
Abstract
Both homo- (causing autosomal-recessive Gaucherʼs disease; GD) and heterozygous mutations in the glucocerebrosidase gene (GBA) are associated with Parkinson’s disease (PD), and represent the most robust known genetic susceptibility factors identified in PD. Since the accumulation of α-synuclein has been considered critical to the pathogenesis of PD among several possible pathways through which glucocerebrosidase (GCase) deficiency may promote the pathogenesis of PD, particular attention was given to the reciprocity with α-synuclein levels, lysosomal dysfunction, endoplasmatic reticulum–Golgi trafficking of GCase, dysregulation of calcium homeostasis and mitochondrial abnormalities. The proportion of PD patients that carry GBA mutations is estimated to be approximately between 5 and 10 %. Individual PD patients with or without GBA mutations cannot be discriminated on clinical or pathological grounds. However, GBA mutation carriers may have slightly earlier age at PD onset, more likely have a positive family history for PD, and more prevalent non-motor symptoms when compared to those patients who are not carriers. Establishing the concept of GBA-related PD promoted a search for the pathogenic mechanisms through which GCase deficiency may influence pathogenesis of PD, suggesting that targeting the GCase–lysosomal pathway might be a rational approach for the development of neuroprotective drugs in PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Both homo- (causing autosomal-recessive Gaucherʼs disease; GD) and heterozygous mutations in the glucocerebrosidase gene (GBA) are associated with Parkinson’s disease (PD), and represent the most robust known genetic susceptibility factor identified in PD [1, 2]. Neudorfer et al. [3] in 1996 reported six patients with GD type 1 who developed progressive parkinsonism refractory to conventional antiparkinsonian therapy, although GD patients with some features of parkinsonism were sporadically reported as early as 1939 [4]. In 2004 it has been shown that mutations in the GBA gene were more frequent in PD patients than in controls, and that heterozygous carriers of the GBA mutation might have an increased risk to develop PD [5, 6]. The main confirmation of such association was the extensive multicentric study (5691 PD patients and 4898 controls) that found the odds ratio of 5.43 for detecting any GBA mutation in PD patients compared to the controls [7].
Proposed mechanism for GBA-associated parkinsonism
Glucocerebrosidase (GCase) is a lysosomal enzyme that hydrolyzes β-glycosyl linkage of glucosylceramide (GlcCer) to glucose and ceramide [8, 9]. Different mechanisms have been proposed in attempt to elucidate common pathological links between GBA mutations and PD, including glia-mediated inflammatory response and α-synuclein accumulation. Astrocytosis and microglial activation were described both in human GD brain samples, as well as in Gba-deficient mouse model of neuronopathic GD [10–12]. The latter model demonstrated that glial cells’ activation preceded neuronal loss in affected brain regions and that the level of inflammatory mediators correlated with disease progression [11]. Futhermore, recent reviews provided extensive analysis of the cellular relationship between GCase and α-synuclein (αS) [13–15], but the mechanisms underlying the relation between GBA mutations and the development of PD is still not clear. There is clinical, genetic and experimental evidence for both mutant GBA-mediated loss-of-function or toxic gain-of-function hypotheses, which are not mutually exclusive [1, 16].
The loss-of-function theories mainly focus on altered lipid metabolism and speculate that GBA mutations result in unstable or deficient protein, that, due to a lack of enzymatic activity, contributes to accumulation of GCase substrate, GluCer, within the lysosome, altering the cell membrane sphingolipid composition. Membrane binding is a key biological feature of αS and many functions of αS occur in association with lipid membranes. For example, in Lewy bodies (LB) αS layers are formed around central lipid core [17]. Hence, accumulation of GluCer can change lipid homeostasis, with subsequent alterations in αS processing. In support to this theory, null GBA alleles have been reported in PD patients and chemical inhibition of GBA could lead to accumulation of αS [18], a finding that was later replicated in different models with GBA mutations [16, 19–21]. Even in brains of PD patients without GBA mutations (PD-non-GBA), the reduction of GCase enzymatic activity was associated with increased αS levels [22].
The gain-of-function theories elaborate the effect of misfolded mutant GCase. Most GBA mutations are missense mutations, resulting in a misfolded protein. The presence of mutant GCase was confirmed in a significant proportion of αS inclusions in brain samples of GD and PD patients [1, 21]. The abnormal conformation can affect lysosomal function and cellular proteostasis systems and lead to αS accumulation, either due to increased formation of αS aggregates, or decreased clearance. Studies on fibroblasts derived from patients with GD showed that mutant GCase failed to fold correctly and was therefore retained in the endoplasmic reticulum (ER), overwhelming ubiquitin–proteasome system [23–25]. As a result, lysosomal GCase levels were significantly decreased, with subsequent accumulation of GluCer. Indeed, most mutant alleles identified in patients with GBA-associated PD (PD-GBA) were missense mutations that resulted in a misfolded protein. Moreover, mutant GCase was found in LB, suggesting its role in αS oligomerisation or impaired degradation [1].
Glucocerebrosidase, α-synuclein and lysosomes: a vicious circle
Deficient lysosomal GCase activity causes accumulation of GlcCer that in turn, accelerates formation and stabilizes soluble αS oligomers. Increased αS oligomers inhibit ER–Golgi trafficking of GCase and its translocation to the lysosome, resulting in further decrease in GCase lysosomal activity [20, 26, 27]. This amplifies GlcCer accumulation and stabilization of soluble αS oligomers, and results in a stronger inhibition of GCase ER–Golgi trafficking with each pathogenic cycle, resulting in more αS aggregates [20]. This way, bidirectional interaction between αS and GCase creates a positive feedback loop that, after a certain threshold, leads to self-propagating disease. Such reciprocal interaction between GCase and αS may underlie the aggregation of αS in the brain of PD-GBA patients. Data from autopsies of PD-GBA patients revealed elevated levels of oligomeric αS and αS-immunoreactive LB in the cortex and hippocampus [28]. However, some studies failed to demonstrate the effect of GCase pharmacological inhibition on αS accumulation/aggregation via lysosomal dysfunction [29].
In vitro experiments showed that in the lysosome-enriched fractions isolated from brain tissues and cultured neuronal cells, αS directly inhibited the lysosomal GCase activity (oligomers > monomers) [30]. It was shown that lysosomal GCase interacts with the C terminus of αS under acidic conditions, which mimicked the lysosomal lumen [31], and that membrane-bound α-helical form of αS inhibited GCase hydrolytic activity [32]. It is not known why αS oligomers inhibit GCase activity more strongly than the monomers. One possibility is that αS oligomers have a higher binding affinity with GCase and can therefore modulate its enzymatic activity to a greater extent. The colocalization of αS and GCase in LB indicates that aggregated αS can tightly bind to GCase [33]. In cultured neuronal cells, in addition to directly inhibiting GCase, αS oligomers may indirectly reduce GCase activity by blocking its transport from the ER to the lysosomes [20, 27].
GCase is located on the surface of the inner membrane of the lysosome. Under normal conditions, newly synthesized GCase is correctly folded in the ER and then translocated to the lysosomes by the trafficking receptor, lysosomal integral membrane protein-2 (LIMP-2) [34]. Cells overexpressing αS show less GCase delivered to the lysosome, as a result of less binding to LIMP-2 [35]. Since LIMP-2 is crucial for the correct trafficking of GCase and that its malfunction may lead to a reduction in GCase levels and activity, it is interesting to consider possible role of LIMP-2 in the development of PD. In LIMP-2 knock-out mice, lysosomal GCase activity was reduced, resulting in αS accumulation and disturbed lysosomal function, leading to neurotoxicity in dopaminergic neurons [36].
The main lysosomal degradation pathway for wild-type alpha αS is chaperone-mediated autophagy (CMA) [37]. Proteins destined for CMA form a complex with cytosolic chaperone, heat shock cognate protein of 70 kDa (Hsc70), which is targeted to the lysosomal membrane where it interacts with lysosomal associated membrane protein 2A (LAMP-2A), the receptor for CMA of αS, and undergoes translocation to the lysosome, followed by degradation [38]. To transport αS into the lysosome, the LAMP-2A protein must form a multimeric complex on the lysosomal membrane [39].
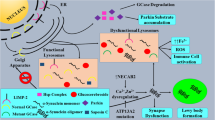
Namely, LAMP-2A exists as a monomer at the lysosomal membrane, in specific membrane lipid microdomains and substrate proteins only bind to monomeric LAMP-2A. To translocate substrate across the lysosomal membrane, it forms a multimeric complex with other proteins, enabling translocation of the substrates into the lysosomal lumen, which only relates to LAMP-2A molecules outside these microdomains [40]. Retention of LAMP-2A within the lipid microdomains may not allow protein-complex formation, which in turn will prevent the translocation of αS, resulting in its accumulation in the cytosol (Fig. 1).

Interaction between glucocerebrosidase, α synuclein, and lysosomes (adapted from Siebert et al. Ref. [13]). 1 After selective recognition by the lysosomal integral membrane protein-2 (LIMP-2), glucocerebrosidase (GCase) is sorted via ER and Golgi to the lysosomes. 2 Inside the lysosomes, GCase is associated with inner face of the lysosomal membrane, and hydrolyzes its substrate, glucocerebroside (GC), thus enabling the maintenance of the lysosomal membrane composition. In addition, α synuclein (αS) monomers interact with lysosomal associated membrane protein 2A (LAMP 2A), and upon formation of multimeric complex, enter the lysosome, where GCase facilitates their breakdown. 3 In case of LIMP-2 deficiency or absence, GCase is not sorted to the lysosomes, and is therefore secreted into the extracellular environment. 4 Impaired GCase activity may affect lysosomal membrane composition, leading to increased density of lipid rafts, affecting the formation LAMP 2A-protein-complexes required for αS translocation across lysosomal membrane, resulting in its accumulation in the cytosol. The increased cytosolic level of soluble monomers facilitates formation of oligomers and fibrils, affecting regular traffic of GCase from ER to Golgi. LIMP-2 lysosomal integral membrane protein-2, GCase glucocerebrosidase, ER endoplasmic reticulum, GC glucocerebroside, αS α synuclein, LAMP 2A lysosomal associated membrane protein 2A, PM plasma membrane, ECE extracellular environment
In a cellular and mouse models of GD, GBA inhibition results in changes of lysosomal membrane composition, including increased concentration of GCase in lipid rafts [41, 42]. It was hypothesized that this process may prevent or reduce the formation of LAMP-2A protein-complexes, which will then reduce the removal of αS by CMA and increase its accumulation in cytosol [43]. In another study, LAMP-2A protein levels were found to be selectively decreased, which directly correlated with increased levels of αS and decreased levels of Hsc70 in the same PD samples, as well as with accumulation of cytosolic CMA substrate proteins, MEF2D and IkΒα [44]. It is possible that alterations in the composition of the lysosomal membrane may affect autophagy-lysosomal pathways in general, hypothetically contributing to PD development.
The role of aging
Aging, an important risk factor for PD, is associated not only with decline in efficiency of the mechanisms responsible for disposal of misfolded proteins in post-mitotic neurons, but also with decline of GCase activity [45]. Age-related increase of oligomeric αS levels in the brains of cynomolgus monkeys was accompanied by a decrease in the expression and activity of GCase [30]. Besides, levels of αS phosphorylated at serine 129, a modification that promotes its oligomerization, also increase with age in the brain and are associated with a reduction in the activity of protein phosphatase 2A (PP2A), an enzyme that facilitates αS dephosphorylation [30]. In the light of described deleterious αS-GCase feedback loop, aging-related αS accumulation may affect GCase activity even in PD-non-GBA patients. Indeed, decreased GCase protein levels and enzymatic activity were recently identified in the substantia nigra (SN) of PD-non-GBA patients [22, 35, 46], that might, unlike general lysosomal inhibition, increase the levels of intracellular αS [18, 47]. Interestingly enough, reduced GCase occured when cellular αS levels were increasing, but before αS deposition in LB, and correlated with reduced LAMP-2A and increased αS levels [22].
Glucocerebrosidase affects α-synuclein cell-to-cell transfer
Although initially thought to be a purely intracellular protein, it gradually became clear that αS could be detected in the conditioned medium of cells and in extracellular fluids, such as plasma and CSF [48, 49]. Recent evidence has pointed towards the significance of cell-to-cell transfer of αS aggregates in PD and other synucleinopathies, as a mechanism of disease propagation [50, 51]. The existence of extracellular αS in rodent and human brain interstitial fluid has been confirmed by microdialysis [52]. Importantly, secreted αS can impact neuronal homeostasis and lead to neuronal death, even at concentrations close to those identified in body fluids [51, 53]. It is conceivable that GCase depletion may increase αS release, while the endolysosomal dysfunction increases its uptake [54, 55]. A recent report described increased neuronal cell-to-cell transmission of endogenous αS in grafted cells lacking GCase [47]. Furthermore, the ectopic expression of wild-type GCase (but not of an activity-deficient GBA mutant) reversed the effects of GBA deletion on the propagation of αS aggregates, indicating that the enzyme hydrolytic activity had a role in cell-to-cell αS transfer [47].
How frequent are GBA mutations among PD patients?
More than 300 different mutations of the GBA have been reported, but the N370S (the most frequent among European populations and Ashkenazi Jews) and L444P (dominated in Asian populations, where N370S was rare) account for the majority of those found in both GD and PD. Both GD patients and asymptomatic heterozygous gene carriers were recognized to be at risk of PD [15]. The proportion of PD patients that carry GBA mutations is estimated to be approximately between 5 and 10 % [7, 56–59] (range 3.2–21 %) [5, 6, 60]. The highest frequency of GBA mutations in PD patients (31.1 %) was reported in Ashkenazi Jews [5], while studies in Norwegian [61] and North African Arab-Berber [62] populations failed to demonstrate different frequencies among PD patients and controls (Table 1).
GBA mutations are important risk factor for developing PD, but most people with mutations will never develop PD [63]. Patients with GD have an almost 20-fold increased life-time risk of developing PD [64], irrespective of either GD severity or enzyme replacement therapy [65]. Carriers of severe (for example L444P) when compared to those with mild GBA mutations (for example N370S) have three to four times higher risk to develop PD, characterized by an earlier age at onset (on average 5 years) and more common cognitive impairment [66, 67].
Risks for PD at the age of 60 and 80 years was 4.7 and 9.1 % for GD patients and 1.5 and 7.7 % for GBA mutations carriers, respectively [68]. Cumulative risk for PD for GBA mutations carriers was 5 % at the age of 60 rising to 15 % at the age of 80 years [58], while Rana et al. [69] reported risks of 2.2 and 10.9 % at the age of 65 and 85 years, respectively. Due to the high cumulative age-specific risk (i.e. penetrance) found in the study by Anhelm et al. [70] (29.7 % at the age of 80 years), it has been suggested that GBA might be considered as an autosomal-dominant PD causing gene with reduced penetrance, although, these results should be interpreted with caution.
It is not surprising that the first-degree relatives of GD patients more frequently have PD, since they are obligate heterozygous GBA carriers [71–73]. PD patients with affected relatives are more frequently carriers of GBA mutations than those with negative family history for PD [57].
Clinical characteristics of Parkinsonʼs disease associated with GBA mutations
Pathological examinations of PD-GBA patients have revealed morphological abnormalities within the spectrum of classical (idiopathic) PD; therefore, this condition is not considered to represent an atypical form of the disease [33, 59]. Autopsy study also found that cortical LB are more frequently seen in PD-GBA patients than in non-carriers [59]. Clinically, GBA mutation carriers have an earlier age at PD onset (between 1.7 and 6.0 years earlier than PD-non-GBA [1, 5, 74–77]; observation not confirmed in several studies) [56, 78, 79], more likely have a positive family history for PD, but also more prevalent non-motor symptoms (NMS) when compared to PD-non-GBA [80].
Both GD patients and asymptomatic GBA mutation carriers may exhibit mild parkinsonian motor signs insufficient to reach the diagnosis of PD that may remain stable after two years of follow-up [81, 82]. In addition, hyposmia, cognitive decline, REM-sleep behavior disorder and depressive symptoms are more frequent among them in comparison to subjects who are not carriers of GBA mutations [81, 83]. We identified a family where both parents of a GD patient (obligate GBA carriers) have stable mild parkinsonian signs during the follow-up of 3 years (Fig. 2).

DaT-SPECT findings in GBA mutation carriers in premotor phase of PD. a Normal DaT-SPECT in a person without parkinsonism and without GBA mutations; b and c show DaT-SPECT findings for parents of GD patient. GD patient doesn’t have any signs of parkinsonism, hence, both parents (obligate carriers of GBA mutations) are in premotor phase of PD. b father of our GD patient (55 years) had no criteria for PD, but expressed hyposmia, REM-sleep behavior disorder, hypomimia, and mild bradykinesia of the left leg, that were stable for a 3 year follow-up and bilateral, but predominantly right-side decrease in DaT-SPECT binding; c mother of the same GD patient (51 years) had only bradykinesia of the right extremities that was stable for 3 year follow-up and reduced striatal DaT binding bilaterally, predominantly on the left side
PD-GBA has typical clinical manifestations: asymmetric parkinsonism with a good response to levodopa [84]. PD phenotype in GD patients and heterozygous GBA mutations carriers is similar [1]. Rarely, PD may precede clinical manifestations of GD for years [56, 85]. Initial symptoms of PD seem to be similar between GBA mutations carriers and non-carriers [5, 86–88]. However, although not confirmed in all studies, there are reports of more common bradykinesia, tremor, weakness and shoulder pain, as well as a lower frequency of rigidity as initial symptoms in PD-GBA [76, 86, 89].
Motor phenotype
When diagnosed, PD-GBA is clinically indistinguishable from PD-non-GBA [57, 76, 77, 79, 88, 90, 91]. However, some studies suggested less prevalent asymmetric onset, more postural instability [7, 56, 87], and more frequent levodopa-induced motor fluctuations and dyskinesia (LID), in PD-GBA patients [57, 92] (conversely, other studies observed that LID occurred with a lower frequency or with a similar prevalence when compared to PD-non-GBA patients) [93–96]. In conclusion, individual PD-GBA patients cannot be discriminated from PD-non-GBA patients on clinical or pathological grounds.
GBA mutations and polymorphisms influence the course of PD [79]. Progression of motor deterioration is more rapid in PD-GBA than in PD-non-GBA patients: GBA mutations carriers have fourfold increased risk of progression to Hoehn and Yahr stage 3 (HY3) [79, 90, 97–99]. Winder-Rhodes et al. [79] estimated that the time of progression to HY3 was 23.5 months for PD-GBA patients, 32 months for PD patients with GBA polymorphisms, and 49 months for non-carriers.
Non-motor features
Prominent NMS appeared to be the key clinical aspect of PD-GBA patients: they suffered more commonly and severely from a variety of NMS than those without GBA mutations [58, 80, 87]. PD-GBA patients show more rapid progression of cognitive decline and have >5fold increased risk of progression to dementia when compared PD-non-GBA patients [78, 79, 96, 98]. Winder-Rhodes et al. calculated projected median time to dementia to be 46 months for PD-GBA patients, 96 months for PD patients with GBA polymorphisms, while <50 % of those without GBA mutations developed dementia over the median follow-up period of 82 months [79].
Visual hallucinations, delusions and psychosis occured in PD-GBA, probably due to an extension of the LB pathology to the temporal lobe [55, 94, 95]. Depression, apathy, indifference, and anxiety disorder have been, although not uniformly, [89, 98, 101–103] reported to have higher prevalence in PD-GBA [58, 80, 87].
Autonomic dysfunction (e.g. orthostatic hypotension, constipation, sweating, urinary, bowel and sexual dysfunction), as well as sleep disturbances, fatigue, and unexplained pain, were also found to be more frequent in PD patients with than those without GBA mutations [58, 80, 87, 104, 105].
Therapeutic considerations
While several reports suggested poor response to levodopa, majority of the studies found an excellent therapeutic effect of levodopa and dopamine agonists in PD-GBA [5, 56, 78, 84, 90]. DBS also had favorable outcome in PD-GBA patients, although these patients might require application of the procedure earlier in the course of the disease and might have faster cognitive decline and axial impairment after DBS [65, 99, 106, 107]. The enzyme replacement therapy for GD neither prevented development of PD, nor modified severity of symptoms and PD progression, probably due to a lack of passage of the enzyme across the blood–brain barrier [108–110]. Alternative therapy for GD is with miglustat, an iminosugar which inhibits the biosynthesis of macromolecular substrates that accumulate pathologically in glycosphingolipidoses [111]. Miglustat is able to cross blood–brain barrier but no clear data are available on the impact of miglustat on parkinsonism in GD patients [110, 112, 113].
Establishing the concept of GBA-related PD promoted a search for the pathogenic mechanisms through which GCase deficiency may influence pathogenesis of PD, suggesting that targeting the GCase–lysosomal pathway might be a rational approach for the “development of neuroprotective drugs in PD” [15].
References
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11(11):986–998. doi:10.1016/S1474-4422(12)70190-4
Brockmann K, Berg D (2014) The significance of GBA for Parkinson’s disease. J Inherit Metab Dis 37(4):643–648. doi:10.1007/s10545-014-9714-7
Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E et al (1996) Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89(9):691–694. doi:10.1093/qjmed/89.9.691
Guimarães J, Amaral O, Sá Miranda MC (2003) Adult-onset neuronopathic form of Gaucher’s disease: a case report. Parkinsonism Relat Disord 9(5):261–264. doi:10.1016/S1353-8020(02)00096-2
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351(19):1972–1977. doi:10.1056/NEJMoa033277
Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E (2004) Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 81(1):70–73. doi:10.1016/j.ymgme.2003.11.004
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER et al (2009) Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N Engl J Med 361(17):1651–1661. doi:10.1056/NEJMoa0901281
Brady RO, Kanfer JN, Shapiro D (1965) Metabolism of glucocerebrosides II. Evidence of an enzymatic deficiency in Gaucher’s disease. Biochem Biophys Res Commun 18(2):221–225
Grabowski GA (2008) Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 372(9645):1263–1271. doi:10.1016/S0140-6736(08)61522-6
Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK et al (2004) Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 82(3):192–207. doi:10.1016/j.ymgme.2004.04.011
Vitner EB, Farfel-Becker T, Eilam R, Biton I, Futerman AH (2012) Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 135(Pt 6):1724–1735. doi:10.1093/brain/aws095
Burrow TA, Sun Y, Prada CE, Bailey L, Zhang W, Brewer A et al (2015) CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: clinical, histopathologic, and biochemical findings. Mol Genet Metab 114(2):233–241. doi:10.1016/j.ymgme.2014.08.011
Siebert M, Sidransky E, Westbroek W (2014) Glucocerebrosidase is shaking up the synucleinopathies. Brain 137:1304–1322. doi:10.1093/brain/awu002
Sardi SP, Cheng SH, Shihabuddin LS (2015) Gaucher-related synucleinopathies: the examination of sporadic neurodegeneration from a rare (disease) angle. Prog Neurobiol 125:47–62. doi:10.1016/j.pneurobio.2014.12.001
Schapira AH (2015) Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci 66:37–42. doi:10.1016/j.mcn.2015.03.013
Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ et al (2011) Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol 69(6):940–953. doi:10.1002/ana.22400
Gai WP, Yuan HX, Li XQ, Power JT, Blumbergs PC, Jensen PH (2000) In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp Neurol 166(2):324–333. doi:10.1006/exnr.2000.7527
Manning-Boğ AB, Schüle B, Langston JW (2009) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30(6):1127–1132. doi:10.1016/j.neuro.2009.06.009
Argyriou A, Dermentzaki G, Papasilekas T, Moraitou M, Stamboulis E, Vekrellis K et al (2012) Increased dimerization of alpha-synuclein in erythrocytes in Gaucher disease and aging. Neurosci Lett 528(2):205–209. doi:10.1016/j.neulet.2012.08.069
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA et al (2011) Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 146(1):37–52. doi:10.1016/j.cell.2011.06.001
Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM et al (2011) CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci USA 108(29):12101–12106. doi:10.1073/pnas.1108197108
Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E et al (2014) Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 137(Pt 3):834–848. doi:10.1093/brain/awt367
Ron I, Horowitz M (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14(16):2387–2398. doi:10.1093/hmg/ddi240
Bendikov-Bar I, Ron I, Filocamo M, Horowitz M (2011) Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis 46(1):4–10. doi:10.1016/j.bcmd.2010.10.012
Bendikov-Bar I, Horowitz M (2012) Gaucher disease paradigm: from ERAD to comorbidity. Hum Mutat 33(10):1398–1407. doi:10.1002/humu.22124
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B et al (2006) Alpha-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 313(5785):324–328. doi:10.1126/science.1129462
Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC et al (2010) Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell 21(11):1850–1863. doi:10.1091/mbc.E09-09-0801
Choi JH, Stubblefield B, Cookson MR, Goldin E, Velayati A, Tayebi N et al (2011) Aggregation of α-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol Genet Metab 104(1–2):185–188. doi:10.1016/j.ymgme.2011.06.008
Dermentzaki G, Dimitriou E, Xilouri M, Michelakakis H, Stefanis L (2013) Loss of β-Glucocerebrosidase Activity Does Not Affect Alpha-Synuclein Levels or Lysosomal Function in Neuronal Cells. PLoS ONE 8(4):e60674. doi:10.1371/journal.pone.0060674
Liu G, Chen M, Mi N, Yang W, Li X, Wang P et al (2015) Increased oligomerization and phosphorylation of α-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol Aging 36(9):2649–2659. doi:10.1016/j.neurobiolaging.2015.06.004
Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N et al (2011) Alpha-synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 286(32):28080–28088. doi:10.1074/jbc.M111.237859
Yap TL, Velayati A, Sidransky E, Lee JC (2013) Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol Genet Metab 108(1):56–64. doi:10.1016/j.ymgme.2012.11.010
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E (2010) Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol 120(5):641–649. doi:10.1007/s00401-010-0741-7
Reczek D, Schwake M, Schröder J, Hughes H, Blanz J, Jin X et al (2007) LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 131(4):770–783. doi:10.1016/j.cell.2007.10.018
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW et al (2012) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neuro 72(3):455–463. doi:10.1002/ana.23614
Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lüllmann-Rauch R et al (2014) LIMP-2 expression is critical for beta-glucocerebrosidase activity and alpha-synuclein clearance. Proc Natl Acad Sci USA 111(43):15573–15578. doi:10.1073/pnas.1405700111
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305(5688):1292–1295. doi:10.1126/science.1101738
Arias E, Cuervo AM (2011) Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 23(2):184–189. doi:10.1016/j.ceb.2010.10.009
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28(18):5747–5763. doi:10.1128/MCB.02070-07
Kaushik S, Massey AC, Cuervo AM (2006) Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J 25(17):3921–3933. doi:10.1038/sj.emboj.7601283
Hein LK, Duplock S, Hopwood JJ, Fuller M (2008) Lipid composition of microdomains is altered in a cell model of Gaucher disease. J Lipid Res 49(8):1725–1734. doi:10.1194/jlr.M800092-JLR200
Hattersley KJ, Hein LK, Fuller M (2013) Lipid composition of membrane rafts, isolated with and without detergent, from the spleen of a mouse model of Gaucher disease. Biochem Biophys Res Commun 442(1–2):62–67. doi:10.1016/j.bbrc.2013.11.009
Gan-Or Z, Dion PA, Rouleau GA (2015) Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 11(9):1443–1457. doi:10.1080/15548627.2015.1067364
Murphy KE, Gysbers AM, Abbott SK, Spiro AS, Furuta A, Cooper A et al (2015) Lysosomal-associated membrane protein 2 Isoforms Are Differentially Affected in Early Parkinson’s Disease. Mov Disord 30(12):1639–1647. doi:10.1002/mds.26141
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P et al (2015) Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol 2(4):433–438. doi:10.1002/acn3.177
Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M et al (2015) Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener 10:15. doi:10.1186/s13024-015-0010-2
Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC et al (2014) Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 5:4755. doi:10.1038/ncomms5755
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ et al (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 17(13):1945–1947. doi:10.1096/fj.03-0098fje
Lee HJ, Patel S, Lee SJ (2005) Intravesicular Localization and Exocytosis of alpha-Synuclein and its Aggregates. J Neurosci 25(25):6016–6024. doi:10.1523/JNEUROSCI.0692-05.2005
Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S et al (2010) Direct Transfer of alpha-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies. J Biol Chem 285(12):9262–9272. doi:10.1074/jbc.M109.081125
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH et al (2010) Cell-Produced alpha-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J Neurosci 30(20):6838–6851. doi:10.1523/JNEUROSCI.5699-09.2010
Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC et al (2011) Assessment of alpha-Synuclein Secretion in Mouse and Human Brain Parenchyma. PLoS ONE 6(7):e22225. doi:10.1371/journal.pone.0022225
Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T et al (2014) The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis 63:1–11. doi:10.1016/j.nbd.2013.11.002
Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ et al (2011) Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 42(3):360–367. doi:10.1016/j.nbd.2011.01.029
Lee HJ, Bae EJ, Lee SJ (2014) Extracellular alpha–synuclein- a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 10(2):92–98. doi:10.1038/nrneurol.2013.275
Kumar KR, Ramirez A, Göbel A, Kresojević N, Svetel M, Lohmann K et al (2013) Glucocerebrosidase mutations in a Serbian Parkinson’s disease population. Eur J Neurol 20(2):402–405. doi:10.1111/j.1468-1331.2012.03817.x
Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C et al (2011) Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet 20(1):202–210. doi:10.1093/hmg/ddq454
McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH (2012) A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry 83(8):853–854. doi:10.1136/jnnp-2012-302402
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH et al (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132(Pt 7):1783–1794. doi:10.1093/brain/awp044
Choi JM, Kim WC, Lyoo CH, Kang SY, Lee PH, Baik JS et al (2012) Association of mutations in the glucocerebrosidase gene with Parkinson disease in a Korean population. Neurosci Lett 514(1):12–15. doi:10.1016/j.neulet.2012.02.035
Toft M, Pielsticker L, Ross OA, Aasly JO, Farrer MJ (2006) Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian. Neurology 66(3):415–417
Nishioka K, Vilariño-Güell C, Cobb SA, Kachergus JM, Ross OA, Wider C et al (2011) Glucocerebrosidase mutations are not a common risk factor for Parkinson disease in North Africa. Neurosci Lett 477(2):57–60. doi:10.1016/j.neulet.2009.11.066
Zokaei N, McNeill A, Proukakis C, Beavan M, Jarman P, Korlipara P et al (2014) Visual short-term memory deficits associated with GBA mutation and Parkinson’s disease. Brain 137(Pt 8):2303–2311. doi:10.1093/brain/awu143
Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S et al (2010) The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis 33(2):167–173. doi:10.1007/s10545-010-9055-0
Chetrit EB, Alcalay RN, Steiner-Birmanns B, Altarescu G, Phillips M, Elstein D et al (2013) Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol Dis 50(3):218–221. doi:10.1016/j.bcmd.2012.11.011
Gan-Or Z, Giladi N, Orr-Urtreger A (2009) Differential phenotype in Parkinson’s disease patients with severe versus mild GBA mutations. Brain 132(Pt 10):e125. doi:10.1093/brain/awp161
Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A et al (2015) Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84(9):880–887. doi:10.1212/WNL.0000000000001315
Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C et al (2014) Comparison of Parkinson Risk in Ashkenazi Jewish Patients With Gaucher Disease and GBA Heterozygotes. JAMA Neurol 71(6):752–757. doi:10.1001/jamaneurol.2014.313
Rana HQ, Balwani M, Bier L, Alcalay RN (2013) Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 15(2):146–149. doi:10.1038/gim.2012.107
Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F et al (2012) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78(6):417–420. doi:10.1212/WNL.0b013e318245f476
Giraldo P, Capablo JL, Alfonso P, Garcia-Rodriguez B, Latre P, Irun P et al (2011) Neurological manifestations in patients with Gaucher disease and their relatives, it is just a coincidence? J Inherit Metab Dis 34(3):781–787. doi:10.1007/s10545-011-9298-4
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E (2004) Parkinsonism among Gaucher disease carriers. J Med Genet 41(12):937–940. doi:10.1136/jmg.2004.024455
Halperin A, Elstein D, Zimran A (2006) Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis 36(3):426–428. doi:10.1016/j.bcmd.2006.02.004
Tan EK, Tong J, Fook-Chong S, Yih Y, Wong MC, Pavanni R et al (2007) Glucocerebrosidase Mutations and Risk of Parkinson Disease in Chinese Patients. Arch Neurol 64(7):1056–1058. doi:10.1001/archneur.64.7.1056
Wu YR, Chen CM, Chao CY, Ro LS, Lyu RK, Chang KH et al (2007) Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J Neurol Neurosurg Psychiatry 78(9):977–979. doi:10.1136/jnnp.2006.105940
Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED et al (2007) Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69(12):1270–1277. doi:10.1212/01.wnl.0000276989.17578.02
Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA et al (2009) Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72(4):310–316. doi:10.1212/01.wnl.0000327823.81237.d1
Setó-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A et al (2012) Glucocerebrosidase Mutations Confer a Greater Risk of Dementia during Parkinson’s Disease Course. Mov Disord 27(3):393–399. doi:10.1002/mds.24045
Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T et al (2013) Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136(Pt 2):392–399. doi:10.1093/brain/aws318
Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T et al (2011) GBA-associated PD presents with nonmotor characteristics. Neurology 77(3):276–280. doi:10.1212/WNL.0b013e318225ab77
Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH (2015) Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol 72(2):201–208. doi:10.1001/jamaneurol.2014.2950
McNeill A, Duran R, Proukakis C, Bras J, Hughes D, Mehta A et al (2012) Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord 27(4):526–532. doi:10.1002/mds.24945
Gan-Or Z, Mirelman A, Postuma RB, Arnulf I, Bar-Shira A, Dauvilliers Y et al (2015) GBA mutations are associated with Rapid Eye Movement Sleep Behavior Disorder. Ann Clin Transl Neurol 2(9):941–945. doi:10.1002/acn3.228
Sato C, Morgan A, Lang AE, Salehi-Rad S, Kawarai T, Meng Y et al (2005) Analysis of the glucocerebrosidase gene in Parkinson’s disease. Mov Disord 20(3):367–370. doi:10.1002/mds.20319
Machaczka M, Rucinska M, Skotnicki AB, Jurczak W (1999) Parkinson’s Syndrome Preceding Clinical Manifestation of Gaucher’s Disease. Am J Hematol 61(3):216–217. doi:10.1002/(SICI)1096-8652(199907)61:3<216:AID-AJH11>3.0.CO;2-E
Kresojević N, Janković M, Petrović I, Kumar KR, Dragašević N, Dobričić V et al (2015) Presenting symptoms of GBA-related Parkinson’s disease. Parkinsonism Relat Disord 21(7):804–807. doi:10.1016/j.parkreldis.2015.04.028
Wang C, Cai Y, Gu Z, Ma J, Zheng Z, Tang BS et al (2014) Clinical profiles of Parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol Aging 35(3):725.e1–725.e6. doi:10.1016/j.neurobiolaging.2013.08.012
Mao XY, Burgunder JM, Zhang ZJ, An XK, Zhang JH, Yang Y et al (2010) Association between GBA L444P mutation and sporadic Parkinson’s disease from Mainland China. Neurosci Lett 469(2):256–259. doi:10.1016/j.neulet.2009.12.007
Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, Bar-Shira A et al (2008) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70(24):2277–2283. doi:10.1212/01.wnl.0000304039.11891.29
Pulkes T, Choubtum L, Chitphuk S, Thakkinstian A, Pongpakdee S, Kulkantrakorn K et al (2014) Glucocerebrosidase mutations in Thai patients with Parkinson’s disease. Parkinsonism Relat Disord 20(9):986–991. doi:10.1016/j.parkreldis.2014.06.007
Socal MP, Bock H, Michelin-Tirelli K, Hilbig A, Saraiva-Pereira ML, Rieder CR et al (2009) Parkinson’s disease and the heterozygous state for glucocerebrosidase mutations among Brazilians. Parkinsonism Relat Disord 15(1):76–78. doi:10.1016/j.parkreldis.2008.01.019
Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E (2008) The spectrum of Parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol 65(10):1353–1357. doi:10.1001/archneur.65.10.1353
Huang CL, Wu-Chou YH, Lai SC, Chang HC, Yeh TH, Weng YH et al (2011) Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson’s disease in Taiwan. Eur J Neurol 18(10):1227–1232. doi:10.1111/j.1468-1331.2011.03362.x
Li Y, Sekine T, Funayama M, Li L, Yoshino H, Nishioka K et al (2014) Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol Aging 35(4):935.e3–935.e8. doi:10.1016/j.neurobiolaging.2013.09.019
Aharon-Peretz J, Badarny S, Rosenbaum H, Gershoni-Baruch R (2005) Mutations in the glucocerebrosidase gene and Parkinson disease: phenotype-genotype correlation. Neurology 65(9):1460–1461. doi:10.1212/01.wnl.0000176987.47875.28
Oeda T, Umemura A, Mori Y, Tomita S, Kohsaka M, Park K et al (2015) Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging 36(12):3306–3313. doi:10.1016/j.neurobiolaging.2015.08.027
Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C (2015) Variants in GBA, SNCA, and MAPT Influence Parkinson Disease Risk, Age at Onset, and Progression. Neurobiol Aging 37:209.e1–209.e7. doi:10.1016/j.neurobiolaging.2015.09.014
Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W et al (2015) GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 30(3):407–411. doi:10.1002/mds.26071
Angeli A, Mencacci NE, Duran R, Aviles-Olmos I, Kefalopoulou Z, Candelario J et al (2013) Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov Disord 28(10):1370–1375. doi:10.1002/mds.25535
Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J et al (2009) Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 66(5):571–576. doi:10.1001/archneurol.2009.72
Alcalay RN, Caccappolo E, Mejia-Santana H, Tang M, Rosado L, Orbe Reilly M et al (2012) Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78(18):1434–1440. doi:10.1212/WNL.0b013e318253d54b
Malec-Litwinowicz M, Rudzińska M, Szubiga M, Michalski M, Tomaszewski T, Szczudlik A (2014) Cognitive impairment in carriers of glucocerebrosidase gene mutation in Parkinson disease patients. Neurol Neurochir Pol 48(4):258–261. doi:10.1016/j.pjnns.2014.07.005
Kresojević N, Mijajlović M, Perić S, Pavlović A, Svetel M, Janković M et al (2013) Transcranial sonography in patients with Parkinson’s disease with glucocerebrosidase mutations. Parkinsonism Relat Disord 19(4):431–435. doi:10.1016/j.parkreldis.2012.12.006
Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E (2008) The spectrum of Parkinsonian Manifestations associated with glucocerebrosidase mutations. Arch Neurol 65(10):1353–1357. doi:10.1001/archneur.65.10.1353
Böttcher T, Rolfs A, Meyer B, Grossmann A, Berg D, Kropp P et al (2013) Clinical, genetic, and brain sonographic features related to Parkinson’s disease in Gaucher disease. J Neurol 260(10):2523–2531. doi:10.1007/s00415-013-7011-2
Weiss D, Brockmann K, Srulijes K, Meisner C, Klotz R, Reinbold S et al (2012) Long-term follow-up of subthalamic nucleus stimulation in glucocerebrosidase-associated Parkinson’s disease. J Neurol 259(9):1970–1972. doi:10.1007/s00415-012-6469-7
Kono S, Ouchi Y, Terada T, Ida H, Suzuki M, Miyajima H (2010) Functional brain imaging in glucocerebrosidase mutation carriers with and without parkinsonism. Mov Disord 25(12):1823–1829. doi:10.1002/mds.23213
Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR et al (2011) The incidence of parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol Dis 46(1):95–102. doi:10.1016/j.bcmd.2010.10.006
Bembi B, Zambito Marsala S, Sidransky E, Ciana G, Carrozzi M, Zorzon M et al (2003) Gaucher’s disease with Parkinson’s disease: clinical and pathological aspects. Neurology 61(1):99–101
Kraoua I, Stirnemann J, Ribeiro MJ, Rouaud T, Verin M, Annic A et al (2009) Parkinsonism in Gaucher’s disease type 1: ten new cases and a review of the literature. Mov Disord 24(10):1524–1530. doi:10.1002/mds.22593
Cox TM, Aerts JM, Andria G, Beck M, Belmatoug N, Bembi B et al (2003) The role of the iminosugar N-butyldeoxynojirmycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis 26(6):513–526. doi:10.1023/A:1025902113005
Goker-Alpan O, Sidransky E (2008) Treating patients with Gaucher disease and parkinsonism: misrepresentation in a title. Parkinsonism Relat Disord 14(1):81–82
Hughes DA, Ginsberg L, Baker R, Goodwin S, Milligan A, Richfield L et al (2007) Effective treatment of an elderly patient with Gaucher’s disease and parkinsonism: a case report of 24 months’ oral substrate reduction therapy with miglustat. Parkinsonism Relat Disord 13(6):365–368. doi:10.1016/j.parkreldis.2006.07.010
Clark LN, Nicolai A, Afridi S, Harris J, Mejia-Santana H, Strug L et al (2005) Pilot association study of the beta-glucocerebrosidase N370S allele and Parkinson’s disease in subjects of Jewish ethnicity. Mov Disord 20(1):100–103. doi:10.1002/mds.20320
De Marco EV, Annesi G, Tarantino P, Rocca FE, Provenzano G, Civitelli D et al (2008) Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov Disord 23(3):460–463. doi:10.1002/mds.21892
Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC et al (2008) Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol 65(3):379–382. doi:10.1001/archneurol.2007.68
Kalinderi K, Bostantjopoulou S, Paisan-Ruiz C, Katsarou Z, Hardy J, Fidani L (2009) Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci Lett 452(2):87–89. doi:10.1016/j.neulet.2009.01.029
Bras J, Paisan-Ruiz C, Guerreiro R, Ribeiro MH, Morgadinho A, Januario C et al (2010) Complete screening for glucocerebrosidase mutations in parkinson disease patients from Portugal. Neurobiol Aging 30(9):1515–1517. doi:10.1016/j.neurobiolaging.2007.11.016
Sun QY, Guo JF, Wang L, Yu RH, Zuo X, Yao LY et al (2010) Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson’s disease in Chinese population. Mov Disord 25(8):1005–1011. doi:10.1002/mds.23009
Hu FY, Xi J, Guo J, Yu LH, Liu L, He XH et al (2010) Association of the glucocerebrosidase N370S allele with Parkinson’s disease in two separate Chinese Han populations of mainland China. Eur J Neurol 17(12):1476–1478. doi:10.1111/j.1468-1331.2010.03097.x
Lesage S, Condroyer C, Hecham N, Anheim M, Belarbi S, Lohman E et al (2011) Mutations in the glucocerebrosidase gene confer a risk for parkinson disease in north Africa. Neurology 76(3):301–303. doi:10.1212/WNL.0b013e318207b01e
Moraitou M, Hadjigeorgiou G, Monopolis I, Dardiotis E, Bozi M, Vassilatis D et al (2011) β-Glucocerebrosidase gene mutations in two cohorts of Greek patients with sporadic Parkinson’s disease. Mol Genet Metab 104(1–2):149–152. doi:10.1016/j.ymgme.2011.06.015
Noreau A, Rivière JB, Diab S, Dion PA, Panisset M, Soland V et al (2011) Glucocerebrosidase mutations in a French-Canadian Parkinson’s disease cohort. Can J Neurol Sci 38(5):772–773
Emelyanov A, Boukina T, Yakimovskii A, Usenko T, Drosdova A, Zakharchuk A et al (2012) Glucocerebrosidase Gene Mutations Are Associated with Parkinson’s Disease in Russia. Mov Disord 27(1):158–159. doi:10.1002/mds.23950
Guimarães Bde C, Pereira AC, Rodrigues Fda C, dos Santos AV, Campos M Jr, dos Santos JM et al (2012) Glucocerebrosidase N370S and L444P mutations as risk factors for Parkinson’s disease in Brazilian patients. Parkinsonism Relat Disord 18(5):688–689. doi:10.1016/j.parkreldis.2011.11.028
Wang Y, Liu L, Xiong J, Zhang X, Chen Z, Yu L, Chen C et al (2012) Glucocerebrosidase L444P mutation confers genetic risk for Parkinson’s disease in central China. Behav Brain Funct 8:57. doi:10.1186/1744-9081-8-57
Zhang X, Bao QQ, Zhuang XS, Gan SR, Zhao D, Liu Y et al (2012) Association of common variants in the glucocerebrosidase gene with high susceptibility to Parkinson’s disease among chinese. Chin J Physiol 55(6):398–404. doi:10.4077/CJP.2011.AMM076
González-Del Rincón Mde L, Monroy Jaramillo N, Suárez Martínez AI, Yescas Gómez P, Boll Woehrlen MC, López López M et al (2013) The L444P GBA mutation is associated with early-onset Parkinson’s disease in Mexican Mestizos. Clin Genet 84(4):386–387. doi:10.1111/cge.12084
Yu Z, Wang T, Xu J, Wang W, Wang G, Chen C et al (2015) Mutations in the glucocerebrosidase gene are responsible for Chinese patients with Parkinson’s disease. J Hum Genet 60(2):85–90. doi:10.1038/jhg.2014.110
Asselta R, Rimoldi V, Siri C, Cilia R, Guella I, Tesei S et al (2014) Glucocerebrosidase mutations in primary parkinsonism. Parkinsonism Relat Disord 20(11):1215–1220. doi:10.1016/j.parkreldis.2014.09.003
Han F, Grimes DA, Li F, Wang T, Yu Z, Song N et al (2016) Mutations in the glucocerebrosidase gene are common in patients with Parkinson’s disease from Eastern Canada. Int J Neurosci 126(5):415–421. doi:10.3109/00207454.2015.1023436
Acknowledgments
This work was supported by a grant of Ministry of Education, Science and Technological Development of the Republic of Serbia (ON 175090). The author would like to thank Dr. Sonja Misilić Denčić (Institute of Medical and Clinical Biochemistry, Faculty of Medicine, University of Belgrade) for her assistance in preparation of Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Ivanka Marković and Nikola Kresojević report no disclosures. Vladimir Kostić received honoraria for lectures from Boehringer and Novartis and grants from Ministry of Education and Science, Republic of Serbia (Project #ON175090); Serbian Academy of Science and Arts, Novartis, Boehriner, Glaxo, Pfizer and Swisspharm. On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Marković, I., Kresojević, N. & Kostić, V.S. Glucocerebrosidase and parkinsonism: lessons to learn. J Neurol 263, 1033–1044 (2016). https://doi.org/10.1007/s00415-016-8085-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-016-8085-4