
Abstract
Corpus callosum abnormality (CCA) outcomes are quite unpredictable and variable, from asymptomatic forms to mild or severe neurodevelopment disorders. The aim of this study was to examine clinical outcomes in CCA patients. The study included 61 children and adolescents in whom brain magnetic resonance imaging (MRI) scans showed CCA, isolated or associated to other central nervous system lesions. All patients underwent anamnesis, physical and neurological examination, routine laboratory tests, electroencephalogram (EEG), and MRI scans. In all participants, the intelligence quotient (IQ) was determined. We divided the participants into two subgroups: the first subgroup included patients with an isolated CCA, and the second subgroup included patients with CCA associated with extra-callosal brain lesions (complex CCA). We found that CCA were associated with elevated frequency to intellectual disability (ID), other neurodevelopment disorders, epilepsy, and isolated EEG anomalies. Mild ID (p = 0.003) was more frequent in the isolated subgroup, while epilepsy (p = 0.036) and pre-perinatal risk factors (p = 0.023) were more frequent in the complex CCA subgroup. Although the role of the CC in the interhemispheric communication is known, neurological and neurodevelopment outcomes of CCA are extremely variable and unpredictable. The presence of extra-callosal brain anomalies is one of the major prognostic factor, and probably, they have an important impact on the clinical outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The corpus callosum (CC) is a unique placental mammal structure. It is the largest of the interhemispheric white matter tracts in the brain, connecting neocortex areas between the two cerebral hemispheres [1–3].
The CC is divided into four anatomically defined regions: the rostrum, the genu, the body, and the splenium. It consists of approximately 200 million axons (2–3 % of all cortical fibers) topographically organized, establishing homotopic and heterotopic connections between several neocortex areas [4, 5]. The anterior sections of the CC connect more anterior regions of the cortex (prefrontal association areas, premotor, supplementary areas, and anterior inferior parietal regions); the more posterior sections connect areas of the parietal, temporal and occipital lobes [6]. Callosal connections are both excitatory and inhibitory, but the majority of fibers are excitatory [7].
In humans, CC development begins by week 8 of fetal life [8]. The number of callosal fibers is more or less determined at birth, but structural changes continue throughout post-natal development, most of all during childhood and adolescence. The CC fibers originate from neurons of layers II/III, V, and VI of the neocortex [9], while pioneering axons originate from the cingulate cortex [10]. Recent neuroimaging and neuroembryology studies show that at 13–14 weeks of post-conceptional age (PCA), callosal fibers begin to cross the midline, and then, they grow bidirectionally during weeks 18 and 19. By 20 weeks of PCA, CC shows its final shape, although exuberant axonal growth continues until 2 months after birth. Usually, post-natal maturation of the splenium precedes the genu one [3, 11, 12].
CC development is influenced by a series of complex and highly regulated events (cellular proliferation, migration, post-migrational maturation, myelinization, and axons pruning). Corpus callosum abnormalities (CCA) are the result of an anomaly of these events caused by intrauterine exposure to teratogens, metabolic disorders, genetic aberrations and several syndromes [13–16]. About 20 % of CCA are caused by single or multiple genes mutations or chromosomal aberrations [3, 15]. Autosomal dominant, autosomal recessive, and X-linked causes of CCA have been described. Many cases are apparently sporadic, so it is possible that a significant proportion of CCA cases are caused by de novo mutation [17]. New genetic microarray analysis improved the possibility to detect specific loci associated with agenesis of the CC, and recently about 30 new genomic loci have been described [18]. Because of phenotypic variability, the identification of a specific genetic cause of CCA is quite difficult.
CCA have an estimated prevalence of 0.3–0.7 % in patients undergoing neuroimaging in the general population [19–21].
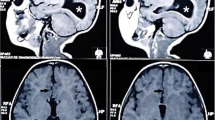
Structural CCA can be classified by the images of magnetic resonance imaging (MRI) scans and include the agenesis of the CC and the hypoplasia of the CC. The total agenesis of the CC (tACC) is a complete absence from birth of all the anatomically regions of the CC; the partial agenesis of the CC (pACC) is the partial absence from birth of at least one region of the CC; the hypoplasia of the CC (HCC) is a CC thinner but with a normal extent; hypoplasia may interest one or all regions of the CC (respectively, partial or harmonic hypoplasia) [22, 23]. Other CCA include hyperplasia and dysgenesis. Hyperplasia refers to a CC thicker than normal and it results from reduced postnatal axonal pruning; dysgenesis refers to a CC developed but malformed in one or more regions [24].
CCA may be isolated or occur in association with additional cerebral lesions and/or congenital anomalies (cortical dysplasia, neuronal migration disorders, and brainstem anomalies) [16, 21, 25, 26].
Although prenatal imaging (ultrasound) may help with a CCA diagnosis, clinical outcomes in patients with CCA are quite unpredictable and variable, from asymptomatic forms to mild or severe neurodevelopment disorders [6, 27, 28]. The aim of this study was to examine clinical outcomes in CCA patients.
Methods
Population
In this observational study, we retrospectively analyzed a sample of 61 children and adolescents admitted to the Child and Adolescent Neuropsychiatric Unit of Bari and the Department of Pediatrics of the University Chieti and of the University of L’Aquila, during the period between 2009 and 2016, because of neuropsychiatric symptoms and in whom magnetic resonance imaging (MRI) scans showed CCA, isolated or associated to other SNC lesions.
We divided the participants into two subgroups: the first subgroup included patients with an isolated CCA and the second subgroup included patients with CCA associated with extra-callosal brain lesions (complex CCA). This study was approved by the Local Ethic Committee of Azienda Ospedaliero-Universitaria Policlinico di Bari, all children were recruited after obtaining written informed consent by their parents; in addition, informed consent was also obtained from the patients who could understand the content and aim of study.
Assessment
All patients underwent anamnesis (familiar, physiological, pre-perinatal, pathological, and academic), physical and neurological examination, routine laboratory tests, including blood count, liver, and renal functions, metabolic laboratory tests (ammonemia, aminoacidemia and aminoaciduria), sleep and awake electroencephalogram (EEG). MRI study was performed with a 1.5 T magnet and 8 channels coil for brain. In all participants, the intelligence quotient (IQ) was determined by Wechsler Intelligence Scale for Children–Fourth Edition (WISC-IV) [29]; Leiter International Performances Scale Revised-Visualization and Reasoning battery (Leiter-R) [30] was administered, as an alternative to WISC-IV, to subjects with verbal disorders.
Statistical analysis
The demographical and clinical variables underwent statistical analysis. We calculated proportions and differences between the subgroup of patients with isolated CCA and the subgroup including patients with complex CCA using a Fisher’s exact test and Mann–Whitney U test (e.g., age) to calculate p values. Results having p < 0.05 were considered significant. Calculations were performed using SPSS version 20.0 (IBM SPSS Statistics, IBM Inc., NY, USA).
Results
The patients involved in this study were 61 (male 47.5 %; female 52.5 %). The mean age was 8.8 ± 4.3 years (range 1–17 years). Three patients (4.9 %) showed a complete agenesis of the CC (tACC), seven patients (13 %) showed a partial agenesis (pACC), and 49 patients (82 %) showed a hypoplasia interesting uniformly its portions (HCC). Table 1 are summarized clinical and demographical data about all the participants. Intellectual disability (ID) was evident in 44 patients (72 %): a mild ID was found in 16 patients (26.2 %), a moderate ID was revealed in 15 patients (24.6 %), and a severe ID was found in 13 patients (21.3 %). A single neurodevelopment disorder was present in 28 patients (47.5 %): language disorders were present in seven patients (11.5 %), autism spectrum disorders (ASD) were found in four patients (6.6 %), attention deficit hyperactivity disorder (ADHD) was present in two patients (3.3 %), and developmental coordination disorder was present in one patient (1.6 %). More than one neurodevelopment disorder was found in 21 patients (35.6 %). Epilepsy was present in 22 patients (36 %): a partial epilepsy in 10 patients (45.4 %), a partial with secondary generalization epilepsy in 6 patients (27.3 %), and a generalized epilepsy in 6 patients (27.3 %). Pre/perinatal risk factors were present in 26 patients (42.6 %). Isolated EEG anomalies were present in 14 patients (23 %), with variable patterns. Cerebral palsy was detected in two patients (3.3 %). A syndromic diagnosis was found in ten patients (16.4 %): a Noonan syndrome in one patient, a Sotos syndrome in one patient, an Arnold–Chiari malformation in one patient, a Dandy–Walker malformation in one patient, an incontinentia pigmenti in two patients, a Smith–Lemli–Opitz syndrome in two patients, and an unknown plurimalformative syndrome in two patients.
Isolated CCA vs. complex CCA
In 16 patients (26.2 %), the CCA was isolated; in 45 patients (73.8 %), the CCA was associated with other SNC lesions, including cerebral cortex malformations (cortical dysplasia, neuronal migration disorders, Arnold–Chiari malformation, and Dandy–Walker malformation), cerebellum hypoplasia, and perinatal hypoxic-ischemic damages. The two subgroups were homogeneous by age (p = 0.266) and sex (p = 0.524). Clinical characteristics of each subgroup are summarized in Table 2. The isolated CCA was found only in the hypoplasia of the CC. ID was present in the 81.3 % of the isolated CCA subgroup (mild ID was present in the 56.6 %, moderate ID was present in the 18.8 %, and severe ID was present in the 12.5 %) and in the 68.9 % of the complex CCA subgroup (mild ID was present in the 15.6 %, moderate ID in the 26.7 %, and severe ID in the 24.4 %). The isolated CCA subgroup was more characterized by mild ID level (p = 0.003) compared with complex CCA subgroup. We found also a statistical significant difference in presence of epilepsy (p = 0.036) and pre-perinatal risk factors (p = 0.023) between subgroups. Epilepsy and pre-perinatal risk factors were more frequent in the complex CCA compared with isolated CCA subgroup. No statistical significant difference was found for the other emerged data including isolated EEG anomalies, moderate and severe ID and other neurodevelopment disorders. Table 2 shows comparison and differences between the two subgroups.
Discussion
CCA are frequently associated with neurological conditions (epilepsy, cerebral palsy, and movement disorders) and neurodevelopment disorders (ID, ASD, developmental coordination disorder, language disorders, learning disorders, and ADHD) [6, 24, 31]. Clinical consequences of CCA are quite unpredictable and wide ranging. The co-presence of other brain anomalies and the etiology may influence the clinical outcome [27, 28]. Even when the neuroradiological and neuroanatomical findings are similar, the clinical consequences of CCA are variable [21]. Therefore, this variability could be influenced by differences in neuronal compensatory plasticity, precise anatomy, presence of other clinical comorbidities, genetic background, or by environmental factors.
In our sample, we found that CCA were associated in high frequency to intellectual disability, other neurodevelopment disorders, epilepsy, and isolated EEG anomalies.
The role of the CC in the cognitive functions is well documented in literature. Recent studies show that the absence of a complete development of the CC interferes with the intra-interhemispheric functional interactions between brain areas involved in some cognitive processes (executive functions, processing speed and problem solving abilities) [31]. However, CCA do not seem to have actually a dramatic or direct impact on general cognitive abilities. Recently, some authors [27] report that neurodevelopment outcome of individuals diagnosed antenatally with an ACC (with no additional postnatal MRI anomalies) can range in a normal development in about 75 % of cases to different levels of intellectual disability; in this series, about 12 % of individuals had severe intellectual disability. Moreover, the neuropsychological outcome is not clearly linked to the severity of the CCA; individuals with CCA have more consistently deficits in complex information processing abilities, such as “cognitive information processing” (the ability to automatically perform previously learnt cognitive tasks), complex attention and memory skills, and specific academic skills, especially mathematics [24]. However, the patients with CCA, even with a normal IQ, have subtle behavioral and social problem [31–33]. In this study, the intellectual disability was present in 72.1 % of all participants. Mild ID prevailed in the isolated CCA subgroup with a statistical significant difference; moderate and severe ID prevailed in the subgroup of the complex CCA, with no statistical significant difference. Therefore, the associated brain lesions may be responsible of the severity of ID. Other neurodevelopment disorders were frequent in our cohort (47.5 % of patients had at least one disorder), with a higher incidence in the complex CCA subgroup. The more frequent disorders were language disorders, ASD, ADHD, developmental coordination disorder, and cerebral palsy. The association of CCA and neurodevelopment disorders is well described in literature. First, studies focused on inter-hemispheric transfer suggested a relationship between CC malformations, language disorders, and motor coordination disorder. This hypothesis was based on the observation that several children with CCA showed also language impairment and motor coordination deficits [34]. More recent functional neuroimaging studies support the hypothesis that, in patients with CCA, there is a correlation between the alterations of the integration of sensorial and motor processes realized by the interhemispheric transmission of the CC and the development of both language disorders and developmental coordination disorders [6]. The involvement of the CC in autism is supported by several literature data. Many studies show that in ASD, there is a reduction of the total or partial volume of the CC. Structural meta-analysis studies [35] and diffusion tensor imaging (DTI) with tractographic reconstruction meta-analysis studies [36, 37] in autistic patients support the hypothesis of a CC structural connectivity dysfunction [6, 38]. First, children with ADHD and dyslexia were detected a significant reduction of genu and splenium of the CC [39]. Successive studies confirmed also these findings in children and adolescent with isolated ADHD [40, 41]. These alterations of the splenium of the CC in ADHD patients were also reported in recent studies of functional neuroimaging based on MRI with DTI technique [42, 43].
Structural alterations of the CC are well reported in epilepsy syndromes with childhood onset [41, 44–46], such as temporal lobe [47] and neocortical epilepsy [48]. In our study, epilepsy was present in the 36 % of the participants and it was more frequent in the complex CCA subgroup than in the isolated CCA subgroup with a statistical significant difference. As reported in a recent review, CCA are not indicative for seizure disorders; in fact, seizures generally hint to an additional pathology. Since white matter structures include no firing neurons they cannot act as epileptic foci [26]. Therefore, the extra-callosal lesions, such as the cortical dysplasia, may be the origin of the epileptic focus and the CC may be involved only in the diffusion of the abnormal electrical activity from one hemisphere to the contralateral hemisphere [26, 49]. A recent study confirmed the genu of the CC as the major pathway for seizure generalization [50]. We found isolated EEG anomalies not associated to epilepsy in the 22 % of our patients and these findings may confirm the role of the CC in the diffusion of interhemispheric abnormal electrical activity. On the other hand, in both subgroups, these EEG anomalies did not present specific patterns as already reported in the literature [49]. Furthermore, pre-perinatal risk factors were present in the 42.6 % of the participants and they were more frequent in the complex CCA subgroup than in the isolated CCA subgroup, with a statistical significant difference. According to literature data, children born pre-term and with a low birth weight have frequently widespread damages of white matter, including a reduction of the total volume of the CC and microstructural alterations of the CC [51–54].
Bias of our data may be linked to small sample sizes, short follow‐up time, lack of consistency in neuropsychological measures, heterogeneity of sampled individuals, and lack of appropriate control subgroups.
Conclusions
The development of the CC is the result of sophisticated processes and regulated events, from the proliferation of the precursor cells to migration and post-migration maturation pathways. These complex mechanisms may be interrupted in different stages of the CC development by several factors (genetic anomalies or environmental factors), resulting in different kind of CCA. However, it remains unclear whether the CCA follow an initial failure of connections to establish correct organization, or a later loss of successfully formed brain connections. Even if the role of the CC in the interhemispheric communication is known, neurological and neurodevelopment outcomes of CCA are extremely variable and unpredictable. The presence of extra-callosal brain anomalies is one of the major prognostic factor, and probably, they have an important impact on the clinical outcome. Future clinical, genetic, and neuroimaging studies may be helpful to better understand CCA clinical outcomes.
Abbreviations
- ACC:
-
Agenesis of the corpus callosum
- ADHD:
-
Attention deficit hyperactivity disorder
- ASD:
-
Autism spectrum disorders
- CC:
-
Corpus callosum
- CCA:
-
Corpus callosum abnormalities
- DTI:
-
Diffusion tensor imaging
- EEG:
-
Electroencephalogram
- HCC:
-
Hypoplasia of the corpus callosum
- ID:
-
Intellectual disability
- IQ:
-
Intelligence quotient
- MRI:
-
Magnetic resonance imaging
- pACC:
-
Partial agenesis of the corpus callosum
- PCA:
-
Post-conceptional age
- tACC:
-
Total agenesis of the corpus callosum
- WISC-IV:
-
Wechsler intelligence scale for children–fourth edition, Italian version
References
Innocenti GM (1986) Postnatal development of corticocortical connections. Ital J Neurol Sci Suppl 5:25–28
Gazzaniga MS (2000) Cerebral specialization and interhemispheric communication: does the corpus callosum enable the human condition? Brain J Neurol 123(Pt 7):1293–1326
Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ (2014) Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain J Neurol 137(Pt 6):1579–1613. doi:10.1093/brain/awt358
Wahl M, Lauterbach-Soon B, Hattingen E, Jung P, Singer O, Volz S, Klein JC, Steinmetz H, Ziemann U (2007) Human motor corpus callosum: topography, somatotopy, and link between microstructure and function. J Neurosci Off J Soc Neurosci 27(45):12132–12138. doi:10.1523/JNEUROSCI.2320-07.2007
Wahl M, Strominger Z, Jeremy RJ, Barkovich AJ, Wakahiro M, Sherr EH, Mukherjee P (2009) Variability of homotopic and heterotopic callosal connectivity in partial agenesis of the corpus callosum: a 3T diffusion tensor imaging and Q-ball tractography study. AJNR Am J Neuroradiol 30(2):282–289. doi:10.3174/ajnr.A1361
Paul LK (2011) Developmental malformation of the corpus callosum: a review of typical callosal development and examples of developmental disorders with callosal involvement. J Neurodev Disord 3(1):3–27. doi:10.1007/s11689-010-9059-y
Bloom JS, Hynd GW (2005) The role of the corpus callosum in interhemispheric transfer of information: excitation or inhibition? Neuropsychol Rev 15(2):59–71. doi:10.1007/s11065-005-6252-y
Rakic P, Yakovlev PI (1968) Development of the corpus callosum and cavum septi in man. J Comp Neurol 132(1):45–72. doi:10.1002/cne.901320103
Fame RM, MacDonald JL, Macklis JD (2011) Development, specification, and diversity of callosal projection neurons. Trends Neurosci 34(1):41–50. doi:10.1016/j.tins.2010.10.002
Rash BG, Richards LJ (2001) A role for cingulate pioneering axons in the development of the corpus callosum. J Comp Neurol 434(2):147–157
Hofer S, Frahm J (2006) Topography of the human corpus callosum revisited—comprehensive fiber tractography using diffusion tensor magnetic resonance imaging. NeuroImage 32(3):989–994. doi:10.1016/j.neuroimage.2006.05.044
Luders E, Thompson PM, Toga AW (2010) The development of the corpus callosum in the healthy human brain. J Neurosci Off J Soc Neurosci 30(33):10985–10990. doi:10.1523/JNEUROSCI.5122-09.2010
Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D (1999) X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet 86(4):331–337
Sztriha L, Johansen JG, Al-Gazali LI (2005) Extreme microcephaly with agyria-pachygyria, partial agenesis of the corpus callosum, and pontocerebellar dysplasia. J Child Neurol 20(2):170–172
Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, Sherr EH (2007) Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci 8(4):287–299. doi:10.1038/nrn2107
Schell-Apacik CC, Wagner K, Bihler M, Ertl-Wagner B, Heinrich U, Klopocki E, Kalscheuer VM, Muenke M, von Voss H (2008) Agenesis and dysgenesis of the corpus callosum: clinical, genetic and neuroimaging findings in a series of 41 patients. Am J Med Genet Part A 146A(19):2501–2511. doi:10.1002/ajmg.a.32476
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367(20):1921–1929. doi:10.1056/NEJMoa1206524
O’Driscoll MC, Black GC, Clayton-Smith J, Sherr EH, Dobyns WB (2010) Identification of genomic loci contributing to agenesis of the corpus callosum. Am J Med Genet Part A 152A(9):2145–2159. doi:10.1002/ajmg.a.33558
Grogono JL (1968) Children with agenesis of the corpus callosum. Dev Med Child Neurol 10(5):613–616
Jeret JS, Serur D, Wisniewski KE, Lubin RA (1987) Clinicopathological findings associated with agenesis of the corpus callosum. Brain Dev 9(3):255–264
Al-Hashim AH, Blaser S, Raybaud C, MacGregor D (2016) Corpus callosum abnormalities: neuroradiological and clinical correlations. Dev Med Child Neurol 58(5):475–484. doi:10.1111/dmcn.12978
Hanna RM, Marsh SE, Swistun D, Al-Gazali L, Zaki MS, Abdel-Salam GM, Al-Tawari A, Bastaki L, Kayserili H, Rajab A, Boglarka B, Dietrich RB, Dobyns WB, Truwit CL, Sattar S, Chuang NA, Sherr EH, Gleeson JG (2011) Distinguishing 3 classes of corpus callosal abnormalities in consanguineous families. Neurology 76(4):373–382. doi:10.1212/WNL.0b013e318208f492
Neal JB, Filippi CG, Mayeux R (2015) Morphometric variability of neuroimaging features in children with agenesis of the corpus callosum. BMC Neurol 15:116. doi:10.1186/s12883-015-0382-5
Palmer EE, Mowat D (2014) Agenesis of the corpus callosum: a clinical approach to diagnosis. Am J Med Genet Part C Semin Med Genet 166C(2):184–197. doi:10.1002/ajmg.c.31405
Tang PH, Bartha AI, Norton ME, Barkovich AJ, Sherr EH, Glenn OA (2009) Agenesis of the corpus callosum: an MR imaging analysis of associated abnormalities in the fetus. AJNR Am J Neuroradiol 30(2):257–263. doi:10.3174/ajnr.A1331
Unterberger I, Bauer R, Walser G, Bauer G (2016) Corpus callosum and epilepsies. Seizure 37:55–60. doi:10.1016/j.seizure.2016.02.012
Sotiriadis A, Makrydimas G (2012) Neurodevelopment after prenatal diagnosis of isolated agenesis of the corpus callosum: an integrative review. Am J Obstet Gynecol 206(4):337. e331–337. e335. doi:10.1016/j.ajog.2011.12.024
Santo S, D’Antonio F, Homfray T, Rich P, Pilu G, Bhide A, Thilaganathan B, Papageorghiou AT (2012) Counseling in fetal medicine: agenesis of the corpus callosum. Ultrasound Obstet Gynecol Off J Int Soc Ultrasound Obstet Gynecol 40(5):513–521. doi:10.1002/uog.12315
Wechsler D (2004) The Wechsler intelligence scale for children—fourth edition. Pearson, London
Roid GH, Miller LJ (1997) Leiter international performance scale—revised: examiner’s manual. Stoelting Co, Wood Dale
Hinkley LB, Marco EJ, Findlay AM, Honma S, Jeremy RJ, Strominger Z, Bukshpun P, Wakahiro M, Brown WS, Paul LK, Barkovich AJ, Mukherjee P, Nagarajan SS, Sherr EH (2012) The role of corpus callosum development in functional connectivity and cognitive processing. PLoS One 7(8):e39804. doi:10.1371/journal.pone.0039804
Siffredi V, Anderson V, Leventer RJ, Spencer-Smith MM (2013) Neuropsychological profile of agenesis of the corpus callosum: a systematic review. Dev Neuropsychol 38(1):36–57. doi:10.1080/87565641.2012.721421
Lau YC, Hinkley LB, Bukshpun P, Strominger ZA, Wakahiro ML, Baron-Cohen S, Allison C, Auyeung B, Jeremy RJ, Nagarajan SS, Sherr EH, Marco EJ (2013) Autism traits in individuals with agenesis of the corpus callosum. J Autism Dev Disord 43(5):1106–1118. doi:10.1007/s10803-012-1653-2
Njiokiktjien C, Valk J, Ramaekers G (1988) Malformation or damage of the corpus callosum? A clinical and MRI study. Brain Dev 10(2):92–99
Frazier TW, Hardan AY (2009) A meta-analysis of the corpus callosum in autism. Biol Psychiatry 66(10):935–941. doi:10.1016/j.biopsych.2009.07.022
Travers BG, Adluru N, Ennis C, Tromp do PM, Destiche D, Doran S, Bigler ED, Lange N, Lainhart JE, Alexander AL (2012) Diffusion tensor imaging in autism spectrum disorder: a review. Autism Res 5(5):289–313. doi:10.1002/aur.1243
Aoki Y, Abe O, Nippashi Y, Yamasue H (2013) Comparison of white matter integrity between autism spectrum disorder subjects and typically developing individuals: a meta-analysis of diffusion tensor imaging tractography studies. Mol Autism 4(1):25. doi:10.1186/2040-2392-4-25
Barbeau EB, Lewis JD, Doyon J, Benali H, Zeffiro TA, Mottron L (2015) A greater involvement of posterior brain areas in interhemispheric transfer in autism: fMRI, DWI and behavioral evidences. NeuroImage Clin 8:267–280. doi:10.1016/j.nicl.2015.04.019
Hynd GW, Hall J, Novey ES, Eliopulos D, Black K, Gonzalez JJ, Edmonds JE, Riccio C, Cohen M (1995) Dyslexia and corpus callosum morphology. Arch Neurol 52(1):32–38
Valera EM, Faraone SV, Murray KE, Seidman LJ (2007) Meta-analysis of structural imaging findings in attention-deficit/hyperactivity disorder. Biol Psychiatry 61(12):1361–1369. doi:10.1016/j.biopsych.2006.06.011
Hutchinson E, Pulsipher D, Dabbs K, y Gutierrez AM, Sheth R, Jones J, Seidenberg M, Meyerand E, Hermann B (2010) Children with new-onset epilepsy exhibit diffusion abnormalities in cerebral white matter in the absence of volumetric differences. Epilepsy Res 88(2–3):208–214. doi:10.1016/j.eplepsyres.2009.11.011
Pastura G, Doering T, Gasparetto EL, Mattos P, Araujo AP (2015) Exploratory analysis of diffusion tensor imaging in children with attention deficit hyperactivity disorder: evidence of abnormal white matter structure. Atten Defic Hyperact Disord. doi:10.1007/s12402-015-0185-y
Dougherty CC, Evans DW, Myers SM, Moore GJ, Michael AM (2016) A comparison of structural brain imaging findings in autism spectrum disorder and attention-deficit hyperactivity disorder. Neuropsychol Rev 26(1):25–43. doi:10.1007/s11065-015-9300-2
Taylor M, David AS (1998) Agenesis of the corpus callosum: a United Kingdom series of 56 cases. J Neurol Neurosurg Psychiatry 64(1):131–134
Groppel G, Gallmetzer P, Prayer D, Serles W, Baumgartner C (2009) Focal lesions in the splenium of the corpus callosum in patients with epilepsy. Epilepsia 50(6):1354–1360. doi:10.1111/j.1528-1167.2008.01800.x
Kim SE, Lee JH, Chung HK, Lim SM, Lee HW (2014) Alterations in white matter microstructures and cognitive dysfunctions in benign childhood epilepsy with centrotemporal spikes. Eur J Neurol 21(5):708–717. doi:10.1111/ene.12301
Meng L, Xiang J, Kotecha R, Rose D, Zhao H, Zhao D, Yang J, Degrauw T (2010) White matter abnormalities in children and adolescents with temporal lobe epilepsy. Magn Reson Imaging 28(9):1290–1298. doi:10.1016/j.mri.2010.03.046
Kim H, Harrison A, Kankirawatana P, Rozzelle C, Blount J, Torgerson C, Knowlton R (2013) Major white matter fiber changes in medically intractable neocortical epilepsy in children: a diffusion tensor imaging study. Epilepsy Res 103(2–3):211–220. doi:10.1016/j.eplepsyres.2012.07.017
Ji GJ, Zhang Z, Xu Q, Zang YF, Liao W, Lu G (2014) Generalized tonic-clonic seizures: aberrant interhemispheric functional and anatomical connectivity. Radiology 271(3):839–847. doi:10.1148/radiol.13131638
Wieshmann UC, Milinis K, Paniker J, Das K, Jenkinson MD, Brodbelt A, Crooks D, Keller SS (2015) The role of the corpus callosum in seizure spread: MRI lesion mapping in oligodendrogliomas. Epilepsy Res 109:126–133. doi:10.1016/j.eplepsyres.2014.10.023
Peterson BS, Vohr B, Staib LH, Cannistraci CJ, Dolberg A, Schneider KC, Katz KH, Westerveld M, Sparrow S, Anderson AW, Duncan CC, Makuch RW, Gore JC, Ment LR (2000) Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA 284(15):1939–1947
Nosarti C, Nam KW, Walshe M, Murray RM, Cuddy M, Rifkin L, Allin MP (2014) Preterm birth and structural brain alterations in early adulthood. NeuroImage Clin 6:180–191. doi:10.1016/j.nicl.2014.08.005
Rademaker KJ, Lam JN, Van Haastert IC, Uiterwaal CS, Lieftink AF, Groenendaal F, Grobbee DE, de Vries LS (2004) Larger corpus callosum size with better motor performance in prematurely born children. Semin Perinatol 28(4):279–287
Counsell SJ, Edwards AD, Chew AT, Anjari M, Dyet LE, Srinivasan L, Boardman JP, Allsop JM, Hajnal JV, Rutherford MA, Cowan FM (2008) Specific relations between neurodevelopmental abilities and white matter microstructure in children born preterm. Brain J Neurol 131(Pt 12):3201–3208. doi:10.1093/brain/awn268
Acknowledgments
We thank the families for their contribution to this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the Local Ethic Committee of Azienda Ospedaliero-Universitaria Policlinico of Bari, all children were recruited after obtaining written informed consent by their parents; in addition, informed consent was also obtained from the patients who could understand the content and aim of study.
Conflicts of interest
The authors declare that they have no conflict of interests.
Funding
No fundings were provided for this study. The authors declare that they have no financial relationship with any sponsoring organization.
Rights and permissions
About this article

Cite this article
Margari, L., Palumbi, R., Campa, M.G. et al. Clinical manifestations in children and adolescents with corpus callosum abnormalities. J Neurol 263, 1939–1945 (2016). https://doi.org/10.1007/s00415-016-8225-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8225-x