
Abstract
Background
Despite recent progress in the field of genetics, sporadic late-onset (> 40 years) cerebellar ataxia (SLOCA) etiology remains frequently elusive, while the optimal diagnostic workup still needs to be determined. We aimed to comprehensively describe the causes of SLOCA and to discuss the relevance of the investigations.
Methods
We included 205 consecutive patients with SLOCA seen in our referral center. Patients were prospectively investigated using exhaustive clinical assessment, biochemical, genetic, electrophysiological, and imaging explorations.
Results
We established a diagnosis in 135 (66%) patients and reported 26 different causes for SLOCA, the most frequent being multiple system atrophy cerebellar type (MSA-C) (41%). Fifty-one patients (25%) had various causes of SLOCA including immune-mediated diseases such as multiple sclerosis or anti-GAD antibody-mediated ataxia; and other causes, such as alcoholic cerebellar degeneration, superficial siderosis, or Creutzfeldt–Jakob disease. We also identified 11 genetic causes in 20 patients, including SPG7 (n = 4), RFC1-associated CANVAS (n = 3), SLC20A2 (n = 3), very-late-onset Friedreich’s ataxia (n = 2), FXTAS (n = 2), SCA3 (n = 1), SCA17 (n = 1), DRPLA (n = 1), MYORG (n = 1), MELAS (n = 1), and a mitochondriopathy (n = 1) that were less severe than MSA-C (p < 0.001). Remaining patients (34%) had idiopathic late-onset cerebellar ataxia which was less severe than MSA-C (p < 0.01).
Conclusion
Our prospective study provides an exhaustive picture of the etiology of SLOCA and clues regarding yield of investigations and diagnostic workup. Based on our observations, we established a diagnostic algorithm for SLOCA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebellar ataxia (CA) is a disorder of balance and coordination due to cerebellum and/or cerebellar pathways dysfunction that may impact activities of daily living [1]. The causes underlying this heterogeneous neurological disorder are numerous, and vary according to the age of onset, the rapidity of installation, the clinical picture, and the presence of familial medical history [2].
Sporadic late-onset cerebellar ataxias (SLOCA), defined by the occurrence of CA after 40 years of age without family history of CA, represent a challenging subgroup, since the diagnosis remains unknown in many cases [3], despite the recent advances in genetic diagnosis techniques, the increasing availability of next-generation sequencing, and the discovery of new CA-associated genes such as RFC1 [4] or SPG7 [5]. Some hierarchical diagnostic approaches and diagnostic algorithm have been proposed [6, 7], although no study aiming to prospectively assess the relevance and the performance of the different explorations has been reported so far.
The objective of the present study was to describe the etiology of SLOCA in a cohort of consecutive patients recruited in Strasbourg University Hospital and to evaluate the relevance of the different investigations performed, including imaging, electrophysiology, and genetic testing.
Materials and methods
Population
Patients successively seen for SLOCA in our referral center for neurogenetic disease and movement disorders were included in a prospective study aiming to investigate the underlying etiology. The inclusion criteria were (i) CA beginning after 40 years, (ii) a chronic and progressive evolution for at least 3 months, and (iii) the absence of documented family history of CA. Patients with a follow-up below 1 year were excluded from the analysis. Informed consent was obtained from all individual participants included in the study. Strasbourg University Hospital local ethics committee approved the study.
Clinical examination
A thorough general and neurological examination was performed at baseline then every 6 months including assessment of SARA (Scale for the Assessment and Rating of Ataxia) [8], UPDRS-III (United Parkinson Disease Rating Scale) [9], and SDFS (Spinocerebellar Degeneration Functional Score) [10]. We measured the SARA, UPDRS-III, and SDFS scores at inclusion, and calculated the SARA/DD, UPDRS/DD, and SDFS/DD by dividing the respective scores by the estimated years of disease duration at inclusion to estimate disease progression rate. We also calculated the ∆SARA as the difference between the most recent SARA score available and the SARA score at inclusion divided by the duration between the two scores. We calculated the ∆UPDRS and ∆SDFS in the same way.
Dysautonomia was evaluated through anamnesis (erectile dysfunction, urinary urgencies, or incomplete bladder emptying, etc.…), bladder scan (abnormal if urine residual volume > 100 mL), and orthostatic hypotension test (considered positive if diminution of at least 20 mmHg of systolic blood pressure or of at least 10 mmHg of diastolic blood pressure). We also paid attention for diminution of a least 30 mmHg of systemic blood pressure or of at least 15 mmHg of diastolic blood pressure within de first 3 min of the test to diagnose probable MSA. Clonidine test was applied to search for an absence of growth hormone peak as previously described [11]. Clinical examination also included the screening of extrapyramidal symptoms, oculomotor disorders, dysphagia, dysarthria, pyramidal syndrome, and peripheral neuropathy. Cognitive decline was also searched for using Montreal Cognitive Assessment (MoCA) or Mini-Mental State Examination (MMSE).
Imaging and electrophysiology
Brain MRI was performed at baseline then every year using 1,5-T or 3-T MR scanners using sagittal T1-weighted, axial T2-weighted, axial T2-proton density-weighted, axial T2 FLAIR-weighted, axial DWI-weighted, and axial T2*-weighted sequences, as previously described [12]. (I123)-FP-CIT-SPECT was performed at baseline then every year as previously described [13]. Body CT-scan was performed at baseline and repeated every 6 months if disease duration was under 3 years, to investigate a hypothetical paraneoplastic syndrome. If the suspicion of paraneoplastic syndrome was strong and the body-CT-scan negative, a PET-scan was further performed. EMG and EEG were performed at least once during the first year.
Laboratory
Biological explorations included copper, ceruloplasmin, homocysteine, vitamins B1, B6, B9 and B12, vitamin E, cholestanol, very-long-chain fatty acid (VLCFA), hexosaminidase, arylsulfatase, alpha-foeto-protein (AFP), phytanic acid dosages, search for autoimmune disease by assay of antineuronal (DOT-blot for anti-Hu, anti-Yo, anti-Ri, anti-CV2, anti-amphiphysine, anti-Ma2 antibodies), antinuclear, antiganglioside, antiphospholipid, anti-glutamic acid decarboxylase (GAD), anti-thyroid peroxidase (TPO), and anti-transglutaminase antibodies in serum. Lumbar puncture with search for oligoclonal bands, protein 14-3-3, antineuronal antibody, and PCR for T. whipplei was done in all patients at inclusion. If diagnosis was not established 1 year after the inclusion and if the phenotype was compatible with a mitochondrial disorder, a muscular biopsy was performed.
Genetic analysis
The following genes were investigated for all patients: FMR1 (Fragile X tremor and ataxia syndrome: FXTAS) and FXN (Friedreich ataxia). In case of early death of one parent or if the clinical and imaging picture was compatible, testing for genes associated with SCA 1, 2, 3, 6, 7, 17 and DRPLA were performed. According to the phenotype, we also searched for mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy with ragged-red fibers (MERRF), and genes implicated in Primary Familial Brain Calcifications (PFBC), i.e., XPR1, PDGFB, PDGFBR, SLC20A2, and MYORG.
If this first-tier analysis was negative, we performed the sequencing of a panel including 392 genes (whose list can be found in Online Resource 1) related to CA and movement disorders and we screened patients for biallelic expansion in RFC1 [14].
We calculated for each ancillary exploration the number of abnormal exams divided by the number of performed exams and estimated the relevance based on the number of patients whose exam had contributed to the diagnosis divided by the number of performed exams (the latter being defined as the yield of investigation).
Statistical analysis
We used pvalue.io software (Medistica) to perform the statistical analysis. For categorical variables, we used Chi-2 and Fisher’s exact test to test for differences between groups. For quantitative data, we used Welch test to test for differences between groups, except for of MMSE and MoCA for which we calculated the median and used therefore Mann–Whitney test to compare the groups because of non-normal distribution. The p values < 0.05 were considered as statistically significant. After adjustment with Bonferroni–Holm correction, the p values < 0.001 were considered as statistically significant.
Results
From January 2013 to December 2019, 205 patients were included in this monocentric, prospective study. Out of them, 76 patients were previously reported by Gebus et al. with shorter follow-up [15]. The mean duration of follow-up was 32.3 ± 20.8 months.
The mean age of onset was 57.9 ± 9.7 years. The mean age at inclusion was 62.6 ± 9.4 years with a mean disease duration at inclusion of 4.7 ± 5.2 years. Sex ratio M/F was 1.30. There was no history of consanguinity among our patients.
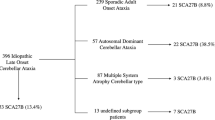
Diagnosis was established for 135 patients (66%) with 26 different diseases; 84 patients (41%) with multiple system atrophy cerebellar type (MSA-C) according to the 2008 criteria [16] (including 37 (18%) with probable and 47 (23%) with possible MSA-C) and 51 (25%) with other diagnosis. The 70 (34%) remaining patients without diagnosis despite investigations were gathered in the idiopathic late-onset cerebellar ataxia (ILOCA) group (Fig. 1).

Diagnosis distribution. MSA-C multiple system atrophy cerebellar type; ILOCAs : Idiopathic late-onset cerebellar ataxias, CJD Creutzfeldt–Jakob disease, PSP-C progressive supranuclear palsy cerebellar type, MD mitochondrial disease, PFBC primary familial brain calcifications, SCA spinocerebellar ataxia; FXTAS Fragile X tremor/ataxia syndrome, vLOFA very-late-onset Friedreich ataxia, ACD alcoholic cerebellar degeneration
The main clinical and explorations outcomes as well as the comparison between MSA-C, ILOCA, and patients with a genetic diagnosis, including Bonferroni–Holm corrections, are detailed in Table 1.
The diagnosis, main clinical, and explorations outcome of the patients are summarized in Table 2.
Three patients were diagnosed with biallelic expansions of RFC1. Those patients were positive for AAGGG PCR and negative for AAAAG and AAAGG PCR. One patient underwent videonystagmography showing bilateral symmetric vestibular hyporeflexia. Two patients had sensitive neuropathy compatible with a ganglionopathy and one had sensorimotor neuropathy with predominant-sensitive involvement. Ninety-five patients were tested with our gene panel for late-onset cerebellar ataxia. We found pathogenic variants in 4 patients (4 SPG7 with biallelic mutations, thus included in the “genetic” group) and variants of uncertain significance (VUS) in 7 patients. There were also possibly causative variants in 24 patients which could not be retained as pathogenic, because the segregation analysis of asymptomatic relatives excluded variants pathogenicity or the clinical picture was incompatible with the expected phenotype related to the mutated gene. VUS can be found on Online Resource 2.
We diagnosed a single patient with cerebellar type of Progressive Supranuclear Palsy despite the lack of both well-validated criteria and pathological confirmation, because the clinical and imaging data were compatible with this diagnosis [17].
Out of the two patients diagnosed with Sjogren’s syndrome, one fulfilled AECG criteria of 2002 [18]. The other patient received Sjogren’s syndrome diagnosis because of positive salivary gland biopsy, positive antinuclear antibody at 1/1280, and dry mouth. Three patients were diagnosed with alcoholic cerebellar degeneration, although one of those patients also presented anti-MAG neuropathy, which could have participated to the ataxic phenotype. Four patients were diagnosed with functional neurological disorders, because investigations were negative and especially because of atypical features such as distractibility or sway of the trunk with stable lower limbs when performing Romberg test.
Two patients were positive for anti-GAD antibody in serum but only one also had intrathecal anti-GAD antibody and thus diagnosed with anti-GAD encephalitis.
We showed in Fig. 2 some brain MRI features associated with CA-causes found in our cohort. We detailed in Table 3 the results, relevance, and diagnostic yields of laboratory investigation, imaging, muscle biopsy, and genetic analysis.

Brain MRI abnormalities identified in our cohort. MSA-C (a–d). Axial FLAIR-weighted MRI: Hot-cross bun sign (white arrow) and middle cerebellar peduncle hyperintensity (white dotted arrow) (a). Axial T2*-weighted MRI: bilateral putaminal hypointensity (black arrow) (b). Sagittal FLAIR-weighted MRI: severe atrophy of the pons and the cerebellum (c). Axial T2-weighted MRI: putaminal rim sign (black arrows) (d). PSP-C: Sagittal FLAIR-weighted MRI showing the humming bird sign (white arrow) (e). vLOFA: Sagittal FLAIR-weighted MRI showing the atrophy of cervical spinal cord (black arrow) (f). DRPLA: axial FLAIR-weighted MRI showing diffuse cerebral atrophy and extensive leukopathy (g). SCA3: Axial proton density-weighted MRI showing the hot-cross bun sign (h). FXTAS: Axial FLAIR-weighted MRI showing hyperintensity of the middle cerebellar peduncles (white dotted arrow) (i) and hyperintensity of the splenium of the corpus callosum (j). PFBC: Axial T2*-weighted MRI showing calcifications as hypointensity of dentate nuclei (k) and the basal ganglia (l). Superficial siderosis: Axial T2*-weighted MRI showing hemosiderin deposits by peri-axial hypointensity (m). SPG7: Axial FLAIR-weighted MRI showing bilateral hyperintensity of dentate nuclei (white arrow) (n). Progressive multiple sclerosis: axial FLAIR-weighted MRI showing multiple periventricular white matter lesions (o). CANVAS: sagittal FLAIR-weighted MRI showing cerebellar atrophy (p)
Discussion
We reported here the diagnosis work of 205 patients with SLOCA. Eighty-four patients (41%) were diagnosed with MSA-C, which was the most frequent cause by far. In a previous study, Gebus et al.[15] reported 36% (n = 29) of patients suffering from MSA-C including probable (n = 16) and possible (n = 13) MSA-C. Of the 13 possible MSA-C reported by Gebus et al., nine converted to probable MSA-C in this study. This result was in accordance with the gathering of possible MSA-C with probable MSA-C in our study. The proportion of probable MSA-C remained similar in this study (18%). Giordano et al. found about 40% of probable MSA-C in a multicentric cohort of 295 patients with SLOCA after the exclusion of established acquired causes of ataxia, which explain the smaller proportion of probable MSA-C in our cohort [19]. In the same article by Giordano et al., the annual progression of SARA for MSA-C patients was 3.3 ± 3.2 [19] which is similar to the ∆SARA (2.00 ± 3.05) but not to the SARA/DD (6.05 ± 4.95) in our study. This could be explained by the fact that SARA/DD is based on the disease duration at inclusion as reported by the patient. On the other hand, the difference between two SARA scores for one patient does not only depend on the progression of the disease, but also for instance of the inter- or intra-operator variability, the fluctuation of the patient’s condition according to the fatigue or even the effect of physical therapy. These two ways of measuring the progression of MSA-C patients are of interest and seem reliable, since they both indicate a faster progression in MSA-C patients than in ILOCA patients.
Twenty patients had a genetic diagnosis. Those patients had a slower disease course with disease duration at inclusion being longer and UPDRS/DD, SARA/DD, and SDFS/DD being lower than MSA-C patients. They were also associated with sensory neuropathy, MRI features including atrophy of cervical spinal cord, hyperintensity of the splenius of corpus callosum, and calcifications when compared with MSA-C or ILOCA patient. These clues are of interest to distinguish possible genetic causes in patients with SLOCA.
Four patients (1.9%) had CA due to SPG7 mutation. SPG7 initially reported as a frequent cause of autosomal recessive hereditary spastic paraparesis was then described in up to 19% of undiagnosed CA notably the p.Ala510Val variant [5]. On brain MRI, three of our patients with SPG7 mutations presented a cerebellar atrophy and one showed a peculiar hyperintensity of the dentate nucleus, both features being previously reported [20]. (I123)-FP-CIT-SPECT can be abnormal SPG7 patients [21], as found in one of our patient, although no parkinsonism was noted, showing the importance of this imaging not only to diagnose MSA-C but also to identify patients affected with SPG7-related CA.
Three patients (1.4%) had biallelic pathogenic expansion of RFC1. Expansion of RFC1 is assumed to be a frequent cause of SLOCA with a prevalence of 1/20 000 in general population and of 0.7% for heterozygous carrier. In a cohort of 911 adult-onset sporadic cerebellar ataxia, Syrina et al. found 29 (3.2%) patients with biallelic pathogenic expansion in RFC1 [4, 22]. Interestingly, two of our patients also had an abnormal (I123)-FP-CIT-SPECT. The phenotype of RFC1-related disorders is expanding with several studies reporting parkinsonism in patients with biallelic expansion of RFC1 [23, 24] and a case report of a dopa-responsive parkinsonism and presynaptic dopaminergic denervation in a patient with biallelic expansion of RFC1[25]. The link between MSA-C and RFC1 biallelic pathogenic mutations is still controversial [26, 27]. Search for RFC1 biallelic expansion should be systematically performed facing a patient presenting with sporadic or presumed recessively inherited late-onset cerebellar ataxia including when dysautonomia is part of the phenotype.
Three patients had a spinocerebellar ataxia (SCA), namely one with SCA3, one with SCA17, and one with DRPLA. For two of these patients, there was a history of early death of one parent but the lack of family history of CA. It is important to specifically search for triplet repeat expansion-related SCA in ataxic patients with a history of early death of a parent or with a suggestive clinical and imaging picture, because genes panel do not allow to detect triplet repeat expansions.
Herein, 70 patients (34%) remained undiagnosed despite investigations (ILOCA group) including numerous gene analysis. The gene panel performed in 95 patients found 4 SPG7. Some of these ILOCA patients could carry mutations in genes that were not explored by our CA gene panel or not yet reported in CA. For such patients, whole-exome or even whole-genome sequencing could be of interest to improve the diagnostic yield.
This study confirms and further precises the results of the previous work of Gebus et al. concerning the relevance of investigations.
The most relevant investigation with the highest yield (38%) was MRI. Some exams also appeared relevant regarding their yields, namely orthostatic hypotension test, bladder scan, and clonidine test, although the interest of the latter remains controversial [11]. (I123)-FP-CIT-SPECT also had a good relevance. We thus consider that (I123)-FP-CIT-SPECT should be performed systematically facing a SLOCA, since it gives hint for MSA-C which is the most common cause of SLOCA, and also for genetic diagnosis. Indeed, it has been shown that dopaminergic denervation can occur in several genetic diseases, such as SPG7, FXTAS, or biallelic expansion of RFC1 [21, 25]. Since parkinsonism is not easily identified in a patient with cerebellar ataxia an abnormal (I123)-FP-CIT-SPECT could also lead to test the efficacy of levodopa.
Despite having lower yields, CSF analysis and EMG remain interesting exams which should be performed in all patients. The first one gives hint for immune-mediated ataxias which belong to the treatable causes of SLOCA and are not to be missed. One may consider that deeper investigations devoted to gluten ataxia with antigliadin antibodies or to the search for primary autoimmune cerebellar ataxia could have been helpful to find more autoimmune causes of SLOCA that could be treated with immunosuppressive drugs [28]. EMG is useful to be aware of axonal sensory neuropathy which could prompt genetic testing as it was found out to be associated with genetic disease in our study.
Furthermore, some exams had a high yield, but were performed only on selected patients, including PET-scan which was performed on patients suspected of paraneoplastic syndrome, or for the genetic testing for MERRF and muscle biopsy which were performed on patients suspected of mitochondrial disease. For the latter exam, the result was often abnormal but only orientated the diagnosis in a few cases.
Regarding laboratory exploration, the analysis of VLCFA, cholestanol, hexosaminidase, arylsulfatase, phytanic acid, vitamin E, AFP, and acanthocytes did not lead to any diagnosis. Except for cholestanol, vitamin E, and phytanic acid which orientate the diagnosis toward scarce but treatable causes of CA, these exams should not be performed systematically. EEG appeared not relevant but for the diagnosis of Creutzfeldt–Jakob disease, and should not be performed systematically.
Based on the elements discussed above, we propose a diagnostic algorithm (Fig. 3). The first-line exams include brain MRI, (I123)-FP-CIT-SPECT, lumbar puncture, laboratory explorations including the treatable causes of SLOCA, search for autonomic failure with orthostatic hypotension test, and bladder scan. If available, a clonidine test can also be proposed in first line of exams, since it has a 75% negative predictive value, although its sensitivity remains low in early stages of MSA-C patients [11]. Body CT-scan and the PET-scan should be performed only if a paraneoplastic syndrome is suspected. This step aims to investigate most of the acquired causes of SLOCA as well as to find arguments for MSA-C or genetic causes.

Algorithm for sporadic late-onset cerebellar ataxia. ANA antinuclear antibodies, TG transglutaminase, ACE angiotensin-converting enzyme, GAD glutamic acid decarboxylase, TPO thyroid peroxidase, CSF Cerebrospinal fluid, HTO orthostatic hypotension test, MS multiple sclerosis, MSA-C multiple system atrophy cerebellar type, ACD alcoholic cerebellar degeneration, PCD paraneoplastic cerebellar degeneration, PFBC primary familial brain calcifications, HCB hot-cross bun, SCC splenius of the corpus callosum
The second line of exams includes genetic testing with search for RFC1/CANVAS, Friedreich ataxia, FXTAS, as well as, if necessary SCA 1, 2, 3, 6, 7, 17 and DPRLA and gene panel to search for SPG7, if there are arguments for a genetic cause based on the features detailed Table 1 or if the criteria for probable MSA-C are not fulfilled. A muscle biopsy should be performed only when facing mitochondrial disease suspicion and if the former explorations are negative.
The strengths of our study include its prospective nature, the large cohort of patients, extensive laboratory, and imaging investigations and genetic analysis as well as a long follow-up period. We included in our cohort many causes of late-onset CA and gave a comprehensive view of the underlying etiologies. Nevertheless, our study has several limitations. First, we do not have pathological data to support the diagnosis of MSA-C. Finally, about 40% of ILOCA patients did not benefit from the CA-related gene panel sequencing.
In conclusion, we diagnosed 135 (66%) of our patients with 26 heterogeneous etiologies of late-onset cerebellar ataxia -MSA being the most frequent by far—including 11 genetic causes that need to be searched for, possibly based on the algorithm we propose. When compared with MSA-C, genetic causes were associated with slower disease evolution, cervical spine atrophy hyperintensity of the splenius of corpus callosum, and calcification on brain MRI and a sensory neuropathy on EMG. Expansion of the cohort and further work are warranted to better clarify the underlying cause of ILOCA.
References
Mahlknecht P, Kiechl S, Bloem BR et al (2013) Prevalence and burden of gait disorders in elderly men and women aged 60–97 years: a population-based study. PLoS ONE 8:e69627. https://doi.org/10.1371/journal.pone.0069627
Muzaimi MB (2004) Population based study of late onset cerebellar ataxia in south east Wales. J Neurol Neurosurg Psychiatry 75:1129–1134. https://doi.org/10.1136/jnnp.2003.014662
Klockgether T (2010) Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol 9:94–104. https://doi.org/10.1016/S1474-4422(09)70305-9
Cortese A, Simone R, Sullivan R et al (2019) Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 51:649–658. https://doi.org/10.1038/s41588-019-0372-4
Pfeffer G, Pyle A, Griffin H et al (2015) SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 84:1174–1176. https://doi.org/10.1212/WNL.0000000000001369
Manto M, Gandini J, Feil K, Strupp M (2020) Cerebellar ataxias: an update. Curr Opin Neurol 33:150–160. https://doi.org/10.1097/WCO.0000000000000774
Lieto M, Roca A, Santorelli FM et al (2019) Degenerative and acquired sporadic adult onset ataxia. Neurol Sci 40:1335–1342. https://doi.org/10.1007/s10072-019-03856-w
Schmitz-Hubsch T, du Montcel ST, Baliko L et al (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66:1717–1720. https://doi.org/10.1212/01.wnl.0000219042.60538.92
Goetz CG, Tilley BC, Shaftman SR et al (2008) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results: MDS-UPDRS: Clinimetric Assessment. Mov Disord 23:2129–2170. https://doi.org/10.1002/mds.22340
Anheim M, Fleury M, Monga B et al (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics 11:1–12. https://doi.org/10.1007/s10048-009-0196-y
Bonnard C, Wirth T, Gebus O et al (2020) Clonidine GH stimulation test to differentiate MSA from idiopathic late onset cerebellar ataxia: a prospective, controlled study. J Neurol 267:855–859. https://doi.org/10.1007/s00415-020-09737-z
Carré G, Dietemann JL, Gebus O et al (2020) Brain MRI of multiple system atrophy of cerebellar type: a prospective study with implications for diagnosis criteria. J Neurol 267:1269–1277. https://doi.org/10.1007/s00415-020-09702-w
Anheim M, Lagier-Tourenne C, Stevanin G et al (2009) SPG11 spastic paraplegia: a new cause of juvenile parkinsonism. J Neurol 256:104–108. https://doi.org/10.1007/s00415-009-0083-3
Montaut S, Diedhiou N, Fahrer P et al (2021) Biallelic RFC1-expansion in a French multicentric sporadic ataxia cohort. J Neurol 268:3337–3343. https://doi.org/10.1007/s00415-021-10499-5
Gebus O, Montaut S, Monga B et al (2017) Deciphering the causes of sporadic late-onset cerebellar ataxias: a prospective study with implications for diagnostic work. J Neurol 264:1118–1126. https://doi.org/10.1007/s00415-017-8500-5
Gilman S, Wenning GK, Low PA et al (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71:670–676. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Höglinger GU, Respondek G, Stamelou M et al (2017) Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria: MDS Clinical Diagnostic Criteria for PSP. Mov Disord 32:853–864. https://doi.org/10.1002/mds.26987
Vitali C (2002) Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 61:554–558. https://doi.org/10.1136/ard.61.6.554
Giordano I, Harmuth F, Jacobi H et al (2017) Clinical and genetic characteristics of sporadic adult-onset degenerative ataxia. Neurology 89:1043–1049. https://doi.org/10.1212/WNL.0000000000004311
Hewamadduma CA, Hoggard N, O’Malley R et al (2018) Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol Genet 4:e279. https://doi.org/10.1212/NXG.0000000000000279
De la Casa-Fages B, Fernández-Eulate G, Gamez J et al (2019) Parkinsonism and spastic paraplegia type 7: expanding the spectrum of mitochondrial Parkinsonism. Mov Disord 34:1547–1561. https://doi.org/10.1002/mds.27812
Aboud Syriani D, Wong D, Andani S et al (2020) Prevalence of RFC1 -mediated spinocerebellar ataxia in a North American ataxia cohort. Neurol Genet 6:e440. https://doi.org/10.1212/NXG.0000000000000440
Traschütz A, Cortese A, Reich S et al (2021) Natural History, phenotypic spectrum, and discriminative features of multisystemic RFC1 disease. Neurology 96:e1369–e1382. https://doi.org/10.1212/WNL.0000000000011528
Matos PCAAP, Rezende TJR, Schmitt GS, et al (2021) Brain structural signature of RFC1‐related disorder. Mov Disord. https://doi.org/10.1002/mds.28711
Silva Schmitt G, Martinez ARM, Graça FF et al (2020) Dopa-responsive parkinsonism in a patient with homozygous RFC1 expansions. Mov Disord 35:1889–1890. https://doi.org/10.1002/mds.28286
Wan L, Chen Z, Wan N et al (2020) Biallelic intronic AAGGG expansion of RFC1 is related to multiple system atrophy. Ann Neurol 88:1132–1143. https://doi.org/10.1002/ana.25902
Sullivan R, Yau WY, Chelban V et al (2020) RFC1 intronic repeat expansions absent in pathologically confirmed multiple systems atrophy. Mov Disord 35:1277–1279. https://doi.org/10.1002/mds.28074
Hadjivassiliou M, Graus F, Honnorat J et al (2020) Diagnostic criteria for primary autoimmune cerebellar ataxia—guidelines from an international task force on immune-mediated cerebellar ataxias. Cerebellum 19:605–610. https://doi.org/10.1007/s12311-020-01132-8
Funding
The authors received a grant from the association “Connaître les syndromes cérébelleux” (CSC).
Author information
Authors and Affiliations
Contributions
All authors contributed to the investigation and writing—review and editing of the study. Project administration and supervision were performed by Christine Tranchant and Mathieu Anheim. Visualization and writing–original draft preparation were performed by Thomas Bogdan, Thomas Wirth, and Mathieu Anheim. Conceptualization and methodology were performed by Thomas Bogdan, Thomas Wirth, Andra Iosif, Christine Tranchant, and Mathieu Anheim. Formal analysis was performed by Thomas Bogdan and Thomas Wirth. Data curation was performed by Thomas Bogdan, Thomas Wirth, and Andra Iosif. Resources were obtained from Audrey Schalk, Jean-Baptiste Chanson, Gaël Nicolas, Jamel Chelly, Michel Koenig, Cécile Cazeneuve, Caroline Bund, Izzie-Jacques Namer, Stéphane Kremer, and Nadège Calmels. Funding acquisition was performed by Mathieu Anheim.
Corresponding author
Ethics declarations
Conflicts of interest/competing interests
The authors declare that they have no conflict of interest.
Ethical approval
Approval was obtained from the local ethics committee of Strasbourg University Hospital.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bogdan, T., Wirth, T., Iosif, A. et al. Unravelling the etiology of sporadic late-onset cerebellar ataxia in a cohort of 205 patients: a prospective study. J Neurol 269, 6354–6365 (2022). https://doi.org/10.1007/s00415-022-11253-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11253-1