
Abstract
Objectives
Our aim was to determine the molecular cause of autosomal dominant familial retinal arteriolar tortuosity (FRAT) in a family with three affected subjects.
Material and methods
Ophthalmologic evaluation included determination of best-corrected visual acuity (BCVA), slit-lamp and dilated fundus inspection, applanation tonometry, fundus photography, and fluorescein retinal angiography (FA). Molecular methods included whole exome sequencing analysis and Sanger sequencing validation of putative causal mutation in DNA from affected individuals.
Results
Typical signs of familial retinal arteriolar tortuosity were observed in all three patients. Exome sequencing identified a heterozygous c.1528G > A (p. Gly510Arg) mutation in COL4A1. Sanger sequencing confirmed that all three patients harbored the same pathogenetic mutation in COL4A1. The p. Gly510Arg variant in COL4A1 was absent in DNA from an available unaffected daughter, from a set of control alleles, and from publicly available databases.
Conclusions
The molecular basis of familial retinal arteriolar tortuosity was identified for the first time, thus expanding the human phenotypes linked to COL4A1 mutations. Interestingly, the COL4A1 p.Gly510Arg mutation has been previously identified in a family with HANAC (Hereditary Angiopathy with Nephropathy, Aneurysm and Cramps), a multisystemic disease featuring retinal arteriolar tortuosity. No cerebral, neurologic, renal, cardiac or vascular anomalies were recognized in the pedigree described here. These data indicate that identical mutations in COL4A1 can originate both eye-restricted and systemic phenotypes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Increased tortuosity of retinal vessels is a frequent ophthalmoscopic finding and it can occur as a localized or generalized anomaly, affecting one or both eyes, and as a sporadic or inherited trait. Familial retinal arterial tortuosity (FRAT, OMIM %180000), first reported by Beyer in 1958, is an uncommon dominant disorder characterized by marked tortuosity of second-order and third-order retinal arteries with normal first-order arteries and venous system [1]. Typically, vascular tortuosity in FRAT is predominantly located at the macular and peripapillary area and develops during childhood or early adulthood [2–5]. Although the disease may be asymptomatic, most FRAT patients complain of variable degrees of transient vision loss due to retinal hemorrhage following physical exertion or minor trauma (reviewed in [6]). The observation of vertical transmission in affected pedigrees, male to male inheritance, and occurrence in parents and their children in the absence of consanguinity strongly suggests autosomal dominant inheritance [7–9]. To date, approximately 18 familial cases have been reported [6, 8, 9]. In most cases, systemic involvement of non-ocular vascular beds has not been demonstrated in FRAT patients, but occasionally other associated vascular abnormalities have been reported, including malformations in the Kieselbach nasal septum, spinal cord vascular mass [3], telangiectasis of bulbar conjunctiva [6], and internal carotid artery aneurysm [10]. While several syndromic genetic entities feature increased retinal arterial tortuosity, isolated FRAT is considered a discrete autosomal dominant entity with an as of yet unknown etiology. In this work, we report the results of exome sequencing analysis in a two-generation family affected with FRAT and provide evidence that a heterozygous missense mutation in COL4A1 gene is responsible for the retinal phenotype in this pedigree.
Methods
Clinical studies
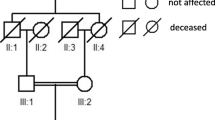
Institutional Review Board (IRB)/Ethics Committee approval was obtained. All patient samples were collected with written informed consent and clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. A two-generation family from Spain was studied (Fig. 1). In all patients, ophthalmologic examinations included determination of best-corrected visual acuity (BCVA), slit-lamp and dilated fundus inspection, applanation tonometry, fundus photography, and fluorescein retinal angiography (FA). To exclude systemic involvement, hemogram, urianalysis, glomerular filtration rate (GFR) and creatine phosphokinase (CPK) levels measurements, kidney ultrasonography, Doppler echography, magnetic resonance angiography (MRA), and clinical neurological examination were performed in all three patients.

Pedigree of the family with autosomal dominant FRAT and segregating a heterozygous p.Gly510Arg mutation. Solid symbols indicate affected subjects; WT indicates a wild type COL4A1 allele
Whole exome sequencing
Exome sequencing was performed by Edgebio (Gaithersburg, MD, USA) on a single FRAT patient (father) from this family. Samples were prepared using Illumina’s protocol TruSeq DNA Sample Preparation Guide. Briefly, samples were sheared to an average size of 300–400 bp using sonication. DNA fragment ends were repaired and phosphorylated using Klenow, T4 DNA Polymerase and T4 Polynucleotide Kinase. Next, an ‘A’ base was added to the 3′ end of the blunted fragments, followed by ligation of Illumina Paired-End adapters via T-A mediated ligation. From here, samples were prepared using the NimbleGen protocol outlined in “NimbleGen SeqCap EZ Exome Library SR User’s Guide” (Version 3.0). The libraries were amplified using LM-PCR and 1ug of amplified sample libraries were hybridized with Nimblegen’s Exome Library baits for 64 h at 47 °C. Captured DNA was then washed and recovered using Streptavidin Dynabeads. The captured DNA was LM-PCR amplified for a total of 17 cycles. The amplified capture DNA library size and concentration were determined using an Agilent Bioanalyzer.
The captured library was then loaded on a HiSeq 2000 platform for sequencing with a mean exome coverage of 30×. Raw image files were processed by Illumina Pipeline v1.7 for base calling. Single nucleotide polymorphisms (SNPs) and indels were called using an in-house developed software. Identified variants were filtered against the Single Nucleotide Polymorphism database (dbSNP 129, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), 1,000 genomes project (www.1000genomes.org), and Exome Variant Server (http://evs.gs.washington.edu/EVS/) databases. We excluded the variants that we don’t considered pathogenic according to the following criteria: 1. Minor allele frequency (MAF) ≥0.01 from 1000 Human Genome Project database; 2. Located in non-coding regions without affecting splicing site; 3. Synonymous variants without affecting splicing site; 4. Homozygous variations (as we assumed an autosomal dominant transmission in this family). All the other variants were considered pathogenic and summarized for validation. Non-excluded missense mutations were tested for mutational effects by using amino acid substitution prediction tools such as PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and PANTHER (http://www.pantherdb.org/tools/).
PCR and Sanger sequencing for familial segregation analysis of COL4A1 mutation
Genomic DNA was extracted from peripheral blood leukocytes using an automated system (Qiacube, Qiagene Mexico, Mexico City, Mexico). The exon number 24 of COL4A1 was amplified by PCR using pairs of primers derived from normal gene sequences (exon 24 Fwd: 5′- CCTTTCTGAGTCCGTCTTGG −3′; Rev: 5′-CACTTACCAGCTCCCACACA −3′ (Ensembl ID ENSG00000187498). Each 25 μl PCR amplification reaction contained 1× buffer, 100 ng of genomic DNA, 0.2 mM of each dNTP, 2 U Taq polymerase, 1 mM of forward and reverse primers, and 1.5 mM MgCl2. PCR products were analyzed in 1.5 % agarose gels, from which the bands with the amplified templates were excised and the DNA was subsequently purified with the help of the MiniElute PCR Purification Kit (Qiagen). Direct automated sequencing of PCR amplicons was performed with the BigDye Terminator Cycle Sequencing kit (Apllied Biosystems, Foster City, CA, USA). All samples were analyzed on an ABI 3130 Genetic Analyzer (Applied Biosystems). Wild-type and mutated COL4A1 sequences were compared manually. Familial segregation of the mutation was analyzed.
Results
Clinical assessment
Case #1 (father)
Case #1 is 53-year old male who was evaluated due to photophobia. He denied any other visual symptom. At examination, best corrected visual acuity was 20/20 in both eyes (OU), and intraocular pressure was 16 mmHg in the right eye (OD) and 17 mmHg in the left eye (OS). Structures of the anterior segment were unremarkable. At funduscopy, marked tortuosity of second-order and third-order arterioles was noted bilaterally. Venous system appeared normal and no evidence of past retinal hemorrhage was noted. In addition, discrete hypopigmentation of the retinal pigment epithelium was observed in both fundi (Fig. 2). Hemogram and urianalysis tests were normal, while glomerular filtration rate (GFR) calculated using the Modification of Diet in Renal Disease (MDRD) Study equation was 91 ml/min/ 1.73 m2 (normal value: > 60 ml /min/ 1.73 m2). Kidney ultrasonogram excluded renal anomalies, while Doppler echography did not identify aortic or renal arterial abnormalities. Magnetic resonance angiography (MRA) revealed a small (3 mm in diameter) internal carotid artery aneurysm and no evidence of leukoencephalopathy. No other intracranial vascular lesion was found. No neurologic symptoms were identified and the patient denied having experienced muscle cramps or migraine. CPK levels were 154 u/L (normal values for men: 55–170 U/L).

Fundus photograph of patient #1 showing the pathognomonic pattern of arteriolar tortuosity in right (a) and left (b) eyes. Red-free retinography (c) shows no evidence of retinal hemorrhage and exhibits the striking tortuosity of second and third order arterioles
Case #2
Case #2 is a 21-year old female, the oldest daughter of case #1. At the age of 15 years, she suffered from an episode of exercise-related mild retinal hemorrhage. Her visual acuity was 20/20 (OD) and 20/32 (OS). No anterior segment anomalies were present at biomicroscopic examination. Funduscopic examination revealed increased tortuosity of second-order and third-order arterioles, several round perifoveal intraretinal hemorrhages OD, and a foveal hemorrhage OS (Fig. 3). As observed in her father’s fundi, she exhibited a generalized RPE hypopigmentation OU. On follow-up evaluations, hemorrhages resolved spontaneously and visual acuity recovered to normal 20/20. She has experienced several self-resolving events of retinal hemorrhages during the last 5 years. Hemogram and urianalysis tests were normal, while GFR was normal at 98 ml/min/1.73 m2. Renal USG results were unremarkable, doppler echography did not identify aortic or renal arterial abnormalities, and MRA did not detect structural anomalies or evidence of leukoencephalopathy. No neurologic symptoms were identified and she denied having experienced muscle cramps or migraine. CPK levels were 115 U/L (normal values for women: 45–135 U/L).

Fundus image of patient #2 showing marked tortuosity of arterioles and intraretinal hemorrhages at perimacular area in right (a) and left (b) eye
Case #3
Case #3 is an 18-year old female, sister of case #2. She suffered from an episode of exercise-related mild retinal hemorrhage at the age of 13 years. Best corrected visual acuity was 20/20 (OD) and 20/200 (OS). Anterior segment structures were normal OU, IOP was 16 mmHg OU, while funduscopy revealed increased tortuosity or retinal arterioles and bilateral intraretinal hemorrhages (Fig. 3). In OS, a large foveal hemorrhage was observed (Fig. 4). The hemorrhage reabsorbed spontaneously after a 2-month period and no additional episodes of retinal hemorrhages had occurred since then. Results of hemogram and urianalysis tests were normal, while GFR was 104 ml/min/1.73 m2, within normal limits. Renal USG was normal, doppler echography did not identify aortic or renal arterial abnormalities, and MRA did not detect structural anomalies or white matter changes suggestive of leukoencephalopathy. No neurologic symptoms were identified and she denied having experienced muscle cramps or migraine. CPK levels were within normal limits (89 U/L).

Fundus photographs of patient #3 demonstrating increased arteriolar tortuosity and intraretinal hemorrhage in right eye (a) and arteriolar tortuosity and subretinal hemorrhage involving the fovea in left eye (b). Left eye hemorrhage showed spontaneous reabsorption after a period of 2 months (c)
Exome sequencing results and mutation validation
Whole exome sequencing identified a total of 234 rare non-synonymous heterozygous variants. From this group, 11 variants were shown to be deleterious by both Polyphen and SIFT algorithms. Among those, a c.1528G > A (p.Gly510Arg) mutation in COL4A1 was selected to be a strong candidate for the disorder in this family, as this gene has been previously implicated in a broad spectrum of vascular anomalies. Sanger sequencing of COL4A1 exon 24 demonstrated that the p.Gly510Arg missense mutation co-segregates with disease status in the family (Fig. 5) and was consistently predicted to be damaging by multiple in silico analyses (Polyphen, SIFT, and PANTHER algorithms). In addition, the p.Gly510Arg mutation was absent from DNA of a healthy daughter of case #1, from a set of 200 ethnically matched control alleles, and from the 8,600 exomes in the NHLBI Exome Variant Server. The remaining 10 potentially deleterious missense mutations in other genes (Supplementary table) did not segregate with the phenotype in the family when assessed by Sanger sequencing or were predicted to be benign by Polyphen, SIFT, and PANTHER algorithms.

Partial nucleotide sequence of COL4A1 exon 24 in DNA from patients #1-#3 (A-C). A heterozygous c.1528G > A mutation (arrow), predicting a p.Gly510Arg missense substitution, was identified in affected individuals
Discussion
Familial retinal arteriolar tortuosity (FRAT) is an uncommon, autosomal dominant condition characterized by a pathognomonic pattern of progressive and pronounced tortuosity of second-order and third-order arterioles in the macular and peripapillary retinal area. Based on the report of a number of families without detectable extraocular anomalies, FRAT is regarded to be a distinct monogenic disease (OMIM %180000) of unknown etiology. In this work, the exome sequencing-mediated identification of a missense COL4A1 mutation in affected FRAT subjects from a Spanish FRAT family is reported.
COL4A1 is the most abundant and ubiquitous basement membrane protein. In the eye, COL4A1 is present in the basal lamina of the conjunctiva, corneal epithelium, corneal endothelium, trabecular meshwork, Schlemm’s canal, lens, ciliary body, retinal inner limiting membrane, Bruch’s membrane and vascular basement membranes [11–14]. The phenotypic spectrum of COL4A1 mutations is wide and includes familial porencephaly [15, 16], cerebral white matter small vessel disease [17], cerebral aneurysms [18], cataract, anterior segment dysgenesis, microcornea [19], nephropathy, muscle cramps [20, 21], and Walker Warburg syndrome [22].
A multisystemic disease featuring retinal arteriolar tortuosity is HANAC (Hereditary Angiopathy with Nephropathy, Aneurysm and Cramps), in which affected patients exhibit retinal arteriolar tortuosity, muscle cramps, and renal disease, whereas the brain phenotype was usually clinically silent [20, 21, 23]. Mutations identified thus far in HANAC patients cluster within a 31 amino acid region (residues 498–528) of the COL4A1 protein that encompasses integrin binding sites [20, 21, 23, 24].
Interestingly, the p.Gly510Arg mutation identified in the FRAT family described in the present work is also located within this critical region, and affects a conserved glycine within the collagenous region of the protein. Pathogenic mutations within the collagenous domain often perturb triple helix assembly and impair secretion of the collagen heterotrimers, and concomitantly, misfolded proteins accumulate within cells [23, 25]. Remarkably, an identical p.Gly510Arg COL4A1 mutation was previously recognized by Plaisier et al. in three subjects from a family suffering from retinal arterial tortuosity (all three patients), Raynaud phenomena (two out of three), migraine (one out of three), and supraventricular arrhythmia (one out of three) and whom were diagnosed as having HANAC syndrome [23]. In the three affected FRAT subjects reported here, no cerebral, neurologic, renal, cardiac or vascular (except for a small carotid aneurysm in one subject) anomalies were recognized, indicating that FRAT could be considered a distinct mild form of COL4A1-related disorder with few (or no) HANAC non-ocular features. Interestingly, HANAC individuals due to the p.Gly510Arg COL4A1 mutation reported by Plaisier et al. [23] and the patients in the present family carrying the same mutation did not exhibit brain anomalies, suggesting an incipient genotype-phenotype correlation (Table 1).
Our data indicates that FRAT can be considered another member of the COL4A1-related group of diseases and that an identical mutation in COL4A1 can result in different clinical spectrum, even within the same family. As previously suggested, besides the location of pathogenic COL4A1 mutations, environmental factors and/or genetic modifiers may influence the phenotypic expression and the severity of the organ involvement in COL4A1-related disease [24]. Further research will be needed to explain why some COL4A1 mutations originate a pleiotropic effect, while in others they are associated with single organ phenotypes as FRAT. Finally, although our results support that a mutation of COL4A1 underlies the molecular cause of the disease in the family described here, the involvement of a different gene(s) in other FRAT cases cannot be excluded at this time. Thus, the molecular analysis of additional FRAT sporadic and familial cases would be of great relevance.
References
Beyer EM (1958) Familiare tortuositas der kleinen netzhautarterien mit makulablutung. Klin Monatsbl Augenheilkd 132:532–539
Goldberg MF, Pollack IP, Green WR (1972) Familial retinal arteriolar tortuosity with retinal hemorrhage. Am J Ophthalmol 73:183–191
Bartlett WJ, Price J (1983) Familial retinal arteriolar tortuosity with retinal hemorrhage. Am J Ophthalmol 95:556–558
Kayazawa F, Machida T (1983) Retinal arteriolar tortuosity with macular hemorrhage. Ann Ophthalmol 15:42–43
Clearkin IG, Rose H, Patterson A, Mody CH (1986) Development of retinal arteriolar tortuosity in previously unaffected family members. Trans Ophthalmol Soc U K 105:568–574
Sutter FK, Helbig H (2003) Familial retinal arteriolar tortuosity: a review. Surv Ophthalmol 48:245–255
Wells CG, Kalina RE (1985) Progressive inherited retinal arteriolar tortuosity with spontaneous retinal hemorrhages. Ophthalmology 92:1015–1024
Sears J, Gilman J, Sternberg P Jr (1998) Inherited retinal arteriolar tortuosity with retinal hemorrhages. Arch Ophthalmol 116:1185–1188
Desmettre T, Moreau JM, Plaisier E (2006) Autosomal dominant syndrome of retinal arterial tortuosity. J Fr Ophtalmol 29:e8
Seo JH, Kim I, Yu HG (2009) A case of carotid aneurysm in familial retinal arterial tortuosity. Korean J Ophthalmol 23:57–58
Ishizaki M, Westerhausen-Larson A, Kino J, Hayashi T, Kao WW (1993) Distribution of collagen IV in human ocular tissues. Invest Ophthalmol Vis Sci 34:2680–2689
Sarthy V (1993) Collagen IV mRNA expression during development of the mouse retina: an in situ hybridization study. Invest Ophthalmol Vis Sci 34:145–152
Qin P, Piechocki M, Lu S, Kurpakus MA (1997) Localization of basement membrane-associated protein isoforms during development of the ocular surface of mouse eye. Dev Dyn 209:367–376
Hann CR, Springett MJ, Wang X, Johnson DH (2001) Ultrastructural localization of collagen IV, fibronectin, and laminin in the trabecular meshwork of normal and glaucomatous eyes. Ophthalmic Res 33:314–324
Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SW (2005) Mutations in COL4A1 cause perinatal cerebral hemorrhage and porencephaly. Science 308:1167–1171
Breedveld G, de Coo IF, Lequin MH, Arts WF, Heutink P, Gould DB, John SW, Oostra B, Mancini GM (2006) Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J Med Genet 43:490–495
Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SW (2006) Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 354:1489–1496
Lanfranconi S, Markus HS (2010) COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke 41:e513–e518
Coupry I, Sibon I, Mortemousque B, Rouanet F, Mine M, Goizet C (2010) Ophthalmological features associated with COL4A1 mutations. Arch Ophthalmol 128:483–489
Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, Marro B, Desmettre T, Cohen SY, Roullet E, Dracon M, Fardeau M, Van Agtmael T, Kerjaschki D, Antignac C, Ronco P (2007) COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 357:2687–2695
Plaisier E, Alamowitch S, Gribouval O, Mougenot B, Gaudric A, Antignac C, Roullet E, Ronco P (2005) Autosomal-dominant familial hematuria with retinal arteriolar tortuosity and contractures: A novel syndrome. Kidney Int 67:2354–2360
Labelle-Dumais C, Dilworth DJ, Harrington EP, de Leau M, Lyons D, Kabaeva Z, Manzini MC, Dobyns WB, Walsh CA, Michele DE, Gould DB (2011) COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet 7:e1002062
Plaisier E, Chen Z, Gekeler F, Benhassine S, Dahan K, Marro B, Alamowitch S, Paques M, Ronco P (2010) Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am J Med Genet A 152A:2550–2555
Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T, Marro B, Ronco P (2009) Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology 73:1873–1882
Persikov AV, Ramshaw JA, Brodsky B (2005) Prediction of collagen stability from amino acid sequence. J Biol Chem 280:19343–19349
Acknowledgments
Financial support was provided by Mexican Research Council CONACYT Grant 169352.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Juan C. Zenteno and Jaume Crespí Contributed equally to this work and should be considered equivalent first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 32 kb)
Rights and permissions
About this article

Cite this article
Zenteno, J.C., Crespí, J., Buentello-Volante, B. et al. Next generation sequencing uncovers a missense mutation in COL4A1 as the cause of familial retinal arteriolar tortuosity. Graefes Arch Clin Exp Ophthalmol 252, 1789–1794 (2014). https://doi.org/10.1007/s00417-014-2800-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-014-2800-6