
Abstract
Immune-mediated myelopathies are a heterogeneous group of inflammatory spinal cord disorders including autoimmune disorders with known antibodies, e.g. aquaporin-4 IgG channelopathy or anti-myelin oligodendrocyte glycoprotein-associated myelitis, myelopathies in the context of multiple sclerosis and systemic autoimmune disorders with myelopathy, as well as post-infectious and paraneoplastic myelopathies. Although magnetic resonance imaging of the spinal cord is still challenging due to the small dimension of the cord cross-section and frequent movement and susceptibility artifacts, recent methodological advances have led to improved diagnostic evaluation and characterization of immune-mediated myelopathies. Topography, length and width of the lesion, gadolinium enhancement pattern, and changes in morphology over time help in narrowing the broad differential diagnosis. In this review, we give an overview of recent advances in magnetic resonance imaging of immune-mediated myelopathies and its role in the differential diagnosis and monitoring of this heterogeneous group of disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immune-mediated myelopathies are a heterogeneous group of inflammatory spinal cord (SC) disorders including autoimmune disorders with known pathogenic autoantibodies, e.g. aquaporin-4 (AQP4) immunoglobulin (Ig)G positive neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein (MOG) IgG associated disease, myelopathies that are thought to be immune-mediated without known specific antibodies (e.g. multiple sclerosis), systemic autoimmune disorders with myelopathy (e.g. sarcoidosis), as well as post-infectious and paraneoplastic myelopathies.
Clinical presentation varies depending on the location and extension of the lesions within the SC and the type of cell affected and comprises a wide spectrum ranging from subtle symptoms such as isolated Lhermitte phenomenon without further sensory or motor deficits to quadriplegia with severe autonomic dysfunction. The rapid recognition of the underlying cause of the myelopathy is essential since the timely initiation of appropriate treatment significantly influences long-term outcome.
MR imaging of the SC is challenging due to the inhomogeneous magnetic field environment of the cord, its small cross-sectional dimensions, and artifacts due to physiological motion of the cord and adjacent structures. The application of appropriate shimming techniques, the choice of suitable pulse sequences and image orientation as well as employment of gating techniques can help to address some of these challenges [1]. Imaging the spinal cord at higher field strength improves the signal to noise, and therefore, allows for higher resolution of the small cross-sectional dimensions per given scanning time. Whether imaging at 3 T confers additional diagnostic or prognostic value in immunemediated myelopathies compared to 1.5 T is, however, still controversial [2,3,4,5].
SC MRI can help in differentiating and characterizing immune-mediated myelopathies in terms of signal change topography, lesion length and width, gadolinium enhancement pattern, and evolution over time to narrow the broad differential diagnosis of these myelopathies. The aim of this review is to give an overview of recent advances in MR imaging of immune-mediated myelopathies and its role in the differential diagnosis of this heterogeneous group of disorders.
Multiple sclerosis
Multiple sclerosis (MS) is the most common immune-mediated disorder of the central nervous system (CNS) with a progressive evolution of demyelinating lesions and atrophy. Severe clinical symptoms, e.g. restricted ambulation and bladder dysfunction, result mainly from SC involvement. More than 80% of patients with newly diagnosed MS show T2-hyperintense SC lesions on sagittal MRI. SC lesions due to demyelination can usually be differentiated from those that are related to other inflammatory disorders or vascular disease, since meningeal involvement, vertically spreading lesions over several segments and horizontally spreading lesions involving a significant part of the SC are atypical for MS [6]. MS lesions are typically short (less than 3 vertebral segments), multifocal and primarily located in the cervical cord (Fig. 1a–c) [7, 8]. However, involvement of the thoracolumbar region can be seen in up to 40% of cases [6, 8, 9]. MS-related SC lesions are mostly located in the dorsal or lateral columns, present rather asymmetrically and do not respect the gray and white matter boundaries [10]. Recommendations of the magnetic resonance imaging in MS (MAGNIMS) Consortium suggest to perform two sets of sagittal images with different contrasts (e.g. dual-echo T2/Proton density or short tau inversion recovery) of the whole cord at a minimum field strength of 1.5 T as part of the diagnostic work-up [2]. An additional axial plane should be added to increase diagnostic certainty when T2-hyperintensities in the sagittal plane are inconclusive [2, 8, 9]. In particular, diffuse hyperintensities, a common feature in patients with primary progressive disease type, are well depicted in the center of the cord cross-section [8, 11, 12]. Ring-like gadolinium enhancement may be present in lesions; however, gadolinium enhancement is also often associated with clinical MS symptoms and cord swelling [13]. Therefore, the application of contrast media in disease monitoring of MS cord changes is a matter of debate. Yet, in the work-up of differential diagnoses (see below) a 2D or 3D T1-weighted (T1-w) scan after contrast media is still mandatory [2, 14]. For a long time, the frequency of focal T1-hypointensities in the SC of MS patients has been underestimated [15]. However, higher field strength and better resolution of 3D images now enable improved detection of chronic T1-hypointense lesions within the cervical cord particularly in patients with progressive MS, with moderate correlation between T1-hypointense lesion count and disability [5]. MRI of the SC shows prognostic value in MS as well. While the presence of SC lesions may be associated with a worse prognosis in relapsing-remitting MS (RRMS) [16,17,18], in radiologically or clinically isolated syndromes the presence of cord lesions predicts the conversion into definite MS [19,20,21]. Nevertheless, the correlation between focal cord demyelination and disability is weak. A more promising biomarker could be SC atrophy that can be easily assessed on conventional 3D T1-w scans. SC atrophy can be detected in all stages of the disease [22]. In patients with RRMS, SC volume loss correlates with the number of relapses [23]. In progressive cases the evaluation of SC atrophy seems to be especially meaningful [24,25,26] with smaller upper cervical cord area (UCCA) and faster atrophy rates in progressive versus relapse-onset patients. Moreover, in patients with progressive MS the extent of atrophy correlates with clinical impairment and acts as an independent predictor of disease progression [25,26,27]. Conventional MRI unfortunately only allows for the quantification of overall SC volume or cross-sectional area [28,29,30,31]. Novel, advanced magnetic imaging techniques, such as phase-sensitive inversion recovery (PSIR) imaging [32] and averaged magnetization inversion recovery acquisition (AMIRA) imaging [33] now allow for improved contrast between gray and white matter in the SC. Application of the former revealed cervical SC gray matter atrophy in MS patients even if signs of white matter atrophy were missing. SC gray matter atrophy correlates well with clinical disability and disease course [34, 35]. Furthermore, quantitative MRI techniques including the measurement of the myelin water fraction and the myelin thickness may give additional information about the disease severity and progression [36, 37]. While SC imaging in MS is currently used for diagnostic work-up at disease onset [2], the value of monitoring disease using it at regular intervals is still debated [38, 39]. In particular, measurements of SC atrophy have the potential to be part of future clinical care and monitoring of disease progression [23].

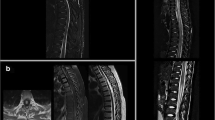
a–c Example of an MS-related myelitis in a 28-year-old patient. a Circumscribed, round T2-hyperintense lesion at the level C4 (arrow). b focal contrast enhancement (arrow). c Asymmetric and eccentric location on axial T2-w images. d–g LETM in a 76-year old patient with AQP4-positive NMOSD. d Diffuse longitudinal T2- hyperintensity and swelling of the SC between C4 and T5 accentuated at the levels T3-5. e The axial view of the SC at the intervertebral disc level T3/T4 shows central cord involvement with (f) contrast enhancement. g Six months follow-up MRI with signs of pronounced SC atrophy, most prominent at the levels T2–4 (arrow). h–j Example of a 66-year old woman with AQP4-positive NMOSD in the context of Sjögren’s syndrome and SLE. h Irregularly shaped longitudinal T2-hyperintense lesions in the thoracic cord with i and j. bright spotty lesions on axial T2-w images. k–n Example of a sarcoidosis-associated myelitis in a 48-year-old patient with longitudinally extensive SC lesions. l, k Longitudinally extensive lesions in the medulla, cervical and thoracic SC with subependymal contrast enhancement (k, arrows). m, n. dorsal subependymal gadolinium enhancement and enhancement of the central canal (n, arrow)
Neuromyelitis optica spectrum disorders
NMOSD represent an evolving group of relapsing or monophasic demyelinating inflammatory diseases of the CNS, which are distinct from MS. They are recognized as an astrocytopathy [40] that classically involves the SC and optic nerve, but can also affect the circumventricular organs, the diencephalon and other brain regions [41]. AQP4-IgG is a pathogenic antibody that targets the astrocytic water channel aquaporin-4 and can be detected in the majority of patients fulfilling the current diagnostic criteria for NMOSD [42,43,44,45,46].
An increasing subgroup of patients diagnosed with NMOSD has, however, a negative AQP4-IgG status, challenging a unified classification of the NMOSD entity [47].
AQP4-positive NMOSD/autoimmune aquaporin-4 channelopathy
AQP4 has been shown to be highly expressed at the astrocyte end-feet in the optic nerve and the SC compared to other compartments of the CNS, explaining the characteristic distribution of NMOSD lesions with predilection sites in the SC, optic nerve, area postrema, brainstem, and diencephalon [42, 48].
One of the classical manifestations of the disease is longitudinal extensive transverse myelitis (LETM), with high risk of recurrence in patients with AQP4-IgG antibodies [49, 50]. In contrast to MS, NMOSD lesions extend over three or more vertebral segments (Fig. 1 D) [42, 51]. Takahashi et al. reported a positive correlation between the length of the SC lesion and the serum level of AQP4-antibodies [52]. The length of the lesion visible on MRI crucially depends on the timing of the imaging study. Short segment myelitis has been described to be present in 14% of antibody-positive patients early in the course of LETM [53] or during recovery. After treatment with steroids, LETM has been shown to change in morphology with the appearance of several shorter lesions in about a quarter of patients [54].
NMOSD SC lesions are predominantly located in the cervical cord, with frequent extension into the thoracic cord or into the brainstem. About 30% of patients show thoracic cord lesions [54]. 60–70% of SC lesions observed in NMOSD occupy more than half of the cord area (Fig. 1e, f). Given the high expression of AQP4 around the central canal and in SC gray matter, lesions predominantly involve the central gray matter [55] (Fig. 1e, f), but frequently extend to the pial surface on axial images [56]. Bright spotty lesions on axial T2-w images are a relatively specific finding in NMOSD [57, 58], and help differentiating NMOSD from MS (Fig. 1i, j). Central hypointensities on T1-w images, cord expansion due to swelling, and gadolinium enhancement are also frequently observed in NMOSD [58, 59]. Ring-like enhancement of lesions (as frequently observed in MS) can be seen in about 30% of patients with NMOSD myelitis [60] and therefore, does not allow for the differentiation between these disorders. In contrast to MS, asymptomatic SC lesions are only rarely reported in NMOSD with a frequency suspected to be less than 5% [61, 62].
Focal or generalized atrophy is seen in up to 50–60% of NMOSD patients with history of myelitis on follow-up MRI [54, 59] (Fig. 1g) and is of high relevance, as SC atrophy correlates well with disability and number of relapses [63]. SC atrophy is even reported in patients with AQP4-IgG positive NMOSD without prior myelitis or SC lesions [64].
While detailed MRI imaging features have been incorporated into the diagnostic criteria of NMOSD [42], an official consensus on imaging protocols for the diagnosis and monitoring of this evolving disease spectrum is still lacking.
AQP4-negative NMO
An increasing subgroup of patients with the typical clinical presentation of neuromyelitis optica have been found to be AQP4-IgG seronegative. Wingerchuk et al. [42] defined clinical and imaging requirements for the diagnosis of NMOSD in these seronegative patients with myelitis.
NMOSD patients lacking antibodies against AQP4 show similar lengths of the SC lesions [50]. Similar to AQP4-IgG positive NMOSD, lesions are most frequently located in the cervical cord [65]. The absence of AQP4-IgG antibodies has been associated with a reduced risk of LETM recurrence compared to seropositive patients [66].
Novel autoantibody-positive disorders with myelitis
Recently, in a subgroup of AQP4-negative patients, a new antibody against the myelin oligodendrocytes glycoprotein (MOG) was detected, binding to the outer surface of the oligodendrocytes [67]. Not only does the clinical course of these patients differ from the classical NMO syndrome as they may have a more favorable outcome, but also the magnetic images of the SC show distinct characteristics [68]. Longitudinally extensive SC lesions occur frequently, but short lesions have been observed in 44% of cases [69]. In MOG-positive myelitis, SC lesions frequently occur in the thoracolumbar region [65, 70] and can predominantly involve the gray matter [71]. Necrosis, cavitation and atrophy of the SC are rarely present [72]. The recurrence of LETM seems to be infrequent [66]. Recently, extended MRI brain and SC lesion criteria were published and help to further differentiate between AQP4 and MOG-positive NMOSD from MS with a 100% sensitivity and 80–90% specificity [73].
Another novel autoimmune neurologic disorder with antibodies against the glial fibrillary acidic protein (GFAP) has been described lately. The GFAP α isoform is considered to be a pan-astrocytic marker, whereas the ε and κ isoforms are found only in neural progenitor cells and immature astrocytes, primarily located in the periventricular region, the hippocampus, and in the central area of the SC [74, 75]. 22% of the patients with GFAP-antibodies showed signs of myelitis or encephalomyelitis [75]. The SC involvement is mostly longitudinal; however, lesions have been observed to be hazier with a thin and linear enhancement along the central canal. The minority of these patients have co-existing AQP4 antibodies [75]. Since an underlying malignant disease was shown to be present in 25% of patients with involvement of the nervous system, the presence of GFAP-antibody may indicate a paraneoplastic autoimmune origin in a subgroup of patients.
Infection-associated autoimmune myelitis
Post-infectious myelitis develops as a delayed immune-mediated response occurring within 4 weeks of a microbial infection in or mostly outside the CNS. Several mechanisms have been postulated to be relevant in the pathophysiology of post-infectious myelitis, such as molecular mimicry, bystander activation and super-antigens, which trigger the immune-mediated attack against SC tissue [76].
Following an infection or vaccination, acute LETM can occur as part of acute disseminated encephalomyelitis (ADEM), a predominantly monophasic inflammatory disorder with multifocal perivascular demyelinating lesions most commonly seen in children with an incidence of 0.4–0.8/100,000 [77]. Typically, it occurs after a preceding mild infection of the upper respiratory tract or an unspecific febrile state, though many viral and bacterial pathogens have been described in association with ADEM [77]. The involvement of the SC with the development of an extensive SC lesion has been reported to occur in about one-third of ADEM cases, sharing similar MRI-characteristics with patients having NMOSD [78, 79]. Rarely, ADEM is followed by the occurrence of NMO (defined as ADEM-NMO) [78]. Interestingly, in children with ADEM, MOG-antibodies were shown to be present in about 25–40% [80] and the prevalence of LETM in this group of patients reached 90% [72]. Postvaccinal ADEM is usually monophasic; however, some patients with MOG-positive postvaccinal ADEM follow a relapsing course [69].
Similarly, although spatially more confined than ADEM, NMOSD can occur in association with preceding infections in up to 30% of the cases [81]. Most commonly it is linked to viral infections caused by varicella zoster, or if bacterial, often caused by Mycobacterium tuberculosis [82]. Typically, it presents as LETM and shows gadolinium enhancement in the cervicothoracic region of the SC.
Myelitis associated with rheumatological diseases/systemic diseases
Myelitis associated with systemic lupus erythematodes
Systemic lupus erythematosus (SLE) is a systemic autoimmune connective tissue disease that may affect the cardiovascular and pulmonary system, the skin, joints, liver, kidneys, and nervous system. Transverse myelitis among SLE patients is rare with a prevalence of about 0.9-2%, but can be potentially serious [83,84,85]. The immunopathological features of SLE associated myelitis are not well characterized with only few pathological reports of fulminant cases available [86, 87] highlighting marked SC vasculitis, secondary infarction and necrosis. However, myelitis can occur in patients with otherwise clinically inactive SLE [88], suggesting that SC disease may be due to an inflammatory demyelinating process rather than a vascular event. The co-existence of SLE and NMOSD has been observed in several cohorts [89]. Given the pathogenicity and high specificity of the AQP4 antibody, AQP4-IgG seropositivity in myelitis patients with SLE indicates a different pathogenesis, which is, however, based on a SLE intrinsic B cell hyperreactivity [42, 89].
Spinal MRI in SLE associated myelitis typically shows LETM accompanied by cord swelling [90]. The cervical–mid-lower thoracic segments are most frequently involved [91]. In severe cases, the lesion may involve the entire SC and spread into the medulla [86, 92]. SLE-associated myelitis may be further differentiated into two groups: gray matter myelitis (defined clinically by flaccidity and hyporeflexia) with a devastating, mostly monophasic course with prominent cord swelling on MRI; and white matter myelitis (defined by spasticity and hyperreflexia) with a less drastic, more frequently recurrent course [93]. 81% of SLE patients with white matter myelitis in this study fulfilled the revised 2006 diagnostic criteria for NMO [94] or presented as NMOSD [89], while only 18% of the patients with gray matter myelitis did. In line with this, AQP4-IgG positivity was observed in only 12.5% with gray matter myelitis. Importantly, gray matter myelitis occurred in the context of SLE disease activity, which was less frequently observed in white matter myelitis [93]. Oiwa and colleagues [95] confirmed in a systematic review (including the cases of Birnbaum and colleagues) a higher rate of AQP4-antibodies in white matter and a higher rate of anti-ds-DNA antibodies in gray matter myelitis, proposing that white matter myelitis may be a complication of NMOSD in a subset of patient, while gray matter myelitis might be a more direct consequence of SLE [95].
Myelitis associated with antiphospholipid syndrome
Antiphospholipid syndrome (APS) is an autoimmune-mediated syndrome characterized by venous and/or arterial thrombosis, recurrent miscarriages and the persistent presence of antiphospholipid antibodies. APS can occur as a primary disease or secondary to other, mostly autoimmune diseases (e.g. SLE). The most common neurological complications are strokes and transient ischemic attacks due to hypercoagulopathy that rarely also involve the SC [96].
Transverse myelitis is a rare complication reported in less than 1% of patients with antiphospholipid syndrome [97], and its pathogenesis is still poorly understood. Lesions most commonly occur in the thoracic cord [98, 99] and may show patchy T2-hyperintensity and white matter degeneration as well as cord swelling [100]. In a recent study, 46% of patients with LETM and 100% of patients with recurrent LETM fulfilling the revised criteria of APS were found to be AQP4-IgG seropositive [101, 102]. Given the high specificity of AQP4-IgG, these results suggest concomitant NMOSD as the primary cause of the myelitis and thus the need of NMOSD specific treatment in these patients [102].
Myelitis associated with Behçet disease
Behçet disease (BD) is a systemic vasculitis that affects both arteries and veins of all vessel sizes and causes venous thrombosis [103]. Myelitis is a rare complication of BD, typically presenting as LETM, sometimes involving the entire length of the SC [104]. Cord swelling and T2-hyperintense lesions are typically present in the acute phase [105]. Contrast enhancement is not a typical feature, being present in some but not all patients. On axial MR images, a central lesion with a hypointense core and hyperintense rim with or without gadolinium contrast enhancement may be observed: the “bagel sign”[106]. Neuro-Behçet may take a relapsing remitting or a progressive course. In patients with progressive BD, SC atrophy is common [107].
Sjögren-associated myelitis
Sjögren’s syndrome (SS) is a systemic autoimmune-mediated disorder that primarily affects the salivary and lacrimal glands through mononuclear infiltration and destruction, causing the typical sicca symptoms. SC involvement is reported in 20–35% of SS patients and may present as acute myelitis or chronic progressing myelopathy [108,109,110]. In about 36% of cases with initial neurological manifestations, myelitis was the presenting symptom [110]. Lesions are typically longitudinally extensive and are located in the cervical cord. Some patients additionally present with optic neuritis and/or cerebral lesions and fulfil the diagnostic criteria of concomitant MS [110]. Similarly to SLE, the association of SS and NMOSD has been reported in various publications: the clinical diagnosis of SS may coexist with NMOSD clinical syndromes in AQP4-IgG positive patients (Fig. 1h–j) [89, 111,112,113].
Sarcoidosis-associated myelitis
Sarcoidosis is a multisystemic granulomatous non-caseous disorder that most commonly affects the respiratory system, skin and lymph nodes. 5–10% of patients show an involvement of the peripheral and/or CNS [114, 115]. Autopsy studies show a high rate of subclinical neurosarcoidosis in 10–27% of cases [116, 117]. Neurosarcoidosis is most commonly associated with granulomatous infiltrates involving the meninges, hypothalamus, pituitary gland and cranial nerves. SC sarcoidosis is relatively rare and can result in intramedullary lesions, intradural extramedullary or extradural lesions, cauda equina syndrome or arachnoiditis [118].
Sarcoidosis-associated myelitis often presents as a LETM that can affect both the cervical and thoracic cord in isolation or as panmyelitis [119, 120]. The typical MRI finding of sarcoidosis-associated myelitis is a longitudinal extensive T2-hyperintense lesion, most commonly located in the dorsal and centrodorsal cord (Fig. 1l). Dorsal subpial enhancement [121] (Fig. 1k) in combination with central canal enhancement (Fig. 1n) can result in a “trident sign” on axial images [122] (Fig. 1m) and can help distinguishing sarcoidosis myelitis lesions from NMOSD lesions [123]. Moreover, an anterior and posterior leptomeningeal enhancement pattern has been described [124]. Spreading to the Virchow–Robin spaces results in parenchymal involvement, which appears as diffuse cord enlargement in MRI [125].
Adequate therapy can lead to improvement of SC lesions [126], however, improvement on imaging may lag behind clinical improvement. SC enhancement often takes several months up to years to resolve, which may also help distinguishing lesions from MS/NMOSD lesions [121]. In untreated cases, repetitive inflammation can result in SC atrophy [125, 126].
Paraneoplastic myelitis
Paraneoplastic neurological disorders are thought to result from an immune response against tumor antigens that are also present in mature cells of the nervous system, including neurons and glia, either intracellularly or in the plasma membrane on the cell surface. While paraneoplastic antibodies targeting cell-surface antigens are thought to be directly pathogenic, the role of antibodies targeting intracellular antigens is less clear [127].
Paraneoplastic myelopathy, though very rare, occurs with various malignancies, most frequently breast and lung cancers [128]. Detection of neural-specific auto-antibodies confirms the diagnosis and helps guide the cancer search. Among the most commonly detected antibodies in paraneoplastic myelopathies are amphiphysin-IgG, anti-neuronal nuclear antibodies (ANNA)-2, -3 and collapsin response mediator protein-5-IgG (CRMP5/anti-CV2) [129]. More recently, paraneoplastic myelopathy has also been recognized in the context of AQP4-IgG antibodies. In a large cohort of AQP4-IgG positive NMOSD, 3.2% showed associated malignancies, the majority being adenocarcinoma of the lung or breast cancer [130]. Female patients and those over the age of 50 years seem to be particular at risk for a paraneoplastic form of NMOSD [131, 132].
SC MRI in paraneoplastic myelitis shows characteristic symmetric, tract-specific T2-hyperintense lesions that extend often over multiple vertebral segments and can be seen best on axial images. Gadolinium enhancement is frequent [127]. Most commonly affected are the lateral columns, but a dorsal column and gray matter involvement has also been described [129]. The symmetric tract-specific and gray matter involvement can sometimes mimic the “owl eye” appearance observed in ischemic lesions [129]. In paraneoplastic anti-AQP4 antibody myelitis, lesions may present as typical NMO LETM lesions with patchy gadolinium enhancement [131]. Tract-specific MRI changes also occur in nutritional deficiencies, (e.g. vitamin B 12 deficiency), however, usually do not show contrast enhancement. Myelopathy can occur simultaneously with the neoplasia or precede any tumor-related symptoms.
Conclusion
The term “immune-mediated myelopathies” comprises a heterogeneous group of evolving disease entities. Recently, several pathogenic autoantibodies have been discovered that can cause longitudinal transverse myelitis along with a spectrum of other CNS manifestations, challenging the traditional concept of syndrome-based disease classification in this field. The novel disease entities that evolve out of these discoveries still need further clinical and paraclinical characterization.
Magnetic resonance imaging is an important tool in the paraclinical in-vivo morphological description of these novel disease entities and helps, together with CSF data and autoantibody markers in the diagnostic work-up and classification of these diseases including those without known pathognomonic antibodies. Moreover, MR imaging is necessary to exclude alternative diagnoses. Table 1 summarizes our current understanding of the morphological MR hallmarks regarding SC lesion location, lesion length and shape, tract involvement, and contrast enhancement pattern of the most common immune-mediated spinal cord disease entities. While for some disease entities distinctive radiographic signs have been proposed, such as, e.g. the Bagel sign in Behcet disease [106] and the trident sign in neurosarcoidosis [122], for the majority of diseases, not one pathognomic sign, but rather a pattern of different morphological characteristics is postulated. Though the different entities might share some similarities on SC MRI despite their distinct origin; numerous unique imaging characteristics remain that help to narrow down the eligible differential diagnoses (Fig. 2). The diagnostic sensitivity and specificity, as well as pathological validation of these radiographic patterns, however, still require further study.

Schematic representations of characteristic myelopathy lesions. a, b MS lesions: asymmetric, located in the dorsal/lateral columns, involving gray and white matter. c NMOSD lesion: occupying a large part of the cord area; centrally located, involving gray and white matter. d Sarcoidosis-associated lesion: “trident sign”, i.e. dorsal subpial enhancement and enhancement of the central canal. e Behçet-associated lesion: “bagel sign”, i.e. central lesion with hypointense core and hyperintense rim. f Paraneoplastic myelitis-associated lesion: tract-specific T2-hyperintensities
References
Stroman PW, Wheeler-Kingshott C, Bacon M et al (2014) The current state-of-the-art of spinal cord imaging: methods. Neuroimage 84:1070–1081
Rovira À, Wattjes MP, Tintoré M et al (2015) MAGNIMS study group. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis-clinical implementation in the diagnostic process. Nat Rev Neurol 11:471–482
Wattjes MP, Harzheim M, Kuhl CK et al (2006) Does high-field MR imaging have an influence on the classification of patients with clinically isolated syndromes according to current diagnostic mr imaging criteria for multiple sclerosis? AJNR Am J Neuroradiol 27:1794–1798
Stankiewicz JM, Neema M, Alsop DC et al (2009) Spinal cord lesions and clinical status in multiple sclerosis: a 1.5 T and 3 T MRI study. J Neurol Sci 279:99–105
Valsasina P, Aboulwafa M, Preziosa P et al (2018) Cervical cord T1-weighted hypointense lesions at MR imaging in multiple sclerosis: relationship to cord atrophy and disability. Radiology 288:234–244
Bot JC, Barkhof F (2009) Spinal-cord MRI in multiple sclerosis: conventional and nonconventional MR techniques. Neuroimaging Clin N Am 19:81–99
Bot JC, Barkhof F, Polman CH et al (2004) Spinal cord abnormalities in recently diagnosed MS patients: added value of spinal MRI examination. Neurology 62:226–233
Weier K, Mazraeh J, Naegelin Y et al (2012) Biplanar MRI for the assessment of the spinal cord in multiple sclerosis. Mult Scler 18:1560–1569
Filippi M, Rocca MA, Ciccarelli O et al (2016) MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol 15:292–303
Tartaglino LM, Friedman DP, Flanders AE et al (1995) Multiple sclerosis in the spinal cord: MR appearance and correlation with clinical parameters. Radiology 195:725–732
Kearney H, Altmann DR, Samson RS et al (2015) Cervical cord lesion load is associated with disability independently from atrophy in MS. Neurology 84:367–373
Kearney H, Miszkiel KA, Yiannakas MC et al (2016) Grey matter involvement by focal cervical spinal cord lesions is associated with progressive multiple sclerosis. Mult Scler 22:910–920
Thorpe JW, Kidd D, Moseley IF et al (1996) Serial gadolinium-enhanced MRI of the brain and spinal cord in early relapsing-remitting multiple sclerosis. Neurology 46:373–378
Wattjes MP, Raab P (2017) Brain and spinal cord MRI in multiple sclerosis: an update. Neurol Int Open 1:e294–e306
Gass A, Filippi M, Rodegher ME et al (1998) Characteristics of chronic MS lesions in the cerebrum, brainstem, spinal cord, and optic nerve on T1-weighted MRI. Neurology 50:548–550
D’Amico E, Patti F, Leone C et al (2016) Negative prognostic impact of MRI spinal lesions in the early stages of relapsing-remitting multiple sclerosis. Mult Scler J Exp Transl Clin 9:2:2055217316631565
Coret F, Bosca I, Landete L et al (2010) Early diffuse demyelinating lesion in the cervical spinal cord predicts a worse prognosis in relapsing-remitting multiple sclerosis. Mult Scler 16:935–941
Dekker I, Sombekke MH, Witte BI et al (2018) Asymptomatic spinal cord lesions do not predict the time to disability in patients with early multiple sclerosis. Mult Scler 24:481–490
Okuda DT, Mowry EM, Cree BA et al (2011) Asymptomatic spinal cord lesions predict disease progression in radiologically isolated syndrome. Neurology 76:686–692
Sombekke MH, Wattjes MP, Balk LJ et al (2013) Spinal cord lesions in patients with clinically isolated syndrome: a powerful tool in diagnosis and prognosis. Neurology 80:69–75
Arrambide G, Rovira A, Sastre-Garriga J et al (2018) Spinal cord lesions: a modest contributor to diagnosis in clinically isolated syndromes but a relevant prognostic factor. Mult Scler 24:301–312
Kearney H, Miller DH, Ciccarelli O (2015) Spinal cord MRI in multiple sclerosis—diagnostic, prognostic and clinical value. Nat Rev Neurol 11:327–338
Tsagkas C, Magon S, Gaetano L et al (2018) Spinal cord volume loss: a marker of disease progression in multiple sclerosis. Neurology 91:e349–e358
Lukas C, Sombekke MH, Bellenberg B et al (2013) Relevance of spinal cord abnormalities to clinical disability in multiple sclerosis: MR imaging findings in a large cohort of patients. Radiology 269:542–552
Aymerich FX, Auger C, Alonso J et al (2018) Cervical cord atrophy and long-term disease progression in patients with primary-progressive multiple sclerosis. AJNR Am J Neuroradiol 39:399–404
Tsagkas C, Magon S, Gaetano L et al (2018) Preferential spinal cord volume loss in primary progressive multiple sclerosis. Mult Scler. https://doi.org/10.1177/1352458518775006
Lukas C, Knol DL, Sombekke MH et al (2015) Cervical spinal cord volume loss is related to clinical disability progression in multiple sclerosis. J Neurol Neurosurg Psychiatry 86:410–418
Losseff NA, Webb SL, O’Riordan JI et al (1996) Spinal cord atrophy and disability in multiple sclerosis. A new reproducible and sensitive MRI method with potential to monitor disease progression. Brain 119:701–708
Pezold S, Amann M, Weier K et al (2014) A semi-automatic method for the quantification of spinal cord atrophy. In: Yao J, Klinder T, Li S (eds) Computational methods and clinical applications for spine imaging, Lecture notes in computational vision and biomechanics, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-07269-2_13
De Leener B, Taso M, Cohen-Adad J et al (2016) Segmentation of the human spinal cord. Magma 29:125–153
Amann M, Pezold S, Naegelin Y et al (2016) Reliable volumetry of the cervical spinal cord in MS patient follow-up data with cord image analyzer (Cordial). J Neurol 263:1364–1374
Papinutto N, Schlaeger R, Panara V et al (2015) 2D phase-sensitive inversion recovery imaging to measure in vivo spinal cord gray and white matter areas in clinically feasible acquisition times. J Magn Reson Imaging 42:698–708
Weigel M, Bieri O (2018) Spinal cord imaging using averaged magnetization inversion recovery acquisitions. Magn Reson Med 79:1870–1881
Schlaeger R, Papinutto N, Panara V et al (2014) Spinal cord gray matter atrophy correlates with multiple sclerosis disability. Ann Neurol 76:568–580
Schlaeger R, Papinutto N, Zhu AH et al (2015) Association between thoracic spinal cord gray matter atrophy and disability in multiple sclerosis. JAMA Neurol 72:897–904
Laule C, Vavasour IM, Zhao Y et al (2010) Two-year study of cervical cord volume and myelin water in primary progressive multiple sclerosis. Mult Scler 16:670–677
Duval T, Le Vy S, Stikov N et al (2016) g-Ratio weighted imaging of the human spinal cord in vivo. Neuroimage 145:11–23
Kearney H (2018) Clinical monitoring of multiple sclerosis should routinely include spinal cord imaging—no. Mult Scler. https://doi.org/10.1177/1352458518770279
Cortese R, Ciccarelli O (2018) Clinical monitoring of multiple sclerosis should routinely include spinal cord imaging—yes. Mult Scler 1:1352458518778010
Fujihara K (2011) Neuromyelitis optica and astrocytic damage in its pathogenesis. J Neurol Sci 306:183–187
Pittock SJ, Lucchinetti CF (2016) Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann N Y Acad Sci 1366:20–39
Wingerchuk DM, Banwell B, Bennett JL et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189
Lennon VA, Wingerchuk DM, Kryzer TJ et al (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112
Lennon VA, Kryzer TJ, Pittock SJ et al (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Wingerchuk DM, Lennon VA, Lucchinetti CF et al (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6:805–815
Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11:535–544
Marignier R, Cobo Calvo A, Vukusic S (2017) Neuromyelitis optica and neuromyelitis optica spectrum disorders. Curr Opin Neurol 30:208–215
Matiello M, Schaefer-Klein J, Sun D et al (2013) Aquaporin 4 expression and tissue susceptibility to neuromyelitis optica. JAMA Neurol 70:1118–1125
Weinshenker BG, Wingerchuk DM, Vukusic S et al (2006) Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 59:566–569
Kitley J, Leite MI, Küker W et al (2013) Longitudinally extensive transverse myelitis with and without aquaporin 4 antibodies. JAMA Neurol 70:1375–1381
Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S et al (2015) MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 84:1165–1173
Takahashi T, Fujihara K, Nakashima I et al (2007) Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130:1235–1243
Flanagan EP, Weinshenker BG, Krecke KN et al (2015) Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 72:81–87
Asgari N, Skejoe HP, Lillevang ST el al (2013) Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): a population-based, descriptive study. BMC Neurol 13:33
Nakamura M, Miyazawa I, Fujihara K et al (2008) Preferential spinal central gray matter involvement in neuromyelitis optica. An MRI study. J Neurol 255:163–170
Kister I, Johnson E, Raz E (2016) Specific MRI findings help distinguish acute transverse myelitis of neuromyelitis optica from spinal cord infarction. Mult Scler Relat Disord 9:62–67
Yonezu T, Ito S, Mori M et al (2014) “Bright spotty lesions” on spinal magnetic resonance imaging differentiate neuromyelitis optica from multiple sclerosis. Mult Scler 20:331–337
Pekcevik Y, Mitchell CH, Mealy MA et al (2016) Differentiating neuromyelitis optica from other causes of longitudinally extensive transverse myelitis on spinal magnetic resonance imaging. Mult Scler 22:302–311
Cassinotto C, Deramond H, Olindo S et al (2009) MRI of the spinal cord in neuromyelitis optica and recurrent longitudinal extensive myelitis. J Neuroradiol 36:199–205
Zalewski NL, Morris PP, Weinshenker BG et al (2017) Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 88:218–225
Zecca C, Disanto G, Sormani MP et al (2016) Relevance of asymptomatic spinal MRI lesions in patients with multiple sclerosis. Mult Scler 22:782–791
Flanagan EP, Weinshenker BG, Krecke KN et al (2015) Asymptomatic myelitis in neuromyelitis optica and autoimmune aquaporin-4 channelopathy. Neurol Clin Pract 5:175–177
Liu Y, Wang J, Daams M et al (2015) Differential patterns of spinal cord and brain atrophy in NMO and MS. Neurology 84:1465–1472
Ventura RE, Kister I, Chung S et al (2016) Cervical spinal cord atrophy in NMOSD without a history of myelitis or MRI-visible lesions. Neurol Neuroimmunol Neuroinflamm 3:e224
Sato DK, Callegaro D, Lana-Peixoto MA et al (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82:474–481
Jitprapaikulsan J, Lopez Chiriboga AS et al (2018) Novel glial targets and recurrent longitudinally extensive transverse myelitis. JAMA Neurol 75:892–895
Pröbstel AK, Rudolf G, Dornmair K et al (2015) Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflamm 12:46
Kitley J, Woodhall M, Waters P et al (2012) Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 79:1273–1277
Jarius S, Ruprecht K, Kleiter I et al (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm 13:280
Kitley J, Waters P, Woodhall M et al (2014) Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 71:276–283
Wang C, Narayan R, Greenberg B (2018) Anti-myelin oligodendrocyte glycoprotein antibody associated with gray matter predominant transverse myelitis mimicking acute flaccid myelitis: a presentation of two cases. Pediatr Neurol 86:42–45
Narayan R, Simpson A, Fritsche K et al (2018) MOG antibody disease: a review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 25:66–72
Bensi C, Marrodan M, González A et al (2018) Brain and spinal cord lesion criteria distinguishes AQP4-positive neuromyelitis optica and MOG-positive disease from multiple sclerosis. Mult Scler Relat Disord 25:246–250
Roelofs RF, Fischer DF, Houtman SH et al (2005) Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia 52:289–300
Flanagan EP, Hinson SR, Lennon VA et al (2017) Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol 81:298–309
Goh C, Desmond PM, Phal PM (2014) MRI in transverse myelitis. J Magn Reson Imaging 40:1267–1279
Sonneville R, Klein I, de Broucker T et al (2009) Post-infectious encephalitis in adults: diagnosis and management. J Infect 58:321–328
Pohl D, Alper G, Van Haren K et al (2016) Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology 87:S38–S45
Callen DJ, Shroff MM, Branson HM et al (2009) Role of MRI in the differentiation of ADEM from MS in children. Neurology 72:968–973
Pröbstel AK, Dornmair K, Bittner R et al (2011) Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 77:580–588
Mealy MA, Wingerchuk DM, Greenberg BM et al (2012) Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol 69:1176–1180
Sellner J, Hemmer B, Mühlau M (2010) The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J Autoimmun 34:371–379
Piga M, Chessa E, Peltz MT et al (2017) Demyelinating syndrome in SLE encompasses different subtypes: do we need new classification criteria? Pooled results from systematic literature review and monocentric cohort analysis. Autoimmun Rev 16:244–252
Saison J, Costedoat-Chalumeau N, Maucort-Boulch D et al (2015) Systemic lupus erythematosus-associated acute transverse myelitis: manifestations, treatments, outcomes, and prognostic factors in 20 patients. Lupus 24:74–81
Kovacs B, Lafferty TL, Brent LH et al (2000) Transverse myelopathy in systemic lupus erythematosus: an analysis of 14 cases and review of the literature. Ann Rheum Dis 59:120–124
Katramados AM, Rabah R, Adams MD et al (2008) Longitudinal myelitis, aseptic meningitis, and conus medullaris infarction as presenting manifestations of pediatric systemic lupus erythematosus. Lupus 17:332–336
Tono T, Nagai T, Hoshiyama T et al (2016) Transverse myelitis extended to disseminated encephalitis in systemic lupus erythematosus: histological evidence for vasculitis. Mod Rheumatol 26:958–962
Li XY, Xiao P, Xiao HB et al (2014) Myelitis in systemic lupus erythematosus frequently manifests as longitudinal and sometimes occurs at low disease activity. Lupus 23:1178–1186
Pittock SJ, Lennon VA, de Seze J et al (2008) Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 65:78–83
Téllez-Zenteno JF, Remes-Troche JM, Negrete-Pulido RO et al (2001) Longitudinal myelitis associated with systemic lupus erythematosus: clinical features and magnetic resonance imaging of six cases. Lupus 10:851–856
Espinosa G, Mendizábal A, Mínguez S et al (2010) Transverse myelitis affecting more than 4 spinal segments associated with systemic lupus erythematosus: clinical, immunological, and radiological characteristics of 22 patients. Semin Arthritis Rheum 39:246–256
Neumann-Andersen G, Lindgren S (2000) Involvement of the entire spinal cord and medulla oblongata in acute catastrophic-onset transverse myelitis in SLE. Clin Rheumatol 19:156–160
Birnbaum J, Petri M, Thompson R et al (2009) Distinct subtypes of myelitis in systemic lupus erythematosus. Arthritis Rheum 60:3378–3387
Wingerchuk DM, Lennon VA, Pittock SJ et al (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66:1485–1489
Oiwa H, Kuriyama A, Matsubara T et al (2018) Clinical value of autoantibodies for lupus myelitis and its subtypes: a systematic review. Semin Arthritis Rheum 14:214–220
Flanagan EP, McKeon A, Weinshenker BG (2014) Anterior spinal artery infarction causing man-in-the-barrel syndrome. Neurol Clin Pract 4:268–269
Cervera R, Piette JC, Font J et al (2002) Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 46:1019–1027
Sherer Y, Hassin S, Shoenfeld Y et al (2002) Transverse myelitis in patients with antiphospholipid antibodies—the importance of early diagnosis and treatment. Clin Rheumatol 21:207–210
Mori A, Nodera H, Nakane S et al (2010) Transverse myelitis and polymyositis associated with antiphospholipid antibody syndrome. Clin Neurol Neurosurg 112:713–716
Rodrigues CE, de Carvalho JF (2011) Clinical, radiologic, and therapeutic analysis of 14 patients with transverse myelitis associated with antiphospholipid syndrome: report of 4 cases and review of the literature. Semin Arthritis Rheum 40:349–357
Miyakis S, Lockshin MD, Atsumi T et al (2006) International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 4:295–306
Guerra H, Pittock SJ, Moder KG et al (2018) Frequency of aquaporin-4 immunoglobulin G in longitudinally extensive transverse myelitis with antiphospholipid antibodies. Mayo Clin Proc 93:1299–1304
Greco A, De Virgilio A, Ralli M et al (2018) Behçet’s disease: New insights into pathophysiology, clinical features and treatment options. Autoimmun Rev 17:567–575
Tang Q, Tian J (2016) Longitudinal myelitis of a neuro-Behçet patient. Arch Rheumatol 31:91–93
Mohamed C, Najib K, Essaadouni L (2015) Radiological findings in Behçet disease. Pan Afr Med J 20:51
Uygunoglu U, Zeydan B, Ozguler Y et al (2017) Myelopathy in Behçet’s disease: the Bagel sign. Ann Neurol 82:288–298
Borhani Haghighi A, Sarhadi S, Farahangiz S (2011) MRI findings of neuro-Behcet’s disease. Clin Rheumatol 30:765–770
Alexander EL, Provost TT, Stevens MB et al (1982) Neurological complications of primary Sjogren’s syndrome. Medicine (Baltimore) 61:247–257
Alexander EL (1986) Central nervous system (CNS) manifestations of primary Sjogren’s syndrome: an overview. Scan J Rheumatol Suppl 61:161–165
Delalande S, de Seze J, Fauchais AL et al (2004) Neurologic manifestations in primary Sjögren syndrome: a study of 82 patients. Medicine (Baltimore) 83:280–291
Kim SM, Waters P, Vincent A (2009) Sjogren’s syndrome myelopathy: spinal cord involvement in Sjogren’s syndrome might be a manifestation of neuromyelitis optica. Mult Scler 15:1062–1068
Jayarangaiah A, Sehgal R, Epperla N (2014) Sjögren’s syndrome and neuromyelitis optica spectrum disorders (NMOSD)—a case report and review of literature. BMC Neurol 14:200
Wingerchuk DM, Weinshenker BG (2012) The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler 18:5–10
Stern BJ, Krumholz A, Johns C et al (1985) Sarcoidosis and its neurological manifestations. Arch Neurol 42:909–917
Judson MA, Boan AD, Lackland DT (2012) The clinical course of sarcoidosis: presentation, diagnosis, and treatment in a large white and black cohort in the United States. Sarcoidosis Vasc Diffuse Lung Dis 29:119–127
Ricker W, Clark M (1949) Sarcoidosis; a clinicopathologic review of 300 cases, including 22 autopsies. Am J Clin Pathol 19:725–749
Manz HJ (1983) Pathobiology of neurosarcoidosis and clinicopathologic correlation. Can J Neurol Sci 10:50–55
Day AL, Sypert GW (1977) Spinal cord sarcoidosis. Ann Neurol 1:79–85
Durel CA, Marignier R, Maucort-Boulch D et al (2016) Clinical features and prognostic factors of spinal cord sarcoidosis: a multicenter observational study of 20 BIOPSY-PROVEN patients. J Neurol 263:981–990
Sohn M, Culver DA, Judson MA et al (2014) Spinal cord neurosarcoidosis. Am J Med Sci 347:195–198
Flanagan EP (2016) Autoimmune myelopathies. In: Pittock SJ, Vincent A (eds) Handbook of clinical neurology, vol 133. Elsevier, Amsterdam, pp 327–351
Zalewski NL, Krecke KN, Weinshenker BG et al (2016) Central canal enhancement and the trident sign in spinal cord sarcoidosis. Neurology 87:743–744
Jolliffe EA, Keegan BM, Flanagan EP (2018) Trident sign trumps aquaporin-4-IgG ELISA in diagnostic value in a case of longitudinally extensive transverse myelitis. Mult Scler Relat Disord 23:7–8
Kumar N, Frohman EM (2004) Spinal neurosarcoidosis mimicking an idiopathic inflammatory demyelinating syndrome. Arch Neurol 61:586–589
Junger SS, Stern BJ, Levine SR, Sipos E et al (1993) Intramedullary spinal sarcoidosis: clinical and magnetic resonance imaging characteristics. Neurology 43:333–337
Christoforidis GA, Spickler EM, Recio MV et al (1999) MR of CNS sarcoidosis: correlation of imaging features to clinical symptoms and response to treatment. AJNR Am J Neuroradiol 20:655–669
Flanagan EP, Keegan BM (2013) Paraneoplastic myelopathy. Neurol Clin 31:307–318
Zalewski NL, Flanagan EP (2018) Autoimmune and paraneoplastic myelopathies. Semin Neurol 38:278–289
Flanagan EP, McKeon A, Lennon VA et al (2011) Paraneoplastic isolated myelopathy: clinical course and neuroimaging clues. Neurology 76:2089–2095
Sepúlveda M, Sola-Valls N, Escudero D et al (2017) Clinical profile of patients with paraneoplastic neuromyelitis optica spectrum disorder and aquaporin-4 antibodies. Mult Scler. https://doi.org/10.1177/1352458517731914
Cai G, He D, Chu L et al (2016) Paraneoplastic neuromyelitis optica spectrum disorders: three new cases and a review of the literature. Int J Neurosci 126:660–668
Ontaneda D, Fox RJ (2014) Is neuromyelitis optica with advanced age of onset a paraneoplastic disorder? Int J Neurosci 124:509–511
Acknowledgements
We thank Dorothee Heinrich for her help and expertise in the graphic representation of different myelopathy lesions and Dr. Matthias Weigel and Prof. Oliver Bieri, Division of Radiological Physics, Department of Radiology, University Hospital Basel, and Department of Biomedical Engineering, University of Basel, Basel, CH for contributing the SC AMIRA image.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Ethical considerations/informed consent
Written consent for the use of MR images was obtained from all patients prior to the inclusion in our review.
Rights and permissions
About this article

Cite this article
Wendebourg, M.J., Nagy, S., Derfuss, T. et al. Magnetic resonance imaging in immune-mediated myelopathies. J Neurol 267, 1233–1244 (2020). https://doi.org/10.1007/s00415-019-09206-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09206-2