
Abstract
The purpose of this study was to describe a pedigree with NEFL N98S mutation associated with a dominant intermediate Charcot–Marie–Tooth disease (DI-CMT) and heterogeneous early-onset phenotype. The pedigree comprised two patients, the proband and her son, aged 38 and 5 years. The proband, evaluated at age 31, showed delayed motor milestones that, as of the second decade, evolved into severe phenotype consisting of sensorimotor neuropathy, pes cavus, clawing hands, gait and kinetic cerebellar ataxia, nystagmus and dysarthria, she being wheelchair bound. By then, a working diagnosis of sporadic early onset cerebellar ataxia with peripheral neuropathy was established. Screening of mutations associated with SCA and autosomal recessive cerebellar ataxias was negative. Her son showed a mild phenotype characterized by delayed motor milestones, and lower-limb hypotonia and areflexia. Electrophysiology in both patients showed nerve conduction slowing in the intermediate range, both in proximal and distal nerve segments, but where compound muscle action potentials exhibited severe attenuation there was conduction slowing down to the demyelinating range. In the proband, cranial magnetic resonance imaging (MRI) showed cerebellar atrophy, electromyography disclosed active denervation in tibialis anterior, and MRI of lower-limb musculature demonstrated widespread and distally accentuated muscle fatty atrophy; furthermore, on water sensitive MRI sequences there was edema of calf muscles. We conclude that the NEFL N98S mutation is associated with a DI-CMT phenotype characterized by early-onset sensorimotor neuropathy delaying motor milestones, which may evolve into a severe and complex clinical picture including cerebellar ataxia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Charcot–Marie–Tooth disease (CMT) is the most frequent form of sensorimotor inherited neuropathy with a prevalence ratio of 28 cases/100,000 inhabitants [1]. CMT was initially classified according to the mode of transmission (autosomal dominant, autosomal recessive or X-linked) and electrophysiological or nerve biopsy features. Characteristically, motor conduction velocities (MCV) in median nerve are below 38 m/s for the demyelinating form (CMT1) and above 38 m/s for the axonal form [2]. For want of a better name, Davis et al. [3] introduced the term intermediate for designating dominant CMT (DI-CMT) families with clinico-electrophysiological and pathological data not fitting into either CMT1 or CMT2. In short, DI-CMT was originally characterized by: (1) absence of clinically observed nerve hypertrophy; (2) median nerve MCV between 25 and 45 m/s (mean in their series, 34.6 m/s); (3) preserved mean compound muscle action potential (CMAP) amplitude (mean, 4.6 mV); and (4) nerve biopsy showing axonal changes, clusters of regenerating myelinated fibres, loss of the larger fibres noted from the unimodal diameter histogram, and onion bulbs with fewer lamellae than in CMT1 [4]. Twelve out of 131 CMT patients in the series by Bouché et al. [5] could no be classified within CMT1 or CMT2 since median nerve MCV ranged between 30 and 40 m/s. As stated by Nicholson and Myers [6], because slow nerve conduction velocity can be found in axonal neuropathies as a result of the loss of large rapidly conduction fibres in distal nerve trunks, it is therefore important to exclude conduction results in which CMAP amplitude is reduced. In this regard, examination of more proximal upper-limb nerve segments may demonstrate that MCV slowing persists in the intermediate limits despite CMAP amplitudes being normal, namely, such conduction slowing cannot be accounted for by axonal degeneration and therefore unequivocally indicate that motor nerve conduction parameters are those of DI-CMT [7, 8].
The number of disease genes identified in CMT has expended rapidly over the last decades, particularly after the development of next-generation sequencing, such that more than 60 CMT-associated genes have now been discovered [9]. Leaving CMTX1 aside [10], DI-CMT encompasses six loci with five cloned genes (DNM2, YARS, MPZ, INF2, and GNB4) [9, 11].
Neurofilament light-chain polypeptide gene (NEFL) mutations may cause either dominant axonal (CMT2E) or dominant demyelinating (CMT1F) phenotype [12–17], and rarely recessive axonal phenotype [18, 19]. NEFL mutations have occasionally been associated with intermediately slowed MCV [16, 20, 21], just one pedigree having been reported under the rubric of DI-CMT [8]. As a whole, NEFL mutations represent between 0.8 and 2 % of all patients with CMT [15, 18, 22–25]. NEFL-associated CMT is a highly variable disorder that comprises more than 18 disease-causing mutations, targeting the head or rod protein domains, with highly variable phenotypic expression, N98S (N97 in the old nomenclature) being reported in five pedigrees with severe early-onset disease phenotype [15, 19, 26, 27].
Here we describe the study of a heterogeneous DI-CMT pedigree, comprising the proband and her affected child, associated to NEFL N98S mutation. Detection of MCVs in the intermediate range was essential for accurate guidance of molecular diagnosis.
Patients and methods
Patient data
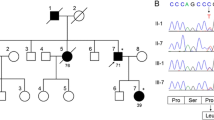
The study is based upon a single Spanish CMT family comprising two affected subjects over two generations (Fig. 1). The hospital ethics committee approved the study; the proband gave informed consent to participate in the study.

a Pedigree of the family. Patients of the first generation died at ages between 40 and 92 years. Case II-2 died, but no further details are available. Genotypes at position c.293 in NEFL are shown below the examined individuals (indicated with horizontal bars). Arrow indicates the proband. b Electropherograms depicting the mutated (MUT) and reference (REF) sequences surrounding the NEFL c.392 position
Electrophysiological study
Conduction studies of median, ulnar, axillary, peroneal, tibial and sural nerves were carried out as reported elsewhere [28, 29] and in accordance with the requirements of the CMT neuropathy score (CMTNS) [30]. Normative terminal motor latency index (TLI) values were taken from Amato et al. [31]. Needle electromyography (EMG) was performed analysing the duration and morphology of the motor unit potentials, the presence of spontaneous activity, and the electromyographic pattern at the maximum voluntary effort. Blink reflex, somatosensory evoked potentials (SEP), brainstem auditory evoked potentials (BAEP) and motor evoked potentials (MEP) were performed according to standard methods [32]. Normative nerve conduction data in patient IV-1 were taken from Cruz-Martínez et al. [33] and García et al. [34].
Imaging study
Non-contrasted magnetic resonance imaging (MRI) of lower-limb musculature was performed in the proband as reported elsewhere [29, 35].
Exome sequencing
Whole exome sequencing was performed on the two affected individuals (III-2 and IV-1). Genomic DNA was sheared to an average size of 150 bp (Covaris) and libraries were prepared using the KAPA HTP Library Preparation Kit for Illumina platforms (KAPA Biosystems). The exomes were captured by the SeqCap EZ Exome v3.0 capture system (Roche) and sequenced on the NextSeq 500 platform (Illumina) using the NextSeq 500 High Output v2 kit (150 cycles PE) as to obtain on average 20-fold coverage of 80 % of the target region. Mapping to the human reference genome (GRCh37) and variant calling were performed with the CLC Genomics Workbench. Subsequent annotation and filtering were executed with GenomeComb [36] (http://genomecomb.sourceforge.net/).
Mutational analysis of NEFL
Genomic DNA was extracted from peripheral blood using standard procedures. The first exon of NEFL (transcript NM_006158.4) was PCR amplified (primers available upon request), purified (ExoSAP-IT®, Affymetrix USB) and bidirectionally sequenced (BigDye® Terminator v3.1 cycle sequencing kit, Applied Biosystems). Fragments were electrophoretically separated on an ABI3730xl DNA Analyzer (Applied Biosystems) and analysed with SeqMan™ II software (DNASTAR Inc.). Mutation nomenclature followed HGVS guidelines (http://www.hgvs.org/mutnomen).
Results
Clinical findings
We present detailed clinical data of the proband (case III-2) and her affected son (IV-1) (Fig. 1). She did not recognize parental consanguinity, her parents being alive and non-affected by history. Examination of her husband, aged 38, was normal; there was no consanguinity between them.
Case III-2 (proband)
In 2008, at age 31, this patient was referred to us to evaluate a complex syndrome comprising progressive early onset cerebellar ataxia and peripheral neuropathy. Her clinical picture was initiated with abnormal motor milestones, independent walking not being possible till the age of 3. In spite of this, she was able to attend her school activities including gym classes. As of age 12 there was a progressive clumsiness and weakness involving lower legs and hands, and dysarthria. By then she also noticed progressive foot and hand deformities, which were unsuccessfully treated with numerous orthopaedic surgical procedures. In the last years she has been wheelchair bound, having great difficulties in typing. In any case, she has continued working as a professional graphic designer. She denied distal sensory symptoms, cramps, deafness, visual loss, sphincter disturbances or cognitive declining. She also denied drug abuse and consumption of alcohol drinks.
On examination, she was able to stand with bilateral support and to walk a few steps with bilateral steppage and marked postural instability. There was bilateral forefoot pes cavus and hand clawing (Fig. 2a, b). There were foot and hand scar sequelae from previous orthophaedic surgical procedures. Palpably enlarged nerves were not observed. There was marked atrophy of lower-leg and hand musculature, and to a lesser degree of thigh and forearm muscles. No fasciculations were observed. Muscle power (Medical Research Scale) was as follows: 0/5 for foot flexors/extensors; 4/5 for thigh and hip muscles; 2/5 for intrinsic hand muscles; 2–4 for forearm muscles, extensors being more weekend than flexors; and 5/5 for shoulder girdle and elbow joint muscles. There was generalized hypotonia. Except for jaw jerk, tendon reflexes were absent; plantar responses were flexor. There was global stocking hypoesthesia (pin, touch and vibration) up to knees, vibration sense being more severely disturbed; to a lesser degree glove hypoesthesia was also noted. Apart from foot and hand post-surgical scars, no skin trophic changes were observed. No ichthyosis was noted. There was ataxic dysarthria. Severe distal weakness made it difficult to carry out testing of co-ordination; be that as it may, the finger-nose test was done in a wavering manner. There were saccadic intrusions during visual smooth pursuit and bilateral horizontal gaze-evoked nystagmus. Saccadic eye movements were not apparently slowed. There were no occular telangiectasiae. Neither facial nor palatal weakness was noted. Ophthalmoscopy showed normal findings; neither retinal striation nor retinitis pigmentosa was noted. CMTNS was of 28 (severe disease).

Clinical and MRI pictures of the proband patient at age 31. a Close-up picture of hands showing marked hand amyotrophy with clawing deformity. b Note bilateral lower-leg amyotrophy with drop feet. c Axial T1 weighted MR image of middle third of thighs showing moderate and diffuse muscle fatty atrophy. d Axial T1 weighted MR image of mid calves showing marked and widespread fatty infiltration of all four muscle compartments. e Axial T2FS weighted MR image, obtained at same level of the previous one, illustrating muscle symmetric hypersignal, involving posterior tibialis, anterior tibialis and gastrocnemious medialis muscles, indicative of edema. f T1 weighted MR image through metatarsal bones showing massive fatty infiltration of foot musculature
Routine laboratory investigations, including serum levels of vitamin E, immunoglobulins, alpha-fetoprotein, cholesterol and albumin, were normal. There was no proteinuria and renal function was normal [37].
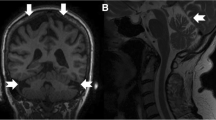
Craniospinal MRI showed cerebellar wasting (Fig. 3) with neither brain nor spinal cord atrophy. MRI of lower-limb musculature findings are illustrated in Fig. 2c–f; in short, there was widespread and distally accentuated muscle fatty atrophy, and lower-leg hypersignal of calf muscles on T2 fat suppressed (T2FS) sequences.

Cranial sagittal T1-weighted MR image in the proband showing severe atrophy of the cerebellar vermis
Table 1 summarizes the results of nerve conduction studies in both patients. In the proband (III-2), MCV for the elbow–wrist segment was 18.3 m/s in median nerve and 29.5 m/s in ulnar nerve, their distal CMAP potential amplitudes being markedly reduced (0.2 and 0.8 mV, respectively). TLI was 0.40 for median nerve (normal, 0.34–0.42) and 0.41 for ulnar nerve (normal, 0.36–0.52). To better define the pattern of median nerve MCV slowing, we investigated the axilla–elbow segment with stimulation at axilla and recording from flexor digitorum superficialis (FDS). Recorded MCV was reduced to the intermediate range for this segment (47.5 m/s; normal value, ≥55), CMAP amplitudes being moderately reduced (see Table 1). Similarly, ulnar nerve MCV for the axilla–elbow segment, while recording from flexor carpi ulnaris muscle, was in the intermediate range (see Table 1). Distal motor latencies (DML) of median and ulnar nerve were prolonged, whereas F waves were unobtainable; in both nerves sensory nerve action potentials (SNAP) were not recordable either. CMAP of axillary nerve was attenuated with motor latency slightly prolonged. Peroneal, tibial and sural nerves were inexcitable; just severe attenuated CMAPs were obtained in tibialis anterior and triceps surae (see Table 1). EMG of tibialis anterior showed absence of voluntary activity and profuse signs of active denervation (fibrillations and positive waves). EMG of deltoid showed a pattern of mild chronic denervation.
Blink reflex showed absence of R1 responses bilaterally, R2 responses being of reduced amplitude. Jaw jerk recording showed delayed latency on both masseter muscles, motor responses being of reduced amplitudes. Somatosensory evoked potentials were absent from lower and upper limb nerve stimulation. Brainstem auditory evoked potentials were normal. In upper limbs, motor evoked potentials (MEP) were so reduced and desynchronized that made it impossible to calculate central motor conduction. In lower limbs, MEPs were absent.
Case IV-1
This patient, born in November 2009, has been serially evaluated since birth. His mother’s pregnancy was normal, no exacerbation of her symptoms having been noticed [38]. He was born of caesarean section at 38 weeks. Examination of the newborn was normal (Apgar score 9/10, weight 2840 g, head circumference 34 cm, and stature 49.5 cm). The only manifestation has been delayed motor milestones, gait being autonomous as of month 22.5. Subsequently, serial examinations have shown lower-limb areflexia and hypotonia. A far as can be assessed, no sensory loss was noted. Neither amyotrophy nor muscle weakness was observed. There was no gait or kinetic ataxia. Romberg sign was negative. Ocular movements were normal with no nystagmus. Social, language and scholar acquisitions have been normal. CMTNS was of 5 (slight disease).
Routine laboratory investigations were normal. The results of nerve conduction studies at age 5 are summarized in Table 1. MCVs of median, ulnar, peroneal and tibial nerves (from 25 to 30.1 m/s) were in the classic CMT demyelinating range [2], just proximal ulnar nerve conduction (axilla–elbow) being in the intermediate range (39.8 m/s). CMAP amplitudes were reduced or in the lower limit of normal (Table 1), their morphology being preserved. DML were systematically prolonged. TLI was not determined. Sensory conduction velocity (SCV) of median nerve, exhibiting normal sensory nerve action potential, was slowed (30.6 m/s; see Table 1). EMG was not done.
Molecular genetic work up
When the proband was initially evaluated in 2008 (note that her affected son was born in 2009), she was the only patient of the pedigree, that is, we were confronted with an apparently sporadic syndrome. By then, our working diagnosis was early onset cerebellar ataxia (EOCA) and sensorimotor polyneuropathy. We therefore planned a molecular diagnosis of autosomal recessive cerebellar ataxias (ARCA) and SCA [39] performed at the “Centre de Diagnòstic Genètic i Molecular (CDGM)-Institut de Recerca Biomèdica de Bellvitge (IDIBELL)”, which is a reference Spanish laboratory for diagnosis of hereditary ataxias. Molecular analyses comprised screening of FRDA, SACS, SETX, SCA1, SCA2, SCA3, SCA7, SCA8, SCA12, SCA17 and DRPLA genes, no mutations being found. We also had planned to screen ATM and APTX, which had remained pending on the laboratory availability.
Three years later and after learning that the proband’s son was affected and that his phenotype was an early-onset hereditary neuropathy, it became clear to us that we were confronted with a heterogeneous DI-CMT syndrome, which, in the proband, included semeiology of the peripheral and central nervous system. Therefore, we investigated the six DI-CMT categories so far reported [8, 9, 11] using whole exome sequencing (WES) of the two affected individuals. The analysis revealed a c.293A>G (p.N98S) missense mutation in NEFL, co-segregating with disease in the proband (III-2) and her affected son (IV-1). The mutation targets highly conserved nucleotide (GERP score = 5.27) and amino acid residues and is predicted to have damaging effects on the NEFL protein (PolyPhen2 score = 1.000 (probably damaging), SIFT score = 0 (deleterious). Moreover, the N98S NEFL mutation has previously been associated with CMT (dbSNP allele: rs58982919 and ClinVar ID: RCV000034136.2). The WES analysis showed no additional mutations in any of the causal genes known to date for CMT or SCA.
Discussion
The current paper describes a heterogeneous DI-CMT pedigree associated with a NEFL N98S mutation, which comprises the proband and her affected son, who was born 1 year after his mother’s evaluation. The propositus, aged 38, presented with delayed motor milestones. As of the second decade, there appeared distal weakness and sensory loss, pes cavus, clawing hands, cerebellar ataxia, nystagmus and dysarthria, she being wheelchair bound with severe CMTNS. Our initial working diagnosis was that of a sporadic neurodegenerative disorder, which could be categorized within EOCA with peripheral sensorimotor neuropathy. Interestingly, initial diagnoses of Friedreich ataxia or spinocerebellar ataxia had previously been established in other NEFL-related CMT patients [16, 20]; furthermore, episodic ataxia or pregnancy-aggravated ataxia may occur in CMT2E patients [17, 38]. The presence of cerebellar pathology, whose clinical manifestations had been largely masked here by her severe polyneuropathic signs, was corroborated by brain MRI showing marked cerebellar atrophy. We therefore decided to screen mutations associated with ARCA and SCA, though we have to admit that intermediately slowed MCV has only been reported in ARSACS [39, 40]. No pathogenic mutations were found. The proband’s affected son, aged 5, showed delayed motor milestones, his examination demonstrating subtle signs that consisted of lower-limb areflexia and hypotonia with mild CMTNS.
As a whole, nerve conduction studies in both patients showed a pattern of conduction slowing that could be classified as demyelinating or intermediate. We have to admit that proband’s median nerve MCV in the segment elbow–wrist was in the demyelinating range (18.3 m/s), a feature that could be accounted for by severe distal CMAP attenuation (0.2 mV) indicating loss of rapidly conduction fibers. Remarkably, prolonged DML was a constant feature that is not reported in CMT2 with decreased MCV due to severe loss of large-diameter fibers [41]. Here, TLI was preserved indicating that a phenomenon of disproportionate distal demyelination was negligible [8, 31]. Wishing to clarify the issue we explored a proximal nerve segment, axilla–elbow, observing that recorded MCV, 47.5 m/s, was within the intermediate range (note that normal value for this segment is ≥55). Intriguingly, comparable differential electrophysiological features between distal versus proximal segments were observed in ulnar nerve, reinforcing the need of exploring proximal upper-limb nerve segments for an accurate definition of pathologic nerve conduction pattern [7, 8].
NEFL-related CMT syndromes have been classified within CMT2E or CMT1F. There are seven papers with figures illustrating sural nerve pathology [15–18, 41–43]. The common pathological hallmark is loss of large myelinated axons with variable signs of regeneration, axonal atrophy, and demyelination secondary to focal swellings (“giant axons”). We wish to focus on the paper by Yum et al. [18] containing nerve transverse ultrathin sections at low magnification and scale bar. Their Fig. 4A (case II-4, aged 16 years) illustrates reduced density of myelinated axons, all of which are small with a maximum diameter of 3 µm. Applying a conversion factor of 6 [44], the conduction velocity of this nerve would be 18 m/s, namely, comparable to MCV of median nerve, 22 m/s (CMAP, 1.2 mV), and ulnar nerve, 17 m/s (CMAP, 0.1 mV). Wisely the authors interpret that the reduced axonal caliber results in markedly slowed conduction velocities well into the “demyelinating range”. The correct formation of an axonal network of neurofilaments is crucial for the establishment and maintenance of axonal caliber and axonal transport, and consequently for the optimization of conduction velocity [45]. The mice model NEFL N98S/+ has revealed multiple inclusions in the cell bodies and proximal axons of spinal cord neurons, whereas the predominant lesions in sciatic nerve are a reduction in the number of neurofilaments and a decrease in the axonal diameters [46]. Conceivably, maldevelopment of neurofilaments may lead to hypotrophy of myelinated fibers manifesting with nerve conduction slowing, which might be in the intermediate or in the demyelinating range. As observed here and in a previous study [8], electrophysiology of median and ulnar nerves may show an intermediate MCV slowing with relative preservation of CMAPs in axilla–elbow segment, and accentuated MCV slowing with severe CMAP attenuation in elbow–wrist segment. Such electrophysiological findings are consistent with our muscle MRI findings (see next paragraph). In short, in DI-CMT probably operates a length-dependent axonopathy, which account for distally accentuated clinico-electrophysiological and imaging changes. Obviously, our interpretation does not exclude the pathogenic role of Schwann cell proliferation and demyelination caused NEFL mutation [16], though NEFL is not prominently expressed by myelinating Schwann cells [18].
On T1-weighted MR images of lower-limb musculature of the proband, there was widespread fatty atrophy, which is categorized as massive in intrinsic foot musculature, marked in lower-leg muscles and moderate in thigh muscles. Such topographic findings strongly suggest a process of length-dependent muscle denervation [8, 35, 47, 48]. On water sensitive sequences (here T2FS) we observed edema of calf muscles, a sign suggestive of subacute denervation [8, 35, 47]. This is in good correlation with electromyography showing active denervation potentials in tibialis anterior. Taken together, such imaging and electrophysiological features strongly suggest that nerve inflammatory changes superimposed on the genetic condition might be pathogenic in the process of denervation [35].
The current heterozygous NEFL N98S missense mutation has been reported in five previous DI-CMT patients [15, 19, 26, 27]; together with our patients, their clinical data appear summarized in Table 2. Six patients exhibited early onset with delayed motor milestones. Severe sensorimotor neuropathy occurred in all patients but one, the proband’s son in the current pedigree, at age 5 showing mild neuropathic signs in lower limbs; the question is whether he, like his mother, will worsen as of the second decade. Median nerve MCV was within the intermediate range except in two patients with severe CMAP attenuation, who showed conduction slowing in the demyelinating range; loss of large myelinated fibers could account for this phenomenon (see above). Additional clinical features include a variable combination of motor delay, mental retardation, hearing loss and nystagmus. The current proband’s phenotype consisting of severe cerebellar ataxia and sensorimotor neuropathy, mimicking some ARCA syndromes, had not previously been reported. There are no autopsy studies to establish clinico-pathological correlations. In N98S knock-in mice showing severe phenotype, pathological changes were widespread involving spinal motor neurons and their axons, cerebellum, cerebral cortex and pons [46].
We conclude that the NEFL N98S mutation is associated with a DI-CMT phenotype characterized by early-onset sensorimotor neuropathy delaying motor milestones, which may evolve into a severe and complex clinical picture including cerebellar ataxia.
References
Combarros O, Calleja J, Polo JM, Berciano J (1987) Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol Scand 75:9–12
Harding AE, Thomas PK (1980) The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 103:259–280
Davis CJ, Bradley WG, Madrid R (1978) The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J Genet Hum 26:311–349
Madrid R, Bradley WG, Davis CJ (1977) The peroneal muscular atrophy syndrome. Clinical, genetic, electrophysiological and nerve biopsy studies. Part 2. Observations on pathological changes in sural nerve biopsies. J Neurol Sci 32:91–122
Bouché P, Gherardi R, Cathala HP, Lhermitte F, Castaigne P (1983) Peroneal muscular atrophy. Part 1. Clinical and electrophysiological study. J Neurol Sci 61:389–399
Nicholson G, Myers S (2006) Intermediate forms of Charcot–Marie–Tooth neuropathy: a review. Neuromolecular Med 8:123–130
Banchs I, Casasnovas C, Montero J, Volpini V, Martínez-Matos JA (2010) Charcot–Marie–Tooth disease with intermediate conduction velocities caused by a novel mutation in the MPZ gene. Muscle Nerve 42:184–188
Berciano J, García A, Peeters K, Gallardo E, De Vriendt E, Pelayo-Negro AL, Infante J, Jordanova A (2015) NEFL E396K mutation is associated with a novel dominant intermediate Charcot–Marie–Tooth disease phenotype. J Neurol 262:1289–1300
Rossor AM, Polke JM, Houlden H, Reilly MM (2013) Clinical implications of genetic advances in Charcot–Marie–Tooth disease. Nat Rev Neurol 9:562–571
Nicholson G, Nash J (1993) Intermediate nerve conduction velocities define X-linked Charcot–Marie–Tooth neuropathy families. Neurology 43:2558–2564
Liu L, Zhang R (2014) Intermediate Charcot–Marie–Tooth disease. Neurosci Bull 30:999–1009
Mersiyanova IV, Perepelov AV, Polyakov AV, Sitnikov VF, Dadali EL, Oparin RB, Petrin AN, Evgrafov OV (2009) A new variant of Charcot–Marie–Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet 67:37–46
De Jonghe P, Mersivanova I, Nelis E, Del Favero J, Martin JJ, Van Broeckhoven C, Evgrafov O, Timmerman V (2001) Further evidence that neurofilament light chain gene mutations can cause Charcot–Marie–Tooth disease type 2E. Ann Neurol 49:245–249
Georgiou DM, Zidar J, Korosec M, Middleton LT, Kyriakides T, Christodoulou K (2002) A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics 4:93–96
Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, Martin JJ, Butler IJ, Mancias P, Papasozomenos SCh, Terespolsky D, Potocki L, Brown CW, Shy M, Rita DA, Tournev I, Kremensky I, Lupski JR, Timmerman V (2003) Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot–Marie–Tooth disease. Brain 126:590–597
Züchner S, Vorgerd M, Sindern E, Schröder JM (2004) The novel neurofilament light (NEFL) mutation Glu397Lys is associated with a clinically and morphologically heterogeneous type of Charcot–Marie–Tooth neuropathy. Neuromuscul Disord 14:147–157
Fabrizi GM, Cavallaro T, Angiari C, Cabrini I, Taioli F, Malerba G, Bertolasi L, Rizzuto N (2007) Charcot–Marie–Tooth disease type 2E, a disorder of the cytoskeleton. Brain 130:394–403
Yum SW, Zhang J, Mo K, Li J, Scherer SS (2009) A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy. Ann Neurol 66:759–770
Abe A, Numakura C, Saito K, Koide H, Oka N, Honma A, Kishikawa Y, Hayasaka K (2009) Neurofilament light chain polypeptide gene mutations in Charcot–Marie–Tooth disease: nonsense mutation probably causes a recessive phenotype. J Hum Genet 54:94–97
Miltenberger-Miltenyi G, Janecke AR, Wanschitz JV, Timmerman V, Windpassinger C, Auer-Grumbach M, Löscher WN (2007) Clinical and electrophysiological features in Charcot–Marie–Tooth disease with mutations in the NEFL gene. Arch Neurol 64:966–970
Pisciotta C, Bai Y, Brennan KM, Wu X, Grider T, Feely S, Wang S, Moore S, Siskind C, Gonzalez M, Zuchner S, Shy ME (2015) Reduced neurofilament expression in cutaneous nerve fibers of patients with CMT2E. Neurology 85:228–234
Lin KP, Soong BW, Yang CC, Huang LW, Chang MH, Lee IH, Antonellis A, Lee YC (2011) The mutational spectrum in a cohort of Charcot–Marie–Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 6:e29393
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 69:22–33
Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MP, Bull K, Ramdharry G, Davis M, Blake JC, Houlden H, Reilly MM (2012) Charcot–Marie–Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83:706–710
Sivera R, Sevilla T, Vílchez JJ, Martínez-Rubio D, Chumillas MJ, Vázquez JF, Muelas N, Bataller L, Millán JM, Palau F, Espinós C (2013) Charcot–Marie–Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81:1617–1625
Yoshihara T, Yamamoto M, Hattori N, Misu K, Mori K, Koike H, Sobue G (2002) Identification of novel sequence variants in the neurofilament-light gene in a Japanese population: analysis of Charcot–Marie–Tooth disease patients and normal individuals. J Peripher Nerv Syst 7:221–224
Baets J, Deconinck T, De Vriendt E, Zimoń M, Yperzeele L, Van Hoorenbeeck K, Peeters K, Spiegel R, Parman Y, Ceulemans B, Van Bogaert P, Pou-Serradell A, Bernert G, Dinopoulos A, Auer-Grumbach M, Sallinen SL, Fabrizi GM, Pauly F, Van den Bergh P, Bilir B, Battaloglu E, Madrid RE, Kabzińska D, Kochanski A, Topaloglu H, Miller G, Jordanova A, Timmerman V, De Jonghe P (2011) Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain 134:2664–2676
García A, Combarros O, Calleja J, Berciano J (1998) Charcot–Marie–Tooth disease type 1A with 17p duplication in infancy and early childhood: a longitudinal clinical and electrophysiologic study. Neurology 5:1061–1067
Berciano J, Gallardo E, García A, Infante J, Mateo I, Combarros O (2006) Charcot–Marie–Tooth disease type 1A duplication with severe paresis of the proximal lower limb muscles: a long-term follow-up study. J Neurol Neurosurg Psychiatry 77:1169–1176
Shy ME, Blake J, Krajewski K, Fuerst DR, Laura M, Hahn AF, Li J, Lewis RA, Reilly M (2005) Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 64:1209–1214
Amato AA, Gronseth GS, Callerame KJ, Kagan-Hallet KS, Bryan WW, Barohn RJ (1996) Tomaculous neuropathy: a clinical and electrophysiological study in patients with and without 1.5-Mb deletions in chromosome 17p11.2. Muscle Nerve 19:16–22
Chiappa KH (1997) Evoked potentials in clinical medicine, 3rd edn. Lippincott-Raven Press Publishers, Philadelphia-New York
Cruz Martínez A, Pérez Conde MC, Del Campo F, Barrio M, Gutiérrez AM, López E (1978) Sensory and mixed conduction velocity in infancy and childhood: I. Normal parameters in median, ulnar and sural nerves. Electromiogr Clin Neurophysiol 18:487–504
García A, Calleja J, Antolín FM, Berciano J (2000) Peripheral motor and sensory nerve conduction studies in normal infants and children. Clin Neurophysiol 111:513–520
Gallardo E, García A, Combarros O, Berciano J (2006) Charcot–Marie–Tooth disease type 1A duplication: spectrum of clinical and magnetic resonance imaging features in leg and foot muscles. Brain 129:426–437
Reumers J, De Rijk P, Zhao H, Liekens A, Smeets D, Cleary J, Van Loo P, Van Den Bossche M, Catthoor K, Sabbe B, Despierre E, Vergote I, Hilbush B, Lambrechts D, Del-Favero J (2011) Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol 18(30):61–68
Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, Huynh Cong E, Arrondel C, Tête MJ, Montjean R, Richard L, Karras A, Pouteil-Noble C, Balafrej L, Bonnardeaux A, Canaud G, Charasse C, Dantal J, Deschenes G, Deteix P, Dubourg O, Petiot P, Pouthier D, Leguern E, Guiochon-Mantel A, Broutin I, Gubler MC, Saunier S, Ronco P, Vallat JM, Alonso MA, Antignac C, Mollet G (2011) INF2 mutations in Charcot–Marie–Tooth disease with glomerulopathy. N Engl J Med 365:2377–2388
Gentile L, Taioli F, Fabrizi GM, Russo M, Stancanelli C, Mazzeo A (2013) Considerable post-partum worsening in a patient with CMT2E. Neurol Sci 34:1813–1814
Berciano J, García A, Infante J (2013) Peripheral nerve involvement in hereditary cerebellar and multisystem degenerative disorders. Handb Clin Neurol 115:907–932
García A, Criscuolo C, de Michele G, Berciano J (2008) Neurophysiological study in a Spanish family with recessive spastic ataxia of Charlevoix-Saguenay. Muscle Nerve 37:107–110
Fabrizi GM, Cavallaro T, Angiari C, Bertolasi L, Cabrini I, Ferrarini M, Rizzuto N (2004) Giant axon and neurofilament accumulation in Charcot–Marie–Tooth disease type 2E. Neurology 62:1429–1431
Elbracht M, Senderek J, Schara U, Nolte K, Klopstock T, Roos A, Reimann J, Zerres K, Weis J, Rudnik-Schöneborn S (2014) Clinical and morphological variability of the E396K mutation in the neurofilament light chain gene in patients with Charcot–Marie–Tooth disease type 2E. Clin Neuropathol 33:335–343
Shin JS, Chung KW, Cho SY, Yun J, Hwang SJ, Kang SH, Cho EM, Kim SM, Choi BO (2008) NEFL Pro22Arg mutation in Charcot–Marie–Tooth disease type 1. J Hum Genet 53:936–940
Waxman SG (1980) Determinants of conduction velocity in myelinated nerve fibers. Muscle Nerve 3:141–150
Perrot R, Berges R, Bocquet A, Eyer J (2008) Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol 38:27–65
Adebola AA, Di Castri T, He C, Salvatierra LA, Zhao J, Brown K, Lin C, Worman HJ, Liem RK (2015) Neurofilament light polypeptide gene N98S mutation in mice leads to neurofilament network abnormalities and a Charcot–Marie–Tooth Type 2E phenotype. Hum Mol Genet 24:2163–2174
Kamath S, Venkatanarasimha N, Walsh MA, Hughes PM (2008) MRI appearance of muscle denervation. Skeletal Radiol 37:397–404
Chung KW, Suh BC, Shy ME, Cho SY, Yoo JH, Park SW, Moon H, Park KD, Choi KG, Kim S, Kim SB, Shim DS, Kim SM, Sunwoo IN, Choi BO (2008) Different clinical and magnetic resonance imaging features between Charcot–Marie–Tooth disease type 1A and 2A. Neuromuscul Disord 18:610–618
Acknowledgments
We are grateful to Dr. Victor Volpini for molecular screening of SCA and ARCA genes, Mrs. Marta de la Fuente for Secretarial assistance, and Mrs. Coro Sánchez for laboratory help, and Miss. Mar Ruiz for technical support. The study was supported by Research Institute of University Hospital “Marqués de Valdecilla” (IDIVAL) Grants BFR 05/10 and WLA 03/12. The Antwerp team was funded in part by the University of Antwerp (TOP BOF 29069); the Fund for Scientific Research-Flanders (FWO) and the Belgian Association against Neuromuscular Disorders (ABMM). K.P. is supported by a Ph.D. fellowship from the Fund for Scientific Research-Flanders (FWO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
We declare no conflicts of interest.
Ethical standard
Ethical approval was obtained from the proband patient and her husband. This investigation was approved by the “Comité Ético de Investigación Clínica de Cantabria”.
Additional information
J. Berciano and K. Peeters have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Berciano, J., Peeters, K., García, A. et al. NEFL N98S mutation: another cause of dominant intermediate Charcot–Marie–Tooth disease with heterogeneous early-onset phenotype. J Neurol 263, 361–369 (2016). https://doi.org/10.1007/s00415-015-7985-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7985-z