
Abstract
Background
To unravel the molecular genetic background responsible for autosomal dominant congenital pulverulent nuclear cataracts in a four-generation Chinese family.
Methods
Family history data were collected, ophthalmological examinations were performed, and genomic DNA was extracted from peripheral blood of the family members. The candidate genes were captured and sequenced by targeted next-generation sequencing, and the results were confirmed by Sanger sequencing. The structure modelling of the protein was displayed based on Swiss-Model Server, and its possible changes in the secondary structure were predicted using Antheprot 2000 software. The chemical dissimilarity and possible functional impact of an amino acid substitution were performed with Grantham score, PolyPhen-2, and SIFT predictions. Protein distributions were assessed by confocal microscopy.
Results
A novel heterozygous c.829C > T transition that led to the substitution of a highly conserved histidine by tyrosine at codon 277 (p.H277Y) in the coding region of connexin50 (Cx50, GJA8) was identified. Bioinformatics analysis showed that the mutation likely altered the secondary structure of the protein by replacing the helix of the COOH-terminal portion with a turn. The mutation was predicted to be moderately conservative by Grantham score and to be deleterious by both PolyPhen-2 and SIFT with consistent results. In addition, when expressed in COS1 cells, the mutation led to protein accumulation and caused changes in Cx 50 protein localization pattern.
Conclusions
This is a novel missense mutation [c.829C > T, (p.H277Y)] identified in exon 2 of Cx50. Our findings expand the spectrum of Cx50 mutations that are associated with autosomal dominant congenital pulverulent nuclear cataract.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Congenital cataract is the leading cause of reversible blindness in childhood and accounts for one tenth of the cases of childhood blindness [1]. Its prevalence, depending on the regional socioeconomic development, is estimated to vary from 0.6 to six cases per 10,000 live births in industrialized countries [2, 3], and from five to 15 per 10,000 in the poorest areas of the world [4]. Nearly one third of the cases have a genetic basis [5]. It can present in an isolated manner or may occur in association with other ocular or systemic diseases [1, 6].
Although X-linked and autosomal recessive transmissions have been reported for congenital cataract, the autosomal dominant trait seems to be the most prevalent mode [7]. To date, at least 30 independent loci and 18 genes on different chromosomes have been associated with isolated congenital or infantile cataract [8–11]. Of the known mutant genes in cataract families, approximately half involve mutations in crystallins (CRYAA, CRYAB, CRYBA, CRYBB, CRYGC, CRYGD, CRYGS), about a quarter involve mutations in lens specific connexins (Cx43, Cx46, and Cx50), and the remainder include: mutations in major intrinsic protein (MIP) or aquaporin-0 (AQP0), beaded filament structural proteins-2 (BFSP2), paired-like homeodomain transcription factor-3 (PITX3), avian musculoaponeurotic fibrosarcoma (MAF), heat shock transcription factor-4 (HSF4), chromatin modifying protein-4B (CHMP4B), lens intrinsic membrane protein 2 (LIM2), and Eph-receptor type-A2 (EPHA2E) [8, 9, 11–16].
In this study, we applied a next-generation sequencing approach to test 60 known inheritable genetic congenital cataract-related genes in a Chinese family. With this approach, we identified a novel heterozygous mutation, the c.829C > T (p.H277Y) mutation, in exon 2 of Cx50, which co-segregated with autosomal dominant congenital pulverulent nuclear cataract. And with in silico prediction and functional study, we further verified the pathogenic effect of this missense mutation, which may represent a possible mechanism for cataract formation in this Chinese family.
Materials and methods
Clinical examination and isolation of genomic DNA
This study was approved by the medical ethics committee of the First People’s Hospital Affiliated to Shanghai Jiao Tong University (No. 2013KY096) and was conducted in accordance with the Declaration of Helsinki of the World Medical Association. A four-generation family originating from the province of Jiangsu, China, with isolated congenital cataracts was recruited to our hospital. Informed written consent was signed by the participants or their legal guardians. Affected status was identified by a history of cataract surgery or ophthalmological examinations, including visual acuity, slit lamp, biometry, and fundus examination. The cataract phenotypes were recorded using anterior segment photography. Corneal size and axial lengths were measured with IOLMaster (Carl Zeiss Meditec AG, Jena, Germany). Genomic DNA from peripheral blood leukocytes derived from the affected individuals and the available unaffected family members was extracted using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany).
Mutation detection
The list of 60 inheritable genetic congenital cataract-related genes, including 18 isolated congenital cataract genes and their subtypes, and seven other systemic congenital cataract genes, in the panel for captured and targeted next-generation sequencing can be seen in Supplementary Table 1 (Online Resource 1). There were two patients who underwent next-generation sequencing of the gene panel (II:5, III:7, Fig. 1a). As described previously, the exon regions of these 60 genes, including the Cx50 gene (gap junction protein alpha 8, GJA8), were specifically enriched using biotinylated capture probe (MyGenostics, Baltimore, MD, USA) [17, 18].

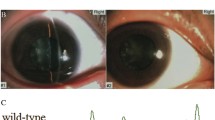
Clinical evaluation of a Chinese pedigree with autosomal dominant congenital cataracts. (a) A Chinese family with autosomal dominant congenital cataracts. The arrow indicates the proband. Squares and circles symbolize males and females, respectively. Black and white denote the status of family members affected or unaffected, respectively, by congenital cataract. Question mark next to the proband’s grandmother indicates the questionable status of family member I:2. (b) Slit-lamp photographs of the affected family member III:5 on the front and lateral sides. The slit-lamp photographs show pulverulent nuclear cataracts affecting the embryonic and fetal nuclei of the lens where the cortex is transparent. R = right; L = left
Short read mapping and alignment were performed using Burrows Wheeler Aligner software. Single-nucleotide polymorphisms (SNP) and insertions/deletions were tested using SOAPsnp software and GATK Indel Genotyper (http://www.broadinstitute.org/gsa/wiki/index.php/; The Genome Analysis Toolkit), respectively. All reference sequences were based on the NCBI37/hg19 assembly of the human genome [19].
To confirm the c.829C > T (p.H277Y) variant identified with next-generation sequencing, genomic DNA samples were amplified with the forward primer 5′-GGGCCACTACTTCCTGTACG-3′ and the reverse primer 5′-CTGCGGCTCTTCTTTTTCAC-3′ for 35 cycles using the following program: 95 °C for 30 s (1 cycle); 95 °C for 30 s, 62 °C for 30 s, 72 °C for 30 s (35 cycles); 72 °C for 4 min (1 cycle).
Bioinformatics analysis
Taking the resolved structure of connexin-26 gap junction as template (Protein Data Bank accession No. 2ZW3), the model structure of homomeric wild-type Cx 50 gap junction channel was modelled by Swiss-Model Server [20] and shown with the PyMOL Molecular Graphic system (Delano Scientific). Comparative secondary structure profiles of wild-type amino acid sequences with the p.H277Y mutant of C x50 were analyzed with Antheprot 2000 software (version 6.0; IBCP, Lyon, France). Additionally, the chemical dissimilarity of codon replacements was predicted by Grantham scores, which categorize into four classes: conservative (0–50), moderately conservative (51–100), moderately radical (101–150), or radical (≥151) according to the classification proposed by Li et al. [21, 22]. Meanwhile, the possible functional impact of an amino acid change was predicted by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) and SIFT (http://sift.jcvi.org/).
Cloning and plasmid construction of wild-type and mutant connexins
Genes coding for Cx50 wild-type, Cx50 p.H277Y mutant were cloned into the eukaryotic gene expression Vector pEGFP-N1 (Invitrogen, Carlsbad, CA, USA) to obtain pEGFP-N1-Cx50 Wild-type, and pEGFP-N1-p.H277Y Mutant, respectively. DNA sequencing confirmed the resultant constructs and verified the target mutation.
Cell culture and transfections
COS1 cells (kidney fibroblasts of African green monkey) were obtained from Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA) containing 10 % fetal bovine serum, 100 mg/ml penicillin and streptomycin, and 22 mM glucose. Cell lines were maintained in a humidified atmosphere containing 5 % CO2 at 37 °C. Transfections were carried out using the transfection reagent Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions.
Western blot analysis
Total proteins were extracted from COS1 cells transfected with pEGFP-N1-Wild-type, pEGFP-N1-p.H277Y Mutant, and EGFP vector plasmids using a RIPA kit (Beyotime, Shanghai, China) as per the manufacturer’s instructions. And total protein concentration was estimated using the BCA kit (Beyotime). After electrophoresis, the proteins were transferred onto nitrocellulose membranes. The target protein was blotted with anti-GAPDH (1:2000; Protein Tech Group, Chicago, IL, USA), anti-GFP (1:2000; Abcam, MA, USA) as primary antibodies, and then incubated with goat anti-mouse IgG-HRP (1:5000, Santa Cruz, CA, USA), goat anti-rabbit IgG-HRP (1:5000, Santa Cruz) as secondary antibodies. GAPDH was used as an internal control.
Immunofluorescence staining
Forty-eight hours after transfection, the cells were permeabilized with 0.2 % Triton X-100, blocked in solution, and the cellular nuclei was labeled by 4’,6-diamidino-2-phenylindole (DAPI; Sigma, Saint Louis, MO, USA). Both wild-type and mutant Cx50 protein distribution and gap junction formation were monitored based on tagged GFP signaling in these cells using a confocal laser scanning microscope (Zeiss LSM 510, Zurich, Switzerland).
Results
Clinical evaluation
There were seven affected individuals with congenital cataract in this four-generation Chinese pedigree (Fig. 1a). The clinical findings of the affected family members who participated in our study are summarized in Table 1. The age of onset was recorded as the age when the disease was first noticed by the patients or their parents, or first documented by a clinician. None of the cases were products of a consanguineous marriage and all affected patients had bilateral congenital cataracts.
The proband (III:7) was diagnosed with bilateral congenital cataract at age 7 years, and she underwent cataract extraction and intraocular lens implantation surgery at age 18 years. Her right eye was diagnosed with posterior capsular opacification at age 22 years, and she did not undergo Nd:YAG capsulotomy. The proband’s fraternal twin sister (III:8) was unaffected. The proband’s grandmother (I:2) had various illnesses and was bed-ridden; hence, she could not be examined with slit-lamp microscopy. Although we were told by her family members that her vision was very poor at an early age, we could not diagnose her condition with so little information. The phenotypes of cataract were pulverulent nuclear cataract affecting the embryonic and fetal nuclei of the lens where the cortex was transparent. These features were similar among all the affected participants (Fig. 1b). The average horizontal corneal diameter was 12.12 mm oculus dexter (OD) and 12.50 mm oculus sinister (OS). The average axial length was 22.82 mm OD and 22.48 mm OS. Fundus examination showed a normal appearance in all participants. Ophthalmic records confirmed that the lens opacities were late-onset and that no history of other ocular or systemic abnormalities was present in this family.
Identification of C x 50 mutations
The 60 inheritable genetic congenital cataract-related genes were captured and sequenced by next-generation sequencing using genomic DNA from two patients (II:5 and III:7). Overall, 58 and 52 variants were found in the two samples, respectively. The variants were filtered out if they were present in the 1000 Genome database (http://www.1000genomes.org/) or the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/). After filtering and samples comparison, one heterozygous change was identified in both patients associated with congenital cataract, C > T, at position 829 (c.829C > T) in exon 2 of Cx50 (GJA8), leading to the replacement of a wild-type histidine with tyrosine at codon 277 (p.H277Y; Fig. 2). All Cx50 exons for both patients were well covered by the next-generation sequencing data. Table 2 presents the coverage statistics. The mutation was further confirmed by Sanger sequencing (Fig. 3). Additional testing proved that this p.H277Y replacement was shown in all affected individuals who participated in our study, and it was not detected in the unaffected family members or in 800 healthy local Chinese controls.

Identification of a heterozygous mutation, c.829C > T, in Cx50 in two Chinese patients (II:5 and III:7) with autosomal dominant congenital cataract using targeted next-generation sequencing. A variant c.804C > T was also found in the same allele; however, it’s a synonymous variant (p.L268L)

The mutation in Cx50 was confirmed with Sanger sequencing. (a) A heterozygous C > T transition at codon 277 in one patient from the family (arrow). (b) Wild-type sequence from an unaffected family member. F = Female
The p.H277Y mutant is located within the intracellular COOH-terminal region of C x 50 as indicated by Maeda’s structural studies of the connexin gap junction channel (Fig. 4a) [23]. Histidine at position 277 is highly conserved in C x 50 according to multiple sequence alignments analyzing ten different species (Fig. 4b).

Schematic diagram showing the human Cx50 mutations reported to date and the relative conservation of a portion of the C x 50 COOH-terminus. (a) Diagram illustrating the topology of the C x 50 protein containing all reported mutations and the locations of the missense mutation (yellow bands |) and frame shift (fs) mutations (◆◆) identified in members of families with inherited cataracts. The red band denotes the mutation p.H277Y reported in our present study. (b) Multiple sequence alignment of a section of the Cx50 amino acid sequence (codons 268–287) from ten different species, indicating that histidine at position 277 (red) is highly conserved among C x 50 members from a variety of species
Bioinformatics analysis
The homology modelling showed the sideview and topview of Cx50 gap junction channel structure (Fig. 5a). The possible effect of the p.H277Y mutation on C x 50 was analyzed with bioinformatics software, which indicated that the wild-type helix in the COOH-terminal portion is likely replaced with a turn in the p.H277Y mutant (Fig. 5b). Moreover, the in silico prediction of the pathogenicity showed a Grantham score of 83, which meant that the variant (p.H277Y) was predicted as being “moderately conservative”. And this mutation was predicted to be “possibly damaging” by PolyPhen-2 with a score of 0.867 (HumVar-trained), and “affect protein function” by SIFT with a score of 0.00.

Structure modelling of Cx50 homomeric channel and comparison of the secondary structure of wild-type and p.H277Y mutant Cx50. (a) Side view and top view of wild-type Cx50 channel in cartoon form, showing the predicted homomeric gap junction channel structure (Swiss-Model Server). Cyans: helix; Magentas: sheet; Dark salmon: loop. (b) Difference in the predicted secondary structure between the wild-type and mutant Cx50 protein indicates that an original helix of the COOH-terminus has been replaced by a turn, labeled by a red arrow and a red circle (Antheprot 2000). Blueberry: helix; Lemon: sheet; Spring: turn; Turquoise (graph only): coil
Functional analysis
N terminus of C x 50 is critical for both V j -gating and unitary conductance [24]. In order to avoid or minimize the effect on structure and function of Cx 50, we used the pEGFP-N1 vector to generate the fusion protein Cx50-GFP. Both GFP-tagged Cx50 wild-type and p.H277Y mutant protein were expressed in COS1 cells, and similar expression level of the wild-type and mutant protein (~70 kDa) was determined based on Western blotting analysis of the cell lysates (Fig. 6).

The expression level of Cx50 wild-type and Cx50 p.H277Y mutant in stably transfected COS1 cells identified by Western blot. The similar expression level indicates that the p.H277Y mutation of Cx50 did not result in instability of the protein. The pEGFP-N1-transfected cells served as controls
Confocal immunocytochemical analysis showed that both GFP-tagged C x50 wild-type and p.H277Y mutant recombinant proteins were localized to the cytoplasm and plasma membrane. The results indicate that the p.H277Y mutant did not affect the subcellular distributions and trafficking to the plasma membrane of Cx50 protein. However, distinct from wild-type, the Cx50 p.H277Y mutant protein showed a different localization pattern with increased accumulation of mutant protein or cytoplasmic inclusions, and reduced linear distribution along appositional membranes (Fig. 7).

Immunofluorescent localization pattern of Cx50 wild-type and p.H277Y mutant. Confocal images of EGFP recombinant proteins showing stably transfected COS1 cells with Cx50 wild-type, p.H277Y mutant, and pEGFP-N1 backbone plasmids (control). DAPI shows nuclear DNA staining. Arrows indicate the aggregation of mutant protein in the cytoplasm and plasma membrane. Stellate symbols denote the linear distribution along appositional membranes corresponding to the gap junction plaques. Scale bar: 50 μm
Discussion
Ultrastructurally, the lens is composed of two distinct types of cells: an anterior layer of organelle-rich cuboidal epithelial cells and a large fiber cell mass making up the bulk of the lens. The cellular architecture and arrangement of the fiber cells, and particularly their sutures, play a pivotal role in light transmission and lens transparency [9]. The normal function and survival of cells in the avascular lens is facilitated by intercellular communication through an extensive network of gap junctions. Each gap junction channel comprises two hemichannels called connexons, which dock in the extracellular space between adjacent cells. Each connexon comprises a hexamer of connexins, which is formed predominantly by three isoforms of connexins (Cx43, C x 46, and Cx50) [12, 25]. Connexin proteins are membrane proteins containing four transmembrane domains (M1, M2, M3, and M4), two extracellular loops (E1 and E2), and three intracellular regions (the NH2-terminus, a cytoplasmic loop, and the COOH-terminus; Fig. 4a) [26].
Gap junctions are membrane specializations that contain clusters of intercellular channels that are permeable to ions and small solutes, including ions (K+, Ca2+), nutrients, small metabolites (e.g. glucose), and second messengers (inositol triphosphate, cAMP, cGMP) (≤1 kDa) [25]. These functions play a critical role in lens metabolic homeostasis and maintenance of transparency of fibers within the ocular lens. The gap channels function is regulated by post-translational modifications of the COOH-terminal cytoplasmic tail. (http://www.genecards.org/cgi-bin/carddisp.pl?gene=GJA8&search=3b0321a9b4cc1de65fcf11360f75f598; The Human Gene Compendium). And recently, Wang et al. also suggested that hemichannels are a pivotal source of autocrine and paracrine messengers [27].
Recent genetic studies showed that Cx50 is essential for lens growth, maturation of lens fiber cells, and lens transparency [28]. Pal et al. found that incorporation of even a single mutant protein molecule into a gap junction in Xenopus oocytes inhibited channel function [29]. Studies of certain connexin mutants linked to congenital cataracts have implicated hemichannels with aberrant voltage-dependent gating or modulation by divalent cations in disease pathogenesis [25]. Further deletion analysis has shown that the fourth transmembrane domain (M4) and a membrane proximal region (codons 231–294) of the cytoplasmic domain are needed for transport from the endoplasmic reticulum and localization to the plasma membrane [30]. However, unlike previously characterized Cx50 mutants that exhibited impaired intracellular transport and subsequent insertion into the plasma membrane and/or lack of function, the Cx50 mutant p.G46V with functional hemichannels and gap junction channels causes cell death even when expressed at minute levels. Biochemical data indirectly suggest a potential novel mechanism by which connexin mutants could lead to cataracts: cytotoxicity owing to enhanced hemichannel function [31].
To date, at least 34 mutations have been identified in human Cx50 (summarized in Fig. 4a, Supplementary Table 2, Online Resource 1). Since mutations in the same gene can result in morphologically different lens opacities and mutations in different gene loci can lead to similar opacities, inherited cataracts lack clear genotype-phenotype correlation rendering both clinical classification and molecular diagnosis challenging [11]. There is only one possible genotype-phenotype correlation that we found after a comprehensive literature search and careful comparison: the Cx50 mutations associated with congenital cataract-microcornea syndrome were exclusively located in first transmembrane domain (M1) and second extracellular loop (E2) (Supplementary Table 2, Online Resource 1). Our current study constitutes the first report that the p.H277Y mutation is associated with congenital pulverulent nuclear cataract. Several amino acid substitutions have been identified near position 277 of Cx50 that are associated with congenital cataract in mice and humans, including p.I247M [32], p.S258F [5], p.S259Y [33], p.S276F [34], and p.L281C [35]. The fact that we identified p.H277Y in this mutational hot spot emphasizes that this residue and surrounding region are critical for Cx50 function.
The overall conformation of wild-type human Cx50 is quite similar to that of the p.H277Y mutant. The most substantive variation was predicted to be in the secondary structure, in which a turn in the mutant likely replaces the wild-type helix in the COOH-terminal subdomain (Fig. 5b). A study about the structure and intermolecular interactions between the Cx40 and Cx43 COOH-terminal and cytoplasmic loop domains suggests that α-helices may be important for stabilizing the COOH-terminal dimer conformation under acidic conditions (i.e. more α-helix leads to a stronger binding affinity for the dimer conformation). Evidence supports involvement of the COOH-terminal domains in regulation of heteromeric channels. Predictions from their sequence indicate that analogous helical structures also occur in wild-type Cx50 COOH-terminus [36].
Furthermore, the novel detected c.829 C > T substitution turned a positively charged histidine into an uncharged tyrosine at position 277, and since it is located in the hydrophobic core of the COOH-terminus as well as in the positively charged cytoplasmic domain [23], it may interfere with the folding of the whole protein. This mutation was predicted to be deleterious by both PolyPhen-2 and SIFT with consistent results. Additionally, such a high degree of conservation at this position (Fig. 4b) argues for functional importance of this amino acid residue as well as that of the nearby residues [37].
Our functional finding further validated the in silico prediction, and the result is consistent with or similar to the reported C x 50 p.P88T [16], p.P88S [38, 39], and p.P88Q [10, 40] mutants, which found a distinct localization pattern and an increased accumulation for mutant protein (Fig. 7). The aberrant Cx 50 p.H277Y mutant protein accumulations likely results from misfolding and/or incomplete/improper oligomerization [41], or may be a result of inadequate degradation capacity of constitutive autophagy [39]. The retention of mutant subunits in the endoplasmic reticulum and the altered degradation of Cx 50 mutants may cause a stress response, which might exhibit alterations for lens homeostasis [27]. Also, the aggregation may cause light scattering, which might account for one of the molecular background underlying cataracts in this family [41].
In addition, our study found that the linear distribution along appositional membranes as expected for large gap junctions plaques is rarely formed for Cx50 p.H277Y mutant (Fig. 7), which may imply the impaired gap channels function or altered ability for entry of ions and small solutes. And the decreased gap junction plaques would result in disarrangement or separation of fiber cells, which may cause metabolic disorder of lens fiber and eventually lead to the formation of cataract. Hence, we consider this p.H277Y variation to be a causative mutation of the disease in this pedigree. Whether the p.H277Y mutant would be a dominant negative inhibitor of wild-type Cx50 remains to be investigated.
In conclusion, this study identified a novel missense mutation [c.829C > T, (p.H277Y)] in exon 2 of Cx50 using targeted next-generation sequencing approach, which further confirms that Cx50 plays a pivotal role in the maintenance of lens transparency, and expands the spectrum of Cx50 mutations that are associated with autosomal dominant congenital cataract. Based on our secondary structure analysis, we speculate that the p.H277Y mutation may not only affect the folding of the whole protein, but also affect the proper assembling of the dimer. Functional study proved a different distribution pattern of the mutant protein, the increased accumulation of mutant protein and decreased gap junction plaques staining may lead to altered function of gap channels and metabolic imbalance of lens fiber, which might be a contributing factor to the cataract formation.
Since there are many different derangements in the cellular/biochemical behavior and function of the Cx50 mutants, further functional impact of the mutant protein on cell growth [16, 42] or hemichannels voltage-dependent gating [30, 43], will improve our understanding of the underlying pathogenesis of cataract formation and illuminate the role of the connexin family in the lens. Also, since this is a dominant disease, it would be really interesting to assess whether the mutant protein dimerize with the wild-type form would impede the proper localization or function of the wild-type [44], which would help to design possible genetic therapies in the future.
References
Wang KJ, Wang S, Cao NQ, Yan YB, Zhu SQ (2011) A novel mutation in CRYBB1 associated with congenital cataract-microcornea syndrome: the p.Ser129Arg mutation destabilizes the βB1/βA3-crystallin heteromer but not the βB1-crystallin homomer. Hum Mutat 32:E2050–E2060
Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT (2004) Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 49:300–315
Holmes JM, Leske DA, Burke JP, Hodge DO (2003) Birth prevalence of visually significant infantile cataract in a defined U.S. population. Ophthalmic Epidemiol 10:67–74
Apple DJ, Ram J, Foster A, Peng Q (2000) Elimination of cataract blindness: a global perspective entering the new millennium. Surv Ophthalmol 45(Suppl 1):S1–S196
Gao X, Cheng J, Lu C, Li X, Li F, Liu C, Zhang M, Zhu S, Ma X (2010) A novel mutation in the connexin 50 gene (GJA8) associated with autosomal dominant congenital nuclear cataract in a Chinese family. Curr Eye Res 35:597–604
Hu S, Wang B, Zhou Z, Zhou G, Wang J, Ma X, Qi Y (2010) A novel mutation in GJA8 causing congenital cataract-microcornea syndrome in a Chinese pedigree. Mol Vis 16:1585–1592
Haargaard B, Wohlfahrt J, Fledelius HC, Rosenberg T, Melbye M (2004) Incidence and cumulative risk of childhood cataract in a cohort of 2.6 million Danish children. Invest Ophthalmol Vis Sci 45:1316–1320
Wang K, Wang B, Wang J, Zhou S, Yun B, Suo P, Cheng J, Ma X, Zhu S (2009) A novel GJA8 mutation (p.I31T) causing autosomal dominant congenital cataract in a Chinese family. Mol Vis 15:2813–2820
Hejtmancik JF (2008) Congenital cataracts and their molecular genetics. Semin Cell Dev Biol 19:134–149
Arora A, Minogue PJ, Liu X, Reddy MA, Ainsworth JR, Bhattacharya SS, Webster AR, Hunt DM, Ebihara L, Moore AT, Beyer EC, Berthoud VM (2006) A novel GJA8 mutation is associated with autosomal dominant lamellar pulverulent cataract: further evidence for gap junction dysfunction in human cataract. J Med Genet 43:e2
Mackay DS, Bennett TM, Culican SM, Shiels A (2014) Exome sequencing identifies novel and recurrent mutations in GJA8 and CRYGD associated with inherited cataract. Hum Genomics 8:19
Santana A, Waiswo M (2011) The genetic and molecular basis of congenital cataract. Arq Bras Oftalmol 74:136–142
Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI (2007) CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet 81:596–606
Shiels A, Bennett TM, Knopf HL, Maraini G, Li A, Jiao X, Hejtmancik JF (2008) The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol Vis 14:2042–2055
Kaul H, Riazuddin SA, Shahid M, Kousar S, Butt NH, Zafar AU, Khan SN, Husnain T, Akram J, Hejtmancik JF, Riazuddin S (2010) Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis 16:511–517
Ge XL, Zhang Y, Wu Y, Lv J, Zhang W, Jin ZB, Qu J, Gu F (2014) Identification of a novel GJA8 (C×50) point mutation causes human dominant congenital cataracts. Sci Rep 4:4121
He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N (2010) Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 464:610–614
Zenteno JC, Crespí J, Buentello-Volante B, Buil JA, Bassaganyas F, Vela-Segarra JI, Diaz-Cascajosa J, Marieges MT (2014) Next generation sequencing uncovers a missense mutation in COL4A1 as the cause of familial retinal arteriolar tortuosity. Graefes Arch Clin Exp Ophthalmol 252:1789–1794
Yang Y, Wu J, Liu H, Chen X, Wang Y, Zhao M, He X (2013) Two homozygous nonsense mutations of GNPTAB gene in two Chinese families with mucolipidosis II alpha/beta using targeted next-generation sequencing. Genomics 102:169–173
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185:862–864
Li WH, Wu CI, Luo CC (1984) Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol 21:58–71
Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T (2009) Structure of the connexin 26 gap junction channel at 3.5 a resolution. Nature 458:597–602
Xin L, Gong XQ, Bai D (2010) The role of amino terminus of mouse C×50 in determining transjunctional voltage-dependent gating and unitary conductance. Biophys J 99:2077–2086
Beyer EC, Berthoud VM (2014) Connexin hemichannels in the lens. Front Physiol 5:20
Yeager M, Harris AL (2007) Gap junction channel structure in the early 21st century: Facts and fantasies. Curr Opin Cell Biol 19:521–528
Wang N, De Bock M, Decrock E, Bol M, Gadicherla A, Vinken M, Rogiers V, Bukauskas FF, Bultynck G, Leybaert L (2013) Paracrine signaling through plasma membrane hemichannels. Biochim Biophys Acta 1828:35–50
Sellitto C, Li L, White TW (2004) Connexin50 is essential for normal postnatal lens cell proliferation. Invest Ophthalmol Vis Sci 45:3196–3202
Pal JD, Berthoud VM, Beyer EC, Mackay D, Shiels A, Ebihara L (1999) Molecular mechanism underlying a C×50-linked congenital cataract. Am J Physiol 276:C1443–C1446
Somaraju Chalasani ML, Muppirala MG, Ponnam SP, Kannabiran C, Swarup G (2012) A cataract-causing connexin 50 mutant is mislocalized to the ER due to loss of the fourth transmembrane domain and cytoplasmic domain. FEBS Open Bio 3:22–29
Minogue PJ, Tong JJ, Arora A, Russell-Eggitt I, Hunt DM, Moore AT, Ebihara L, Beyer EC, Berthoud VM (2009) A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest Ophthalmol Vis Sci 50:5837–5845
Polyakov A, Shagina I, Khlebnikova O, Evgrafov O (2001) Mutation in the connexin 50 gene (GJA8) in a Russian family with zonular pulverulent cataract. Clin Genet 60:476–478
Hansen L, Mikkelsen A, Nürnberg P, Nürnberg G, Anjum I, Eiberg H, Rosenberg T (2009) Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci 50:3291–3303
Yan M, Xiong CL, Ye SQ, Chen YM, Ke M, Zheng F, Zhou X (2008) A novel connexin 50 (GJA8) mutation in a Chinese family with a dominant congenital pulverulent nuclear cataract. Mol Vis 14:418–424
Kumar M, Agarwal T, Khokhar S, Kumar M, Kaur P, Roy TS, Dada R (2011) Mutation screening and genotype phenotype correlation of α-crystallin, γ-crystallin and GJA8 gene in congenital cataract. Mol Vis 17:693–707
Bouvier D, Spagnol G, Chenavas S, Kieken F, Vitrac H, Brownell S, Kellezi A, Forge V, Sorgen PL (2009) Characterization of the structure and intermolecular interactions between the connexin40 and connexin43 carboxyl-terminal and cytoplasmic loop domains. J Biol Chem 284:34257–34271
Yan N, Zhao Y, Wang Y, Xie A, Huang H, Yu W, Liu X, Cai SP (2011) Molecular genetics of familial nystagmus complicated with cataract and iris anomalies. Mol Vis 17:2612–2617
Lichtenstein A, Gaietta GM, Deerinck TJ, Crum J, Sosinsky GE, Beyer EC, Berthoud VM (2009) The cytoplasmic accumulations of the cataract-associated mutant, Connexin50P88S, are long-lived and form in the endoplasmic reticulum. Exp Eye Res 88:600–609
Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM (2011) Autophagy: a pathway that contributes to connexin degradation. J Cell Sci 124:910–920
Vanita V, Singh JR, Singh D, Varon R, Sperling K (2008) A mutation in GJA8 (p.P88Q) is associated with “balloon-like” cataract with Y-sutural opacities in a family of Indian origin. Mol Vis 14:1171–1175
Beyer EC, Ebihara L, Berthoud VM (2013) Connexin mutants and cataracts. Front Pharmacol 4:43
Su D, Yang Z, Li Q, Guan L, Zhang HED, Zhang L, Zhu S, Ma X (2013) Identification and functional analysis of GJA8 mutation in a Chinese family with autosomal dominant perinuclear cataracts. PLoS One 8:e59926
Zhu Y, Yu H, Wang W, Gong X, Yao K (2014) A Novel GJA8 Mutation (p.V44A) Causing Autosomal Dominant Congenital Cataract. PLoS One 9:e115406
Rubinos C, Villone K, Mhaske PV, White TW, Srinivas M (2014) Functional effects of C × 50 mutations associated with congenital cataracts. Am J Physiol Cell Physiol 306:C212–C220
Acknowledgments
We are grateful for all the participating members of the congenital cataract family. We also express our sincere gratitude to Daoyun Zhang, Yunbing Zhang, Chencheng Wang, Zhongqiong Lu, and Qingfu Wang for their technical assistance. This work was supported by grants from the National Science and Technology Pillar Program of the Twelfth Five-year Plan (2011ZX09302-007-02) and the Research Fund for the National Nature Science Funding of China (No. 81273424 and 81170862).
Conflict of interest
All authors certify that they have NO affiliations with or involvement in any organization or entity with any financial interest, or non-financial interest in the subject matter or materials discussed in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 163 kb)
Rights and permissions
About this article

Cite this article
Chen, C., Sun, Q., Gu, M. et al. A novel Cx50 (GJA8) p.H277Y mutation associated with autosomal dominant congenital cataract identified with targeted next-generation sequencing. Graefes Arch Clin Exp Ophthalmol 253, 915–924 (2015). https://doi.org/10.1007/s00417-015-3019-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-015-3019-x