
Abstract
Autosomal dominant ataxia type 14 (SCA14) is a rare usually adult-onset progressive disorder with cerebellar neurodegeneration caused by mutations in protein kinase C gamma. We set out to examine cerebellar and extracerebellar neurochemical changes in SCA14 by MR spectroscopy. In 13 SCA14 patients and 13 healthy sex- and age-matched controls, 3-T single-voxel brain proton MR spectroscopy was performed in a cerebellar voxel of interest (VOI) at TE = 30 ms to obtain a neurochemical profile of metabolites with short relaxation times. In the cerebellum and in additional VOIs in the prefrontal cortex, motor cortex, and somatosensory cortex, a second measurement was performed at TE = 144 ms to mainly extract the total N-acetyl-aspartate (tNAA) signal besides the signals for total creatine (tCr) and total choline (tCho). The cerebellar neurochemical profile revealed a decrease in glutathione (6.12E−06 ± 2.50E−06 versus 8.91E−06 ± 3.03E−06; p = 0028) and tNAA (3.78E−05 ± 5.67E−06 versus 4.25E−05 ± 5.15E−06; p = 0023) and a trend for reduced glutamate (2.63E−05 ± 6.48E−06 versus 3.15E−05 ± 7.61E−06; p = 0062) in SCA14 compared to controls. In the tNAA-focused measurement, cerebellar tNAA (296.6 ± 42.6 versus 351.7 ± 16.5; p = 0004) and tCr (272.1 ± 25.2 versus 303.2 ± 31.4; p = 0004) were reduced, while the prefrontal, somatosensory and motor cortex remained unaffected compared to controls. Neuronal pathology in SCA14 detected by MR spectroscopy was restricted to the cerebellum and did not comprise cortical regions. In the cerebellum, we found in addition to signs of neurodegeneration a glutathione reduction, which has been associated with cellular damage by oxidative stress in other neurodegenerative diseases such as Parkinson’s disease and Friedreich’s ataxia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The spinocerebellar ataxias are autosomal dominantly inherited usually adult-onset movement disorders with progressive cerebellar neurodegeneration. Spinocerebellar ataxia type 14 (SCA14) is caused by missense or deletion mutations in the protein kinase C gamma gene (PRKCG) [1]. Protein kinase C gamma (PKCγ) is highly expressed in cerebellar Purkinje cells and SCA14 mutations are associated with aggregate formation, disrupted synaptic plasticity and Purkinje cell death [2, 3]. Patients present a slowly progressive cerebellar ataxia and age of onset may range from childhood to the seventh decade. Clinical reports on additional symptoms like cognitive decline, hyper-reflexivity and myoclonus might reflect cortical damage [4]. So far, MRI case series have shown mild to severe cerebellar midline atrophy [4–9] with few exceptions of additional pontine, brainstem or cerebral atrophy [10, 11].
Magnetic resonance spectroscopy (MRS) allows the detection of specific biochemical markers of neurodegeneration in brain tissue in vivo, which even may have the potential to evaluate therapeutic interventions [12]. For instance, the widely applied measurement of total N-acetylaspartate (tNAA) resonance allows assessing the level of neuroaxonal integrity at early disease stages. Thus, it represents a useful surrogate marker for neurodegeneration in many diseases and has previously been applied in other spinocerebellar ataxias [13–18]. The group of Öz et al. used a different MRS method applying short relaxation times to find disease-specific patterns of neurochemical changes, even in ataxia patients [19, 20]. With such an MRS approach, a spectrum of additional neurochemical alterations of amino acids relevant to neural transmission, cell structure and cell energy metabolism can be quantified to map the neurometabolic state non-invasively. We set out to examine ataxic individuals with mutations in the PRKCG gene in comparison to age- and gender-matched healthy controls with a similar sensitive single-voxel MRS in the cerebellum and cerebrocortical regions.
Materials and methods
Participants
We conducted a cross-sectional study with matched pair design. Thirteen patients with confirmed mutations in the PRKCG gene and progressive cerebellar ataxia and thirteen pairwise sex- and age-matched healthy controls were prospectively recruited. Based on the mutations and segregation within the pedigree, the diagnosis of SCA14 was confirmed in ten cases and possible in three cases (previously not described genetic variants, segregation analyses not feasible). Inclusion criteria were: age 18–75 years, genetically confirmed missense or deletion mutations in the PRKCG gene, and no condition prohibiting the use of MRI. Exclusion criteria were lack of ability to communicate, and degenerative or inflammatory diseases of the central nervous system, apart from SCA14 in the patient group. There were no clinically evident cognitive abnormalities on neurological examination. However, a cognitive screening test for mild cognitive impairment and early dementia (DemTect) [21] revealed scores below the cutoff in 3/13 patients tested, potentially indicating mild cognitive impairment/dementia.
For all patients, the following clinical data were obtained: age at onset, disease duration, molecular genetic test results, scale for the assessment and rating of ataxia (SARA) scores including subscores (gait and posture (items 1–3); limb kinetic (items 5–8) and inventory of non-ataxia symptoms (INAS) scores [22, 23]. An overview of the demographic and clinical characteristics is given in Table 1.
Magnetic resonance spectroscopy
All spectra were acquired with a clinical 3-T MR Scanner (Siemens Magnetom Trio, Erlangen, Germany), in the standard circular polarized head coil. Sagittal, coronal and transversal T2-weighted turbo spin-echo MR sequence (FOV: 480 × 512, 14 slices transversal, coronal and sagittal, 7 mm slice thickness, TR = 4900 ms, TE = 111 ms, echo number: 20) were used for voxel of interest (VOI) positioning and exclusion of any further relevant pathology than cerebellar atrophy.
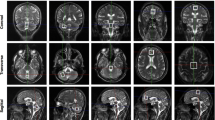
Spectra from the cerebellum and three additional locations with the following VOI dimensions were acquired: VOI: 20 × 30 × 20 mm in the right cerebellar hemisphere, 30 × 20 × 20 mm for prefrontal cortex measurements, 30 × 20 × 20 mm for motor cortex measurements, and 30 × 20 × 20 mm for somatosensory cortex measurements. A sample VOI positioning is shown in Fig. 1.

Spectroscopy voxel positions. Sample voxel positioning in coronal, sagittal and transverse brain sections for cerebellar, prefrontal, motor and somatosensory cortex voxel
We were specifically interested in obtaining a neurochemical profile of the cerebellum as clinically main affected region. Hence, the cerebellar VOI measurement included one with short TE = 30 ms to assess metabolites with short relaxation times. Here, the clearly discernible metabolites glutamine (Gln), glutamate (Glu), glutathione (GSH), inositol (Ins), total choline (tCho), tNAA, total Creatine (tCr), lactate (Lac) and the combined value of glutamate and glutamine (Glx) were included in the analysis.
All four VOIs (cerebellar, prefrontal, somatosensory, and motor cortex) were measured with an echo time of TE = 144 ms to mainly extract the signals from tNAA consisting of the neurotransmitter NAA-glutamate (NAAG) and its precursor NAA, tCr and tCho resonances. This improves the fit quality for tNAA-focused examinations because the signals of other interfering metabolites are already mostly decayed. Please refer for methodological details to supplement 1.
Statistical analysis
MRS metabolite data were normally distributed (Shapiro–Wilk tests). For group comparisons, we used paired sample t tests. To account for the small sample size, we additionally performed Wilcoxon signed ranked tests, which in all cases resulted in similar p values and identical significance levels (not shown). One matched pair was excluded from this analysis because of insufficient MRS data quality in the healthy control measurement. The correlation between SARA and MRS metabolites was analyzed using Spearman’s Rho. Statistical analysis was performed with SPSS version 21 (IBM, Armonk, NY, USA). A p value < 0.05 was considered significant. No correction for multiple testing was applied in this exploratory study.
Results
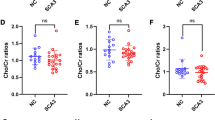
The investigation of the cerebellum as primarily affected region using a scan with short TE generated a neurochemical profile including metabolites with short relaxation times (Table 2; Fig. 2). We found a decrease of GSH and tNAA and trends towards reductions of glutamate and tCr in SCA14 patients. The GSH detection accuracy was limited to a standard deviation of approximately SD ≈ 35 %. This is due to the fact that the GSH resonances interfere with other signals, especially the creatine resonance at 3 ppm. No significant changes were detected in tCho, Glu, Ins or Lac (Table 2; Fig. 2) and none of the metabolites measured at short TE did correlate with the clinical SARA score or subscores (data not shown). NAA changes as sensitive indicator of neurodegeneration were also investigated by tNAA-focused measurements (TE = 144 ms) in the cerebellum. tNAA and NAA but not NAAG, the second component of tNAA, were reduced in SCA14 patients. Likewise, tCr was reduced in patients compared to healthy controls (Table 3; Fig. 3).

Results for short echo time measurements (TE = 30 ms). a Fit results of several metabolites for cerebellar short echo time spectra. Values are shown in arbitrary units for patients in comparison to healthy controls. b Short echo example spectra of the cerebellum. The corresponding fit results are: Patient: tNAA = 2.92e−6 ± 3 %, tCr = 3.78e−5 ± 3 %, tCho = 9.72e−6 ± 3 %, Glu = 2.9e−5 ± 10 %, Gln = 2.59e−5 ± 14 %, GSH = 3.68e−6 ± 34 %, GABA = 6.03e−6 ± 45 %, Lac = 5.26e−6 ± 52 % Control: tNAA = 4.63e−5 ± 5 %, tCr = 4.73e−5 ± 3 % tCho = 1.13e−5 ± 3 %, Glu = 3.75e−5 ± 8 %, Gln = 2.35e−5 ± 15 %, GSH = 6.64e−6 ± 23 %, GABA = 1.18e−5 ± 28 %, Lac = 3.05e−6 ± 101 %. All values are in arbitrary units

Results for long echo time measurements (TE = 144 ms). a tNAA and NAA are reduced in the cerebellar voxel but not in the prefrontal, motor or somatosensory cortex voxels in SCA14 patients compared to controls. b Typical cerebellar and motor cortex spectra examples of a patient with SCA14 compared to a healthy control. The fit results in arbitrary units for the cerebellar measurements are: patient: tNAA = 212 ± 7 %, tCr = 254 ± 3 %, tCho = 78 ± 3 %; Healthy control: tNAA = 369 ± 2 %, tCr = 296 ± 3 %, tCho = 89 ± 3 %. The fit results for the motor cortex measurements are: Patient: tNAA = 366 ± 1 %, tCr = 205 ± 2 %, tCho = 69 ± 2 %; Healthy control: tNAA = 165 ± 5 %, tCr = 106 ± 3 %, 27.5 ± 5 %. All values are in arbitrary units. c NAA and NAAG reduction in SCA14 patients correlates with SARA cerebellar score
While cerebellar tNAA as for the shorter TE did not correlate with SARA score and subscores in SCA14 patients (p = 0.261), its components cerebellar NAA and NAAG showed opposing correlations: NAA decreased as the SARA limb kinetic score increases (Spearman’s Rho = −0.565, p = 0.044) and a similar correlation is seen for SARA posture and gait score (Spearman’s Rho = −0.526, p = 0.065). The correlation between NAAG and SARA score on the other hand showed a direct correlation for SARA limb kinetic (Spearman’s Rho = 0.654, p = 0.015) and SARA posture and gait (Spearman’s Rho = 0.598, p = 0.031) scores. Thus, more severe ataxia in SCA14 patients is accompanied by lower NAA concentrations, but at the same time increased NAAG concentrations (Fig. 3c).
When cerebellar metabolites were normalized to the reduced tCr, we found still a trend towards a GSH reduction in the measurement with short TE = 30 ms, but not for the other metabolites (tNAA, glutamate) (Supplementary Table 4). However, the correlation of NAA and NAAG with SARA subscores remained significant.
In the prefrontal, motor and somatosensory cortical regions, tNAA-focused measurements (TE = 144 ms) revealed no differences for tNAA, tCr, and tCho between SCA14 patients and matched controls (Table 2; Fig. 2) and no correlation existed between SARA score and any metabolite in these regions.
Discussion
In this first controlled imaging study in SCA14, we investigated the cerebellar and cerebrocortical pattern of neurodegeneration using MR spectroscopy. The main findings were (1) the restriction of tNAA deficiency to the cerebellum with NAA and NAAG correlations to SARA score while cerebrocortical regions remained unaffected and (2) the unexpected finding of GSH deficiency in the cerebellum, which may indicate an impaired antioxidative defense system in the cerebellum of SCA14 patients.
The exclusively cerebellar tNAA deficit seen in our study is in line with previous neuroimaging case reports on SCA14 that depicted primarily a pure cerebellar atrophy most pronounced in the vermis [4, 6–9, 11, 24]. Neuropathological case reports showed in accordance a predominant cerebellar Purkinje cell loss without involvement of basal ganglia or cerebral cortex [25, 26].
Voxel-based MR spectroscopy restricts measurements to few predefined regions due to examination time constraints. We focused on the cerebellum and selected cerebrocortical areas. Pathology in other regions or very small tNAA changes in cerebrocortical areas may have escaped detection. To avoid measurement artifacts, we placed the cerebellar voxel in the cerebellar hemisphere and not in the macroscopically most affected vermis, where we may have detected a more pronounced pathology. Given that SCA14 is a rare disorder, only a limited sample size was available. In spite of these limitations, this study revealed robust results for the cerebellum and the identical tNAA optimized MRS technique had previously been capable of detecting cerebrocortical neuronal damage in a similar sample size of SCA1 patients [18]. In concordance with our MRS results, extracerebellar symptoms in our SCA14 patient cohort were limited as reflected by the patients’ low INAS scores. Previous reports described extracerebellar symptoms related to cortical involvement for SCA14 such as stimulus-sensitive myoclonus and cognitive dysfunction to a variable extent. In line with our cohort, these deficits were usually mild compared to the trinucleotide repeat-expansion disorders SCA1, 2 and 3. Reduction of cerebellar NAA is a common finding in SCA1, 2 and 3 and MSA-C [14, 18, 20], but not in SCA 6 [15, 20]. In our study, an inverse correlation of cerebellar NAA with the clinical SARA score was shown, which supports NAA reduction as primary deficit related to clinical severity. The additional direct correlation of NAAG with the SARA score may suggest an imbalance between the two metabolites as a result of a molecular dynamics effect since NAAG is synthesized from NAA and glutamate [27], has multiple proposed functions such as glutamate storage and neurotransmission at metabotropic glutamate receptors [28], and comprises a source of glutamate and NAA, which both were reduced in our study. A cerebellar glutamate reduction, which may indicate disrupted glutamatergic neurotransmission, but overall can be seen as indication for neurodegeneration, has previously been described in SCA1, SCA2 and MSA-C, but not in SCA6 [20].
In the cerebellar neurochemical profile with short TE, cerebellar GSH was decreased in SCA14 patients, which has not been seen in other SCAs before [20]. When normalizing the cerebellar results to the reduced tCr, the GSH reduction still remained as trend; other metabolite changes were no longer detected while the correlations for (normalized) NAA/NAAG with the clinical SARA score remained significant. However, the tCr reduction in SCA14 patients may reflect neurodegenerative changes in brain tissue, since it is known that tCr levels differ between gray and white matter and are related to cell energy pathways. TCr is both the substrate and product of creatine kinase and reflects the energy potential available in brain tissue. In SCA1, SCA2 and MSA-C tCr has been found to be increased in the cerebellar hemisphere and was discussed as gliosis marker [19, 20]. Interestingly in Parkinson’s disease, tCr was reduced along with GSH selectively in the affected substantia nigra pars compacta [29]. As in Parkinson’s disease, the tCr and GSH reduction detected in SCA14 may reflect deficient oxidative metabolism. GSH eliminates the effects of reactive oxygen species under physiological conditions and its lack has been associated with impaired antioxidant defense in neurodegenerative disorders [30]. In general, a decrease of GSH can have two major reasons depending on the relevant pathophysiology. On the one hand, an increased oxidative load can lead to constant GSH utilization and its decrease. On the other hand, a genetic intermediary metabolism anomaly can cause a decrease in GSH, since GSH biochemistry is strongly interweaved with intermediary amino acid metabolism being synthesized of l-cysteine, l-glutamic acid and glycine by several enzymatic reactions. In this case, increased oxidative load is the result and not the cause of decreased GSH.
Interestingly, it has been described for primary GSH deficiencies, that a loss of GSH in the CNS may be associated with cerebellar ataxia and cognitive decline [31]. In ataxia disorders, cerebellar GSH has been evaluated by MR spectroscopy in SCA1 [19] and in Friedreich’s ataxia [32], where it was at control levels. Neuropathological data on GSH in neurodegenerative disorders are scarce and lacking for SCA14. However, single experimental studies on PKCγ knockout mice described increased susceptibility to oxidative stress [33]. GSH was reduced in substantia nigra tissue from Parkinson’s disease patients, whereas MR spectroscopy in vivo gave inconsistent results [30]. Nevertheless, in vivo MR spectroscopy in a Parkinson’s disease patient captured the reversion of GSH decrease after treatment with the precursor N-acetylcysteine [34] demonstrating its usefulness to monitor potential therapeutic interventions. Future studies should aim to assess GSH deficiency in independent samples and to elucidate the role of GSH reduction in SCA14, before it may be considered as disease-specific biomarker or therapeutic target.
Abbreviations
- CHESS:
-
Chemical shift selective suppression
- tCho:
-
Total choline
- tCr:
-
Total creatine
- Glu:
-
Glutamate
- Gln:
-
Glutamine
- GSH:
-
Glutathione
- Glx:
-
The combined value of glutamate and glutamine
- HC:
-
Healthy controls
- Ins:
-
Inositol
- Lac:
-
Lactate
- tNAA:
-
N-Acetylaspartate
- NAAG:
-
NAA-Glutamate
- PRESS:
-
Point-resolved spectroscopy sequence
- SCA14:
-
Spinocerebellar ataxia type 14
- VOI:
-
Volume of interest
References
Chen D-H, Brkanac Z, Verlinde CLMJ et al (2003) Missense mutations in the regulatory domain of pkc: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet 72:839–849
Seki T, Adachi N, Ono Y et al (2005) Mutant protein kinase Cgamma found in spinocerebellar ataxia type 14 is susceptible to aggregation and causes cell death. J Biol Chem 280:29096–29106. doi:10.1074/jbc.M501716200
Shuvaev AN, Horiuchi H, Seki T et al (2011) Mutant PKCγ in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J Neurosci 31:14324–14334. doi:10.1523/JNEUROSCI.5530-10.2011
Chen D-H, Cimino PJ, Ranum LPW et al (2005) The clinical and genetic spectrum of spinocerebellar ataxia 14. Neurology 64:1258–1260. doi:10.1212/01.WNL.0000156801.64549.6B
van de Warrenburg BPC, Verbeek DS, Piersma SJ et al (2003) Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 61:1760–1765. doi:10.1212/01.WNL.0000098883.79421.73
Vlak MHM, Sinke RJ, Rabelink GM et al (2006) Novel PRKCG/SCA14 mutation in a Dutch spinocerebellar ataxia family: expanding the phenotype. Mov Disord 21:1025–1028. doi:10.1002/mds.20851
Wieczorek S, Arning L, Gizewski ER et al (2007) Benign SCA14 phenotype in a German patient associated with a missense mutation in exon 3 of the PRKCG gene. Mov Disord 22:2135–2136. doi:10.1002/mds.21673
Koht J, Stevanin G, Durr A et al (2012) SCA14 in Norway, two families with autosomal dominant cerebellar ataxia and a novel mutation in the PRKCG gene. Acta Neurol Scand 125:116–122. doi:10.1111/j.1600-0404.2011.01504.x
Wedding IM, Koht J, Dietrichs E et al (2013) Cognition is only minimally impaired in Spinocerebellar ataxia type 14 (SCA14): a neuropsychological study of ten Norwegian subjects compared to intrafamilial controls and population norm. BMC Neurol 13:186. doi:10.1186/1471-2377-13-186
Klebe S, Durr A, Rentschler A et al (2005) New mutations in protein kinase Cgamma associated with spinocerebellar ataxia type 14. Ann Neurol 58:720–729. doi:10.1002/ana.20628
Hiramoto K, Kawakami H, Inoue K et al (2006) Identification of a new family of spinocerebellar ataxia type 14 in the Japanese spinocerebellar ataxia population by the screening of PRKCG exon 4. Mov Disord 21:1355–1360. doi:10.1002/mds.20970
Oz G, Vollmers ML, Nelson CD et al (2011) In vivo monitoring of recovery from neurodegeneration in conditional transgenic SCA1 mice. Exp Neurol 232:290–298. doi:10.1016/j.expneurol.2011.09.021
Lopes TM, D’Abreu A, França MC Jr et al (2013) Widespread neuronal damage and cognitive dysfunction in spinocerebellar ataxia type 3. J Neurol 260:2370–2379. doi:10.1007/s00415-013-6998-8
Lirng J-F, Wang P-S, Chen H-C et al (2012) Differences between spinocerebellar ataxias and multiple system atrophy-cerebellar type on proton magnetic resonance spectroscopy. PLoS One 7:e47925. doi:10.1371/journal.pone.0047925
Hadjivassiliou M, Wallis LI, Hoggard N et al (2012) MR spectroscopy and atrophy in Gluten, Friedreich’s and SCA6 ataxias. Acta Neurol Scand 126:138–143. doi:10.1111/j.1600-0404.2011.01620.x
Boesch SM, Schocke M, Bürk K et al (2001) Proton magnetic resonance spectroscopic imaging reveals differences in spinocerebellar ataxia types 2 and 6. J Magn Reson Imaging 13:553–559
Boesch SM, Wolf C, Seppi K et al (2007) Differentiation of SCA2 from MSA-C using proton magnetic resonance spectroscopic imaging. J Magn Reson Imaging 25:564–569. doi:10.1002/jmri.20846
Doss S, Brandt AU, Oberwahrenbrock T et al (2014) Metabolic Evidence for Cerebral Neurodegeneration in Spinocerebellar Ataxia Type 1. Cerebellum 13:199–206. doi:10.1007/s12311-013-0527-2
Oz G, Hutter D, Tkác I et al (2010) Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov Disord 25:1253–1261. doi:10.1002/mds.23067
Oz G, Iltis I, Hutter D et al (2011) Distinct neurochemical profiles of spinocerebellar ataxias 1, 2, 6, and cerebellar multiple system atrophy. Cerebellum 10:208–217. doi:10.1007/s12311-010-0213-6
Kalbe E, Kessler J, Calabrese P et al (2004) DemTect: a new, sensitive cognitive screening test to support the diagnosis of mild cognitive impairment and early dementia. Int J Geriatr Psychiatry 19:136–143. doi:10.1002/gps.1042
Schmitz-Hübsch T, du Montcel ST, Baliko L et al (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66:1717–1720. doi:10.1212/01.wnl.0000219042.60538.92
Jacobi H, Rakowicz M, Rola R et al (2013) Inventory of Non-Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum 12:418–428. doi:10.1007/s12311-012-0421-3
van de Warrenburg BPC, Verbeek DS, Piersma SJ et al (2003) Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 61:1760–1765. doi:10.1212/01.WNL.0000098883.79421.73
Brkanac Z, Bylenok L, Fernandez M et al (2002) A new dominant spinocerebellar ataxia linked to chromosome 19q13.4-qter. Arch Neurol 59:1291–1295. doi:10.1001/archneur.59.8.1291
Yamashita I, Sasaki H, Yabe I et al (2000) A novel locus for dominant cerebellar ataxia (SCA14) maps to a 10.2-cM interval flanked by D19S206 and D19S605 on chromosome 19q13.4-qter. Ann Neurol 48:156–163
Tyson RL, Sutherland GR (1998) Labeling of N-acetylaspartate and N-acetylaspartylglutamate in rat neocortex, hippocampus and cerebellum from [1-13C]glucose. Neurosci Lett 251:181–184
Neale JH, Bzdega T, Wroblewska B (2000) N-Acetylaspartylglutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. J Neurochem 75:443–452
Groger A, Kolb R, Schafer R, Klose U (2014) Dopamine reduction in the substantia nigra of parkinson’s disease patients confirmed by in vivo magnetic resonance spectroscopic imaging. PLoS One. doi:10.1371/journal.pone.0084081
Johnson WM, Wilson-Delfosse AL, Mieyal JJ (2012) Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 4:1399–1440. doi:10.3390/nu4101399
Ristoff E, Larsson A (2007) Inborn errors in the metabolism of glutathione. Orphanet J Rare Dis 2:16. doi:10.1186/1750-1172-2-16
Iltis I, Hutter D, Bushara KO et al (2010) (1)H MR spectroscopy in Friedreich’s ataxia and ataxia with oculomotor apraxia type 2. Brain Res 1358:200–210. doi:10.1016/j.brainres.2010.08.030
Lin D, Barnett M, Lobell S et al (2006) PKCgamma knockout mouse lenses are more susceptible to oxidative stress damage. J Exp Biol 209:4371–4378. doi:10.1242/jeb.02524
Holmay MJ, Terpstra M, Coles LD et al (2013) N-acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin Neuropharmacol 36:103–106. doi:10.1097/WNF.0b013e31829ae713
Acknowledgments
We thank Susan Pikol and Cynthia Kraut for excellent technical support and Hanna Zimmermann, Ella Kadas, Timm Oberwahrenbrock, and Leonora Zange for supporting the study logistics.
Conflicts of interest
SD received financial support for travel and research projects from Teva and Actelion and speakers honoraria from Actelion, SD reports no specific conflict of interest. JLR reports no conflict of interest. TSH received travel grants from Allergan. AUB is cofounder and associate of Motognosis. He received consulting fees, speaker honoraria and research grants from Novartis, Biogen Idec, Teva Pharmaceuticals, Bayer, and Heidelberg Engineering. He reports no conflict of interest in regard to the current study. SP and SM report no conflict of interest. JW serves on advisory boards for Novartis and Biogen Idec. He received a research grant from Novartis, and speaker honoraria from Bayer, Novartis, Teva and Biogen Idec. He is supported by the German ministry of science (BMBF/KKNMS). ME has no direct conflict of interests concerning this manuscript. He has received grant support from AstraZeneca and Sanofi, has participated in advisory board meetings of Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, MSD, Pfizer, Sanofi and has received honoraria from Astra Zeneca, Bayer, Berlin Chemie, Bristol-Myers Squibb, Boehringer-Ingelheim, Desitin, Eisei, Ever, Glaxo Smith Kline, MSD, Novartis, Pfizer, Sanofi, Takeda, Trommsdorff. TK receives/has received research support from the Deutsche Forschungsgemeinschaft (DFG), the Bundesministerium für Bildung und Forschung (BMBF) and the European Union (EU). He has received a lecture honorarium from Lundbeck and from Biogen Idec. He receives/has received royalties for book publications from Thieme, Urban & Schwarzenberg, Kohlhammer, Elsevier, Wissenschaftliche Verlagsgesellschaft Stuttgart and M. Dekker. MM reports no conflict of interest. FP reports to have received grant support from Sanofi, Beyer, Pfizer, and Teva where he sees no direct conflict of interest with this work. FP is supported by the Deutsche Forschungsgemeinschaft (DFG Exc 257), the Bundesministerium für Bildung und Forschung (BMBF Competence Network Multiple Sclerosis), and the European Union (FP 7, combims.eu).
Ethical standard
The study was approved by the local ethics committee of the Charité - Universitätsmedizin Berlin and the University of Bonn and have been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All participants gave informed written consent.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Doss, J. L. Rinnenthal, M. Minnerop, and F. Paul contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Doss, S., Rinnenthal, J.L., Schmitz-Hübsch, T. et al. Cerebellar neurochemical alterations in spinocerebellar ataxia type 14 appear to include glutathione deficiency. J Neurol 262, 1927–1935 (2015). https://doi.org/10.1007/s00415-015-7788-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7788-2