenced the expression of PTN in the progression of fetal rat lung development [40], which supports our results. Cytoskeletal changes are important in proliferative membrane formation. Lee et al. have also proposed that TGF-β1 induces cytoskeletal rearrangement in human RPE cells in the pathogenesis of PVR [41].
The uncontrolled proliferation and migration of RPE cells and cytoskeletal rearrangement are characteristics of the EMT in the process of pathological epiretinal membrane formation [2, 42]. Recent studies have shown that PTN-regulated proliferation and migration is essential in various cancer cells [43]. Our research demonstrated that PTN knockdown significantly inhibited the TGF-β1-induced proliferation and migration of ARPE-19 cells (Fig. 4). We also found that PTN knockdown attenuated TGF-β1-induced cytoskeletal rearrangement in RPE cells (Fig. 3b). We then detected the expression of the cell–cell junction factors claudin-1 and occludin (Fig. 5c–e). Previous studies have shown that claudin-1 and occludin are influential in the EMT process and that claudin-1 induces the EMT [38, 43]. Our study also demonstrated that PTN knockdown led to considerable suppression of cytoskeletal rearrangement and up-regulation of claudin-1 and occludin stimulated by TGF-β1. Claudin-1 and occludin are tight functions, and it has recently become clear that tight junction factors may act as cell signaling molecules, affecting the proliferation, movement, and invasion of cells [44, 45].
Although TGF-β is central to the signaling network of the EMT, the most recent data show that integration and crosstalk with TGF-β signaling can occur with the MAPK and PI3K/Akt signaling pathways, promoting the EMT [7, 46]. The MAPK and the PI3K/Akt pathways have essential roles in various cell activities, including proliferation, migration, inflammation, and death [47]. Here, we investigated the role of PTN in p38 MAPK signaling. As shown in Fig. 5a, a significant reduction was observed in the phosphorylation of p38 in cells treated with PTN-siRNA, suggesting that p38 may be a downstream cellular signaling protein of PTN that can mediate cell functions. In addition, previously published studies have shown that p38 influences the expression of PTN [48]. Thus, PTN may act through the p38 MAPK pathway to mediate the TGF-β1-induced EMT in RPE cells.
To date, more than 25 PVR models have been designed [41]. These models facilitate investigations into the pathogenesis of diseases and the identification of new therapeutic targets [41, 49]. In this study, we demonstrated that PVR formation stimulated by TGF-β1 could be inhibited by the knockdown of PTN in vivo (Fig. 6). Our results show that the PTN-siRNA-treated group exhibited reduced membrane formation. Furthermore, our in vivo data correlated with our in vitro data, which further confirm the involvement of PTN in PVR.
In conclusion, our findings suggest that PTN is a key mediator of the EMT in RPE cells, as well as RPE cell migration and proliferation. PTN might play an important role in the pathogenesis of PVR. These results indicate that PTN could serve as a novel target for therapeutic interventions in PVR.
References
Sadaka A, Giuliari GP (2012) Proliferative vitreoretinopathy: current and emerging treatments. Clin Ophthalmol 6:1325–1333. doi:10.2147/OPTH.S27896
Pennock S, Haddock LJ, Eliott D, Mukai S, Kazlauskas A (2014) Is neutralizing vitreal growth factors a viable strategy to prevent proliferative vitreoretinopathy? Prog Retin Eye Res 40:16–34. doi:10.1016/j.preteyeres.2013.12.006
Machemer R, van Horn D, Aaberg TM (1978) Pigment epithelial proliferation in human retinal detachment with massive periretinal proliferation. Am J Ophthalmol 85:181–191. doi:10.1016/S0002-9394(14)75946-X
Lei H, Rheaume M-A, Kazlauskas A (2010) Recent developments in our understanding of how platelet-derived growth factor (PDGF) and its receptors contribute to proliferative vitreoretinopathy. Exp Eye Res 90:376–381. doi:10.1016/j.exer.2009.11.003
Tosi GM, Marigliani D, Romeo N, Toti P (2014) Disease pathways in proliferative vitreoretinopathy: an ongoing challenge. J Cell Physiol 229:1577–1583. doi:10.1002/jcp.24606
Lee H, O’Meara SJ, O’Brien C, Kane R (2007) The role of gremlin, a BMP antagonist, and epithelial-to-mesenchymal transition in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci 48:4291–4299. doi:10.1167/iovs.07-0086
Yang S, Li H, Li M, Wang F (2015) Mechanisms of epithelial-mesenchymal transition in proliferative vitreoretinopathy. Discov Med 20:207–217
Kita T, Hata Y, Arita R, Kawahara S, Miura M, Nakao S, Ishibashi T (2008) Role of TGF-beta in proliferative vitreoretinal diseases and ROCK as a therapeutic target. Proc Natl Acad Sci U S A 105:17504–17509. doi:10.1073/pnas.0804054105
Kita T, Hata Y, Kano K, Miura M, Nakao S, Noda Y, Ishibashi T (2007) Transforming growth factor-beta2 and connective tissue growth factor in proliferative vitreoretinal diseases: possible involvement of hyalocytes and therapeutic potential of Rho kinase inhibitor. Diabetes 56:231–238. doi:10.2337/db06-0581
Hoerster R, Muether PS, Vierkotten S, Hermann MM, Kirchhof B, Fauser S (2014) Upregulation of TGF-ß1 in experimental proliferative vitreoretinopathy is accompanied by epithelial to mesenchymal transition. Graefes Arch Clin Exp Ophthal 252(1):11–16. doi:10.1007/s00417-013-2377-5
Papadimitriou E, Polykratis A, Hatziapostolou M, Parthymou A, Polytarchou C, Mikelis C (2004) Heparin affin regulatory peptide: a new target for tumour therapy? Curr Cancer Drug Targets 4:471–482. doi:10.2174/1568009043332835
Deuel TF, Zhang N, Yeh H-J, Silos-Santiago I, Wang Z-Y (2002) Pleiotrophin: a cytokine with diverse functions and a novel signaling pathway. Arch Biochem Biophys 397:162–171. doi:10.1006/abbi.2001.2705
Zhu X, Bai Y, Yu W et al (2015) The effects of pleiotrophin in proliferative diabetic retinopathy. PLoS ONE 10:e0115523. doi:10.1371/journal.pone.0115523
Yokoi H, Kasahara M, Mori K et al (2012) Pleiotrophin triggers inflammation and increased peritoneal permeability leading to peritoneal fibrosis. Kidney Int 81:160–169. doi:10.1038/ki.2011.305
Kohashi T, Tateaki Y, Tateno C, Asahara T, Obara M, Yoshizato K (2002) Expression of pleiotrophin in hepatic nonparenchymal cells and preneoplastic nodules in carbon tetrachloride-induced fibrotic rat liver. Growth Factors 20:53–60. doi:10.1080/08977190290023913
Zhang Q, Tao K, Huang W, Tian Y, Liu X (2013) Elevated expression of pleiotrophin in human hypertrophic scars. J Mol Histol 44:91–96. doi:10.1007/s10735-012-9453-8
Pufe T, Bartscher M, Petersen W, Tillmann B, Mentlein R (2003) Expression of pleiotrophin, an embryonic growth and differentiation factor, in rheumatoid arthritis. Arthritis Rheum 48:660–667. doi:10.1002/art.10839
Park TJ, Jeong BR, Tateno C et al (2008) Pleiotrophin inhibits transforming growth factor beta1-induced apoptosis in hepatoma cell lines. Mol Carcinog 47:784–796. doi:10.1002/mc.20438
Bai Y, Yu W, Han N et al (2013) Effects of semaphorin 3A on retinal pigment epithelial cell activity. Invest Ophthalmol Vis Sci 54:6628–6638. doi:10.1167/iovs.13-12625
Huang L, Yu W, Li X et al (2009) Expression of Robo4 in the fibrovascular membranes from patients with proliferative diabetic retinopathy and its role in RF/6A and RPE cells. Mol Vis 15:1057–1069
He S, Chen Y, Khankan R et al (2008) Connective tissue growth factor as a mediator of intraocular fibrosis. Invest Ophthalmol Vis Sci 49:4078–4088. doi:10.1167/iovs.07-1302
Lee J, Choi J-H, Joo C-K (2013) TGF-β1 regulates cell fate during epithelial-mesenchymal transition by upregulating survivin. Cell Death Dis 4:e714. doi:10.1038/cddis.2013.244
Chung EJ, Chun JN, Jung S-A, Cho JW, Lee JH (2011) TGF-β-stimulated aberrant expression of class III β-tubulin via the ERK signaling pathway in cultured retinal pigment epithelial cells. Biochem Biophys Res Commun 415:367–372. doi:10.1016/j.bbrc.2011.10.074
Yang S, Yao H, Li M, Li H, Wang F (2016) Long non-coding RNA MALAT1 mediates transforming growth factor Beta1-Induced epithelial-mesenchymal transition of retinal pigment epithelial cells. PLoS ONE 11:e0152687. doi:10.1371/journal.pone.0152687
Deuel TF (2013) Anaplastic lymphoma kinase: “ligand independent activation” mediated by the PTN/RPTPβ/ζ signaling pathway. Biochim Biophys Acta 1834:2219–2223. doi:10.1016/j.bbapap.2013.06.004
Pastor JC, de la Rúa ER, Martín F (2002) Proliferative vitreoretinopathy: risk factors and pathobiology. Prog Retin Eye Res 21:127–144. doi:10.1016/S1350-9462(01)00023-4
Kalluri R (2009) EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 119:1417–1419. doi:10.1172/JCI39675
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890. doi:10.1016/j.cell.2009.11.007
Perez-Pinera P, Alcantara S, Dimitrov T, Vega JA, Deuel TF (2006) Pleiotrophin disrupts calcium-dependent homophilic cell-cell adhesion and initiates an epithelial-mesenchymal transition. Proc Natl Acad Sci U S A 103:17795–17800. doi:10.1073/pnas.0607299103
Li YS, Milner PG, Chauhan AK et al (1990) Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science 250:1690–1694. doi:10.1126/science.2270483
Yeh HJ, He YY, Xu J, Hsu CY, Deuel TF (1998) Upregulation of pleiotrophin gene expression in developing microvasculature, macrophages, and astrocytes after acute ischemic brain injury. J Neurosci 18:3699–3707
Petersen W, Rafii M (2001) Immunolocalization of the angiogenetic factor pleiotrophin (PTN) in the growth plate of mice. Arch Orthop Trauma Surg 121:414–416. doi:10.1007/s004020000246
Fang W, Hartmann N, Chow DT, Riegel AT, Wellstein A (1992) Pleiotrophin stimulates fibroblasts and endothelial and epithelial cells and is expressed in human cancer. J Biol Chem 267:25889–25897
Li H, Wang H, Wang F, Gu Q, Xu X (2011) Snail involves in the transforming growth factor β1-mediated epithelial-mesenchymal transition of retinal pigment epithelial cells. PLoS ONE 6:e23322. doi:10.1371/journal.pone.0023322
Wang Y, Yuan Z, You C et al (2014) Overexpression p21WAF1/CIP1 in suppressing retinal pigment epithelial cells and progression of proliferative vitreoretinopathy via inhibition CDK2 and cyclin E. BMC Ophthalmol 14:144. doi:10.1186/1471-2415-14-144
Baudouin C, Fredj-Reygrobellet D, Brignole F, Nègre F, Lapalus P, Gastaud P (1993) Growth factors in vitreous and subretinal fluid cells from patients with proliferative vitreoretinopathy. Ophthalmic Res 25:52–59. doi:10.1159/000267221
Parapuram SK, Chang B, Li L et al (2009) Differential effects of TGFbeta and vitreous on the transformation of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 50:5965–5974. doi:10.1167/iovs.09-3621
Suh Y, Yoon C-H, Kim R-K et al (2013) Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene 32:4873–4882. doi:10.1038/onc.2012.505
Kanasty R, Dorkin JR, Vegas A, Anderson D (2013) Delivery materials for siRNA therapeutics. Nat Mater 12:967–977. doi:10.1038/nmat3765
Weng T, Chen Z, Jin N, Gao L, Liu L (2006) Gene expression profiling identifies regulatory pathways involved in the late stage of rat fetal lung development. Am J Physiol Lung Cell Mol Physiol 291:L1027–L1037. doi:10.1152/ajplung.00435.2005
Agrawal RN, He S, Spee C, Cui JZ, Ryan SJ, Hinton DR (2007) In vivo models of proliferative vitreoretinopathy. Nat Protoc 2:67–77. doi:10.1038/nprot.2007.4
Lee J, Ko M, Joo C-K (2008) Rho plays a key role in TGF-beta1-induced cytoskeletal rearrangement in human retinal pigment epithelium. J Cell Physiol 216:520–526. doi:10.1002/jcp.21424
Kojima T, Takano K, Yamamoto T et al (2008) Transforming growth factor-beta induces epithelial to mesenchymal transition by down-regulation of claudin-1 expression and the fence function in adult rat hepatocytes. Liver Int 28:534–545. doi:10.1111/j.1478-3231.2007.01631.x
Turksen K, Troy TC (2011) Junctions gone bad: claudins and loss of the barrier in cancer. Biochim Biophys Acta 1816:73–79. doi:10.1016/j.bbcan.2011.04.001
Stebbing J, Filipović A, Giamas G (2013) Claudin-1 as a promoter of EMT in hepatocellular carcinoma. Oncogene 32:4871–4872. doi:10.1038/onc.2012.591
Chen X-F, Zhang H-J, Wang H-B et al (2012) Transforming growth factor-β1 induces epithelial-to-mesenchymal transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling pathways. Mol Biol Rep 39:3549–3556. doi:10.1007/s11033-011-1128-0
Manna A, De Sarkar S, De S, Bauri AK, Chattopadhyay S, Chatterjee M (2016) Impact of MAPK and PI3K/AKT signaling pathways on Malabaricone-A induced cytotoxicity in U937, a histiocytic lymphoma cell line. Int Immunopharmacol 39:34–40. doi:10.1016/j.intimp.2016.07.004
Polytarchou C, Hatziapostolou M, Poimenidi E et al (2009) Nitric oxide stimulates migration of human endothelial and prostate cancer cells through up-regulation of pleiotrophin expression and its receptor protein tyrosine phosphatase beta/zeta. Int J Cancer 124:1785–1793. doi:10.1002/ijc.24084
Zhao H-M, Sheng M-J, Yu J (2014) Expression of IGFBP-6 in a proliferative vitreoretinopathy rat model and its effects on retinal pigment epithelial cell proliferation and migration. Int J Ophthalmol 7:27–33. doi:10.3980/j.issn.2222-3959.2014.01.05
Acknowledgements
We would like to thank Youzhi Yu for her help with the immunofluorescence assays and Yang Li for suggesting the Western blot analysis.
Author contributions
Research design: D. Xue, Y.J. Bai
Experiments: D. Xue
Data analysis: D. Xue, Y.J. Bai
Manuscript writing: D. Xue, Y.J. Bai
Manuscript review: Y.J. Bai, M.W. Zhao
Grant acquisition: M.W. Zhao
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 81470651 and 81570858) and the Specialized Research Fund for the Doctoral Program of Higher Education for ZMW (20130001110086). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Conflict of interest
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements) or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Xue Ding and Yujing Bai contributed equally to this work.
Rights and permissions
About this article

Cite this article
Ding, X., Bai, Y., Zhu, X. et al. The effects of pleiotrophin in proliferative vitreoretinopathy. Graefes Arch Clin Exp Ophthalmol 255, 873–884 (2017). https://doi.org/10.1007/s00417-016-3582-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-016-3582-9