
Abstract
Granule cell neuronopathy (GCN) is a rare JC virus (JCV)-related disease in immunocompromised patients, characterized by lytic infection of the cerebellar granule cell layer. To enable early diagnosis and intervention, we identify features of GCN and describe possible aspects of disease heterogeneity. We report on two new cases of GCN in HIV-infected patients of whom we retrospectively assessed clinical and radiologic data. In addition, we carried out a literature search and review of clinical, radiologic and histopathologic findings of all published GCN cases. Including the two new cases reported here, a total of 18 GCN cases were included in this study. HIV infection, present in 12 of the cases, was the most common underlying condition, followed by monoclonal antibody treatment which was present in three cases. Cerebellar atrophy was detected in all except two cases. In 12 patients a heterogeneous distribution pattern of white matter changes in the cerebellum and brainstem was observed. Imaging findings in GCN are remarkably heterogeneous; exhibiting cerebellar atrophy, as well as white matter pathology, particularly in the adjacent infratentorial white matter. This suggests an overlap of GCN with other JCV-related diseases, such as progressive multifocal leukoencephalopathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The JC virus (JCV) is a neurotropic polyoma virus first described in the 1970s [1]. After primary infection, JCV establishes a latent and persistent infection in a variety of tissues such as tonsils, kidneys and the human gut and shows constant or periodic replication in some tissues, for instance in the epithelium of the urinary tract. Seroprevalence studies indicate that between 30 and 70 % of individuals are seropositive and seroprevalence increases with age [2]. JCV reactivation occurs frequently in immunocompetent and immunocompromised patients. In immunocompromised patients, JCV may cause a lytic infection of white and grey matter cells of the central nervous systems (CNS). Several JCV-associated diseases have been described, including progressive multifocal leukoencephalopathy (PML), JCV encephalopathy, JCV meningitis and JCV granule cell neuronopathy (GCN) [3].
PML is the most common JCV-associated disease, which is characterized by a lytic infection of oligodendrocytes, astrocytes and neurons, ultimately leading to demyelination and neuronal damage [4]. Most of the knowledge about PML in terms of clinical presentation, imaging characteristics, and prognosis, has been derived from human immunodeficiency virus (HIV)-infected patients. This was especially the case in the pre-highly active anti-retroviral therapy (HAART) era. However, in the past few years other conditions have been associated with PML, including treatment with monoclonal antibodies for autoimmune diseases, such as natalizumab in relapsing multiple sclerosis [5]. Due to its high sensitivity in the detection of PML pathology—even before the onset of clinical symptoms—brain magnetic resonance imaging (MRI) has been incorporated into PML diagnostic criteria and is currently recommended as a screening tool in surveillance programs of immunocompromised patients [6]. Despite its high sensitivity, it has been conclusively demonstrated in monoclonal antibody (e.g., natalizumab) associated PML that MRI findings, particularly at early stages of the disease, are heterogeneous and fluctuating [7–9].
More recently, other JCV-associated disease entities have been described in which lytic infection predominately occurs in grey matter cells. Amongst these, GCN is characterized by a selective infection of the granule cell neurons in the cerebellum, leading to neuronal loss and gliosis and cerebellar atrophy [10]. As in PML, most of the GCN cases have been described in HIV-infected patients [11–16]. However, GCN is even rarer than PML. Recent case reports have also documented GCN in patients treated with monoclonal antibodies, such as rituximab and natalizumab [17–19]. MRI is able to detect GCN related imaging findings such as cerebellar atrophy in GCN patients. In addition to neurodegenerative changes of the cerebellum, white matter changes have been described in GCN patients. This has led to a discussion on whether GCN is a separate disease entity, or just a PML variant with predominant cerebellar grey matter involvement [11, 13, 14, 17, 19–24]. Knowledge about the imaging findings of GCN is crucial to facilitate an early diagnosis. However, the imaging characteristics of all published GCN cases have not been systematically reviewed to date.
The aim of this study is to describe two additional cases of HIV GCN and review the literature in terms of underlying clinical conditions, histopathology and imaging findings in GCN patients.
Patients and methods
Case series
We retrospectively reviewed the medical records and serial imaging files of two HIV-associated GCN patients who were admitted to our clinic, with adherence to the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Literature review
In order to identify all reported GCN cases, we searched the NCBI PubMed database using the search terms: “granule cell neuronopathy”, “granular cell neuronopathy”, “cerebellar JC virus” and “JC virus cerebellum”. This yielded 109 potentially relevant articles. Papers in which both clinical presentation and imaging findings were described in sufficient detail were included. The reference sections of included articles were searched as well. The clinical information, histopathology (if available) and the imaging characteristics of each case were assessed.
Results
Case series
Case 1
A 45-year-old HIV-positive man presented with a 3-month history of gait instability of sub-acute onset. He had been seropositive for HIV for 10 years and HIV viral load was undetectable in plasma at the time of presentation. His treatment regimen consisted of efavirenz, tenofovir and emtricitabine and his absolute CD4+ T cell count fluctuated around 350 cells/mm3. His medical history revealed type 2 diabetes, hyperthyroidism and acute lymphoblastic leukemia for which he had been treated with various chemotherapies, as well as intrathecal methotrexate, cytarabine and dexamethasone. This had resulted in complete remission for the previous 5 years. There was no history of drug use or alcohol abuse.
At presentation, the neurological examination showed cerebellar gait disturbances with a positive Romberg test. In addition, the patient displayed non-fluent pursuit eye movements without nystagmus and decreased light touch sensation in the lower extremities. The finger–nose and heel–knee–shin movements were performed normally and there was no dysdiadochokinesia. Cognitive, motor and speech examinations were normal and deep tendon reflexes were symmetrically low with flexor plantar responses.
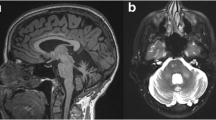
MRI of the brain and spinal cord showed marked cerebellar atrophy, in particular of the vermis, with no gadolinium enhancement or signs of PML or myelopathy (Fig. 1a–e). CSF analysis revealed a normal cell count and normal glucose and protein levels with negative tests for Borrelia and syphilis. Laboratory testing showed normal vitamins B12 and E and paraneoplastic neuronal antibodies (including anti-Hu, anti-Yo, anti-Ri, anti-Tr, anti-amphiphysin, anti-CV2 and anti-Ma2) were negative. EMG showed no evidence of polyneuropathy. Mutation analysis for both Friedreich’s ataxia and spinocerebellar ataxias (SCA) 1, 2, 3, 6 and 7 turned out negative.

MRI obtained from case 1 at the initial presentation (upper row, a–e) and 2 years later after clinical decline (bottom row, f–j). Sagittal FLAIR MR images (top row, a–c) showed mild cerebellar atrophy in the lateral cerebellar hemispheres and already pronounced atrophy at the level of the vermis (closed head arrows). The neurodegeneration progressed substantially during follow-up, with severe atrophy 2 years after the initial presentation. In addition to the neurodegeneration, white matter changes were observed in the atrophic middle cerebellar peduncles visible on the sagittal FLAIR and axial T2-weighted MR images (open head arrows, bottom row, i, j), not visible at the initial presentation (top row, d, e). Please note also the severe atrophy of the brain stem, particularly of the pons (closed head arrow, bottom row, g) with a “hot cross bun sign” on the T2-weighted MR images (closed arrow, bottom row, i)
During follow-up, his unsteady gait worsened and he became wheelchair bound. Upon examination 7 months after presentation, his truncal balance was disturbed and the heel–knee–shin movements were ataxic. Brain MRI showed progressive cerebellar atrophy with new symmetric T2 high signal-intensity lesions in both middle cerebellar peduncles and the transverse pontine fibers (Fig. 1). As these changes in the pons have been described as the “hot cross bun sign” in multiple system atrophy (MSA) patients (Fig. 1f–j), MSA was considered. However, due to the absence of autonomic failure MSA seemed very unlikely. Qualitative PCR for JCV in CSF was positive in three separately obtained samples and the diagnosis JCV GCN was made.
In spite of six cycles of cidofovir, his condition gradually worsened and his speech became affected. He died 4 years after the onset of his complaints due to an unknown cause. No post-mortem examination was performed.
Case 2
A 47-year-old man, in whom HIV infection had been diagnosed 23 years before, presented with progressive drowsiness and an unsteady gait since 1 year. Walking had become increasingly difficult during the final days before admission. In addition, he had increasingly frequent headaches and reported difficulty in speaking. On examination, he was somnolent, bradyphrenic and disoriented in time. There was a marked truncal ataxia, which precluded unsupported standing and he displayed saccadic pursuit eye movements. There was dysdiadochokinesia (left more than right) but finger–nose and heel–knee–shin movements were normal. Motor and sensory examinations and deep tendon reflexes were normal with flexor plantar responses.
His medical history revealed migraine, alcohol abuse and post-encephalitic epilepsy after a smallpox vaccination as an infant for which he used valproic acid, carbamazepine and clonazepam. Although he had been noncompliant with his combination anti-retroviral therapy (cART) medication since 1 year (1 month before admission his absolute CD4+ T cell count was 40 cells/mm3), he had restarted his cART medication 3 weeks before admission.
Brain MRI showed diffuse symmetric atrophy of the cerebellum with adjacent white matter hyperintensities suggestive of gliosis or white matter infection (Fig. 2). CSF analysis showed a normal cell count and normal glucose and protein levels. Qualitative PCR analysis of the CSF was positive for JCV, confirming the diagnosis JCV GCN.

MRI obtained from case 2 at initial presentation. Axial FLAIR (a, b), axial T2-weighted (c) and sagittal FLAIR (d, e) MR images, showing pronounced atrophy of both cerebellar hemispheres (closed head arrow) with adjacent white matter changes (open head arrow)
During admission his complaints decreased and, with physiotherapy, his walking improved. On follow-up 6 weeks later his walking, speaking and bradyphrenia had further improved. His absolute CD4+ T-cell count had increased to 230 cells/mm3 1 month after admission.
Literature review
We retrieved information on a total number of 16 cases diagnosed with GCN [10–24]. In addition, five other HIV-related GCN cases were identified, but as no information on clinical presentation, imaging findings or histopathology were given, we excluded them from this review [25].
Table 1 presents a summary of these 16 cases, plus the two additional cases presented in this report, detailing the underlying clinical condition, imaging findings and histopathology, if available. Twelve patients had HIV, which made it the most common underlying clinical condition, and three patients were using monoclonal antibodies (2 natalizumab and 1 rituximab). Cerebellar atrophy, observed in all but two cases (patients 3 and 13, Table 1), was the most consistent feature on imaging [14, 22]. Another common imaging finding, observed in 12 of the patients, was white matter abnormalities of the cerebellum and brainstem. In six of the seven cases with data available on histopathology, evidence was present of infection of the granule cell neurons in the cerebellum with subsequent loss of the internal granule cell layer. However, in four of the seven patients, white matter changes with JCV-infected or abnormal glial cells were observed.
Discussion
In 2003, Du Pasquier and colleagues described the first case of an HIV-infected patient presenting with PML in both frontal lobes and severe atrophy of the cerebellum. On histopathology, multifocal loss of internal granule cell layer due to JCV-infected granule cell neurons was observed leading to the term granule cell neuronopathy (GCN). Since then, 17 additional cases diagnosed with GCN have been described (see Table 1 for summary of all GCN cases published to date) [10–25].
Infection and loss of the granule cell neurons are the leading histopathological features in these patients. However, in four of the seven patients, JCV-infected or abnormal glial cells were also observed. This suggests that GCN is—at least in a considerable number of patients—not an exclusive grey matter disease as GCN may also affect white matter structures, which is supported by the imaging findings. The most consistent imaging finding in all GCN patients, including the two hereby presented patients, is atrophy of the cerebellum suggestive of neurodegeneration. However, in two-thirds of the cases additional white matter changes in the cerebellum and brainstem, particularly in the middle cerebellar peduncles and the pons, were observed. The underlying pathology of these accompanying white matter abnormalities is not well understood. In some histopathologically confirmed cases there is evidence of infection in astrocytes and oligodendrocytes. In other cases, white matter changes, particularly in the periphery of the cerebellar hemispheres and the cerebellar peduncles, might be secondary to pronounced grey matter loss, suggestive of gliosis. This latter phenomenon can also be observed in other neurodegenerative diseases showing severe focal atrophy, such as frontotemporal lobar degeneration (FTLD) with severe focal atrophy of the frontal and temporal lobes [26, 27].
Another interesting observation in one of our patients (case 1), which has been observed in two cases reported in the literature (patients 7 and 15, Table 1) [17, 20], is the involvement of the pons with linear white matter hyperintensities similar to those seen in patients with MSA, but also in other diseases including PML, coining the term “hot cross bun sign” [28–31]. It remains unclear whether these white matter changes in GCN patients occur from the beginning, or are features of later stages of the disease. However, in case 1 presented in this study, white matter involvement in addition to severe progression of focal atrophy of the cerebellum, also affecting the brain stem, was a feature of the advanced stage of the disease. In an immunocompromised patient with infratentorial white matter lesions and cerebellar atrophy on MRI suggesting GCN, it can be challenging to decide whether these lesions represent infratentorial PML with simultaneous neurodegenerative changes of the cerebellum, or should be classified as GCN with secondary white matter involvement. Indeed, patients with infratentorial PML can show imaging characteristics comparable to GCN with accompanying white matter lesions, and may even show JCV-infected granule cells on histopathology [7, 9, 32–35].
Interestingly, in contrast to PML associated with monoclonal antibody therapy, signs of inflammation such as contrast enhancement, have not been reported in GCN patients at presentation [36, 37]. However, one patient with natalizumab-associated GCN did show contrast enhancement at immune reconstitution syndrome (IRIS) stage (patient 16, Table 1) [19]. Given the simultaneous presence of grey matter and white matter pathology in most of the GCN patients, and the observation that GCN can occur in addition to PML lesions [10, 11], it remains unclear whether GCN has to be considered as a separate entity of JCV-related disease, or a PML subtype with pronounced grey matter involvement including the cerebellar granule cells. The association of GCN with certain mutations of the JCV (located in the VP1 C terminus region of the viral DNA) supports the hypothesis of GCN as a separate disease entity [12, 38]. However, preliminary histopathological data show that granule cells are frequently infected by JCV in PML patients, which suggests that GCN is rather a PML variant—at least in a considerable number of patients [39]. This coincidence of PML and GCN was also present in two GCN cases (patients 1 and 6, Table 1) [10, 11].
A similar phenomenon of a more selective grey matter involvement can be observed in JCV encephalopathy—another JCV-related disease characterized by a predominant infection of cortical pyramidal neurons [40]. However, similar to the frequent involvement of granule cells in PML, an involvement of cortical grey matter—particularly of cortical neurons—can also be observed in PML patients. This calls into question whether JCV encephalopathy is a separate disease entity [41]. Occasionally, the cortical grey matter involvement can be the predominant and leading imaging feature in PML, suggesting a cortical phenotype of PML in some patients and stressing the fact that cortical demyelination and even neuronal infection is a relatively frequent phenomenon in PML [4, 7, 42].
Finally, it is important to note that, similar to PML, GCN may occur not only in HIV patients. Other clinical conditions associated with immunosuppression, such as immunosuppressive therapy with monoclonal antibodies such as natalizumab and rituximab may also lead to GCN [17–19]. As the arsenal of immunosuppressive drugs is constantly growing, more patients with JCV-related diseases might be expected in the near future [35, 43]. Therefore, GCN should be considered in the context of drug surveillance programs to detect opportunistic infections in an early stage. The diagnosis of GCN is challenging since the combination of cerebellar symptoms and cerebellar atrophy has a broad differential diagnosis, even in immunocompromised patients. Knowledge of the broad spectrum and heterogeneity of clinical presentations and imaging characteristics of GCN is crucial in order to enable early diagnosis and intervention by immune reconstitution, which is presumably associated with a more favorable functional outcome.
Conclusion
We show that GCN may present with heterogeneous imaging features, thereby complicating an accurate diagnosis. The most characteristic imaging feature is cerebellar atrophy, which often occurs in combination with white matter changes in the cerebellum and brainstem. Due to the increasing number of patients treated with new generations of immunosuppressive drugs such as monoclonal antibodies and fumarates, a substantial increase of additional cases of GCN could be on the horizon. Therefore, we consider that there is a need for dedicated diagnostic criteria for GCN, which should include imaging characteristics such as cerebellar atrophy with possible concomitant white matter changes in the cerebellum and brainstem.
References
Pinto M, Dobson S (2014) BK and JC virus: a review. J Infect 68(Suppl 1):S2–S8
Hirsch HH, Kardas P, Kranz D, Leboeuf C (2013) The human JC polyomavirus (JCPyV): virological background and clinical implications. APMIS 121:685–727
Brew BJ, Davies NWS, Cinque P et al (2010) Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 6:667–679
Gheuens S, Wüthrich C, Koralnik IJ, Wuthrich C (2013) Progressive multifocal leukoencephalopathy: why gray and white matter. Annu Rev Pathol 8:189–215. doi:10.1146/annurev-pathol-020712-164018
Steiner I, Berger JR (2012) Update on progressive multifocal leukoencephalopathy. Curr Neurol Neurosci Rep 12:680–686
Berger JR, Aksamit AJ, Clifford DB et al (2013) PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology 80:1430–1438
Wattjes MP, Richert ND, Killestein J et al (2013) The chameleon of neuroinflammation: magnetic resonance imaging characteristics of natalizumab-associated progressive multifocal leukoencephalopathy. Mult Scler 19:1826–1840
Wattjes MP, Vennegoor A, Mostert J et al (2014) Diagnosis of asymptomatic natalizumab-associated PML: are we between a rock and a hard place? J Neurol 261:1139–1143. doi:10.1007/s00415-014-7336-5
Wattjes MP, Barkhof F (2014) Diagnosis of natalizumab-associated progressive multifocal leukoencephalopathy using MRI. Curr Opin Neurol 27:260–270
Du Pasquier RA, Corey S, Margolin DH et al (2003) Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology 61:775–782
Bustamante F, Luis Cartier R, Manuel Lavados M (2009) Atrofia cerebelosa por el virus JC en un paciente con SIDA. Rev Chin Neurro-Psiquiat 47:222–227
Dang X, Vidal JE, De Oliveira ACP et al (2012) JC virus granule cell neuronopathy is associated with VP1 C terminus mutants. J Gen Virol 93:175–183. doi:10.1099/vir.0.037440-0
Koralnik IJ, Wüthrich C, Dang X et al (2005) JC virus granule cell neuronopathy: a novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol 57:576–580. doi:10.1002/ana.20431
Otis CN, Moral LA (2005) Images in pathology: granule cell loss in AIDS-associated progressive multifocal leukoencephalopathy. Int J Surg Pathol 13:360
Shin HW, Kang SY, Sohn YH et al (2008) JC viral infection-related cerebellar degeneration as the first manifestation of AIDS. Eur Neurol 59:205–207. doi:10.1159/000114048
Tan IL, Brew BJ (2009) Possible JCV granular cell neuronopathy in a patient with HIV infection. Neurology 73:1598–1599. doi:10.1212/WNL.0b013e3181c0d6cb
Dang L, Dang X, Koralnik IJ, Todd PK (2014) JC polyomavirus granule cell neuronopathy in a patient treated with rituximab. JAMA Neurol 71:487–489. doi:10.1001/jamaneurol.2013.4668
Schippling S, Kempf C, Buchele F et al (2013) JC virus granule cell neuronopathy and GCN-IRIS under natalizumab treatment. Ann Neurol 74:622–626. doi:10.1002/ana.23973
Agnihotri SP, Dang X, Carter JL et al (2014) JCV GCN in a natalizumab-treated MS patient is associated with mutations of the VP1 capsid gene. Neurology 83:727–732. doi:10.1212/WNL.0000000000000713
Granot R, Lawrence R, Barnett M et al (2009) What lies beneath the tent? JC-virus cerebellar granule cell neuronopathy complicating sarcoidosis. J Clin Neurosci 16:1091–1092. doi:10.1016/j.jocn.2008.07.091
Hecht JH, Glenn OA, Wara DW, Wu YW (2007) JC virus granule cell neuronopathy in a child with CD40 ligand deficiency. Pediatr Neurol 36:186–189. doi:10.1016/j.pediatrneurol.2006.10.007
Keith J, Bilbao J, Baskind R (2012) JC virus granular neuronopathy and rhombencephalic progressive multifocal leukoencephalopathy: case report and review of the literature. Neuropathology 32:280–284. doi:10.1111/j.1440-1789.2011.01254.x
Roux D, Bouldouyre MA, Mercier-Delarue S et al (2011) JC virus variant associated with cerebellar atrophy in a patient with AIDS. J Clin Microbiol 49:2196–2199
Shang T, Delgado A, Adams D (2011) JC virus granule cell neuronopathy and hyper-IgE in HIV disease. Neurology 76:1941–1942
Piza F, Fink MC, Nogueira GS et al (2012) JC virus-associated central nervous system diseases in HIV-infected patients in Brazil: clinical presentations, associated factors with mortality and outcome. Braz J Infect Dis 16:153–156
Kitagaki H, Mori E, Hirono N et al (1997) Alteration of white matter MR signal intensity in frontotemporal dementia. AJNR Am J Neuroradiol 18:367–378
Mann DM (1998) Dementia of frontal type and dementias with subcortical gliosis. Brain Pathol 8:325–338
Padmanabhan S, Cherian A, Iype T et al (2013) Hot cross bun sign in HIV-related progressive multifocal leukoencephalopathy. Ann Indian Acad Neurol 16:672–673
Watanabe H, Saito Y, Terao S et al (2002) Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 125:1070–1083
Yadav R, Ramdas M, Karthik N et al (2011) “Hot cross bun” sign in HIV-related progressive multifocal leukoencephalopathy. Neurol India 59:293–294
Wattjes MP (2011) Structural MRI. Int Psychogeriatr 23(Suppl 2):S13–S24
Havla J, Hohlfeld R, Kumpfel T, Kümpfel T (2014) Unusual natalizumab-associated progressive multifocal leukoencephalopathy starting in the brainstem. J Neurol 261:232–234. doi:10.1007/s00415-013-7191-9
Lach B, Connolly B, Wuthrich C, Koralnik IJ (2014) Inflammatory infratentorial progressive multifocal leukoencephalopathy in a patient with rheumatoid arthritis. Neuropathology 34:39–44
Ali K, Amin R, Yoganathan KG, Powell R (2013) Rapidly progressive cerebellar ataxia in West Wales. BMJ Case Rep. 10.1136/bcr-2013-201619
Stoppe M, Thoma E, Liebert UG et al (2014) Cerebellar manifestation of PML under fumarate and after efalizumab treatment of psoriasis. J Neurol 261:1021–1024
Phan-Ba R, Lommers E, Tshibanda L et al (2012) MRI preclinical detection and asymptomatic course of a progressive multifocal leucoencephalopathy (PML) under natalizumab therapy. J Neurol Neurosurg Psychiatry 83:224–226
Wattjes MP, Verhoeff L, Zentjens W et al (2013) Punctate lesion pattern suggestive of perivascular inflammation in acute natalizumab-associated progressive multifocal leukoencephalopathy: productive JC virus infection or preclinical PML-IRIS manifestation? J Neurol Neurosurg Psychiatry 84:1176–1177
Dang X, Koralnik IJ (2006) A granule cell neuron-associated JC virus variant has a unique deletion in the VP1 gene. J Gen Virol 87:2533–2537
Wüthrich C, Cheng YM, Joseph JT et al (2009) Frequent infection of cerebellar granule cell neurons by polyomavirus JC in progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol 68:15–25
Wüthrich C, Dang X, Westmoreland S et al (2009) Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol 65:742–748
Wüthrich C, Koralnik IJ (2012) Frequent infection of cortical neurons by JC virus in patients with progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol 71:54–65
Moll NM, Rietsch AM, Ransohoff AJ et al (2008) Cortical demyelination in PML and MS: similarities and differences. Neurology 70:336–343
Van Oosten BW, Killestein J, Barkhof F et al (2013) PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med 368:1658–1659
Acknowledgments
The authors wish to thank Dr. Alex Rovira for providing Ref. [11] and Phil Riley for critical reading of the manuscript.
Conflicts of interest
Mr. Wijburg, Dr. van Oosten, Dr. Murk and Dr. Karimi report no potential conflicts of interest. Dr. Killestein has accepted consulting fees from Merck-Serono, TEVA, Biogen, Genzyme and Novartis. VU Medical Center has received financial support for research activities from Bayer Schering Pharma, Biogen-Idec, GlaxoSmithKline, Merck Serono, Novartis, and Teva. Dr. Wattjes serves as a consultant for Biogen-Idec. He serves on the editorial board of European Radiology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wijburg, M.T., van Oosten, B.W., Murk, JL. et al. Heterogeneous imaging characteristics of JC virus granule cell neuronopathy (GCN): a case series and review of the literature. J Neurol 262, 65–73 (2015). https://doi.org/10.1007/s00415-014-7530-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7530-5