
Abstract
Probabilistic genotyping permits a comparison of forensic evidence given hypotheses regarding the origin of observed short tandem repeat alleles in a mixed DNA profile. Using the publicly available R package forensim, it has been proposed that mixtures with non-contributors from low genetic diversity populations are more likely to be mistakenly identified as contributors to a mixture than non-contributors from high genetic diversity populations. We hypothesized that these observations are attributed to the unique distribution of alleles in the reference population and may not generalize to other samplings of the same population. We used forensim to simulate 200 US populations (50 each of self-reported African-American, Asian-American, European-American, and Hispanic descent). We compared likelihood ratios for 2400 mixtures to those derived from published data and identified stark differences. A minimum of ten population replicates were required to reduce observed differences relative to published data. Deviations from Hardy–Weinberg equilibrium and allele frequency distributions suggest that simulated populations should be sufficiently evaluated for expectations of population genetic parameters prior to use in DNA mixture modeling experiments. Overall, our findings support the utility of forensim and further describe its suitability to model population genetic parameters but suggest that a single population replicate (directly ascertained or simulated) may be insufficient to make conclusions about a given DNA mixture.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Statistical tools for the interpretation of forensic DNA mixtures are common. While some tools are open-source and freely available to the forensic community, others may be prohibitively costly to the forensic genetics researcher. The R package, forensim, is a statistical tool dedicated to forensic DNA evidence interpretation (1). The forensim package allows for the simulation of DNA data routinely encountered in forensic genetic casework based on prior knowledge of population STR data. Additionally, forensim allows users to compute common statistical calculations for evaluating the weight of the DNA evidence in question: probabilities of exclusion, random match probabilities, likelihood ratios (LRs), and conditional profile probabilities (2). One primary limitation to open-source software in a forensic context is validation (1), including the repeatability of data and reports generated from such tools. Indeed, the stochasticity of Markov chain Monte Carlo algorithms common to many probabilistic genotypic platforms produces minor, yet in some cases statistically and legally meaningful, differences in match statistics (3), 4, 5). It is unclear if forensim produces similar variability and to what extent.
Of tangential importance to the rapid development of improved genotyping and sequencing technologies, it is fundamental to develop tools for simulating largescale genomic data under realistic scenarios (i.e., routine forensic casework) that incorporate complex demographic and environmental factors (6, 7). It was recently demonstrated that non-contributors from low genetic diversity populations may be over-represented as contributors to DNA mixtures relative to non-contributors from high genetic diversity populations (8). It is unclear whether this conclusion reflects a generalizable trend. We hypothesize that this observation is an artifact of (i) the mixtures generated or (ii) the population allele frequencies used to calculate match statistics.
Simulation studies in forensic genetics play a vital role in establishing interpretation thresholds, statistical parameters, and varying expectations when interpreting and applying statistical support to forensic biological evidence of varying quantity and quality (9, 10, 11, 12, 13). When simulating data, it is important to verify that the modeled data appropriately represent the input data. This becomes especially important considering the ethical and legal consequences of match statistics reported for individuals of highly admixed backgrounds (an important topic studied extensively by Ceberio et al.) (8). There is much data demonstrating that using inappropriate or mismatched population allele frequencies bias match statistics (14), but it is unclear whether this bias exists among repeated samplings of the same population. We hypothesize that this within-population variation per locus may have large implications for match statistic stability.
In this study, we used forensim genotype simulations and DNA mixture modeling to compare locus heterozygosities and other population genetic parameters between replicate outputs of forensim back to Novroski et al. allele frequency data for four major population groups in the USA (15). We demonstrate a wide range of allele frequencies in simulated population data, used to create mock mixtures, that translates to appreciable differences in minor contributor match statistics relative to the published population frequencies. This variation suggests that mixture simulation studies can better present minor contribute match rates by using a range of population frequencies rather than a single sampling.
Methods and materials
Data description
STR length–based allele frequencies for this study were borrowed from Novroski et al. with respect to major population groups in the USA: European-Americans (EUA; Nindividuals = 210), African-Americans (AFA; Nindividuals = 200), Asian-Americans (ASA; Nindividuals = 169), and Hispanic-Americans (HIS; Nindividuals = 198) (15). DNA sequencing was performed for STR amplicons using the ForenSeq™ DNA Signature Prep Kit (Verogen, San Diego, CA) and the MiSeq FGx® platform (Verogen), as described in Novroski et al. (15). Sample sizes were matched for all simulated populations modeled by forensim.
Forensim R package
Simulated populations were generated with the R package forensim, a statistical package designed to analyze and interpret forensic DNA mixtures (1, 2). The forensim package permits the generation of genetic data commonly encountered in forensic DNA casework including population statistics and DNA mixtures (1). Forensim is freely available from: https://forensim.r-forge.r-project.org.
Simulated population genetic parameters
To assess the reliability of forensim to model population genetic information from the original (i.e., “truth”) data, we produced 50 iterations of each of four US populations (i.e., N = 200 total populations and N = 155,400 individuals). Our random number seed in R was set to 1,234,567. Population genetic parameters were compared to those from the original parameter from Novroski et al. (15). Allele frequencies were defined as \(p= \frac{n}{2N}\), where n is the number of times each allele was observed, and N is the population sample size. Because this study aimed to quantify the variability in match statistics following population simulation, no minor allele frequency adjustments were applied to STR loci.
Forensim constructs genotypes by randomly combining sampled alleles with replacement such that the resulting population should satisfy Hardy–Weinberg equilibrium (HWE) expectations; however, it is common to assess this feature of a population prior to using it for statistical analyses (16). For each STR with alleles {1…n}, the ith of which observed at frequency pi in a given population, expected heterozygosity of that locus was defined as \({H}_{e}=1-{\sum }_{i=1}^{n}{p}_{i}^{2}\). Hardy–Weinberg equilibrium p-values were computed using χ2 goodness of fit tests.
Mixed DNA profiles
Matched ancestry mixtures (e.g., > 1 EUA individuals mixed together) with two (Nmixtures = 200), three (Nmixtures = 200), and four contributors (Nmixtures = 200) were created with forensim. Using these profiles, we evaluated LR distributions using (i) “truth” allele frequencies from Novroski et al. (15) and (ii) allele frequencies observed across simulated populations.
All LRs were calculated using defense (Hd) and prosecution (Hp) hypotheses of simple mixture events such that the observed alleles at a locus are best represented by the contribution of one known contributor (i.e., a victim) and N other contributors. Here, \(LR=\frac{Pr\left(E|{H}_{p}\right)}{Pr\left(E|{H}_{d}\right)}\), where the probability of the evidence under Hp (numerator) is fixed at one assuming all contributors are known. No assumptions were made in this study regarding drop-in, drop-out, or theta correction. In forensim, these parameters were set to zero.
Statistical considerations
To better capture the magnitude of LR differences across analyses, we present standardized mean differences (SMD) which quantify effect sizes in measurements of unit difference. SMDs were used to compare the distribution of log10(LRs) across 200 mixtures calculated with allele frequencies from one simulated population and Novroski et al. (15). Here, \(\mathrm{SMD}=\frac{{\overline{X} }_{i}-{\overline{X} }_{j}}{{\sigma }_{ij}}\), where xi and xj are the mean log10(LR) for 200 mixtures calculated using allele frequencies from populations i and j, respectively, and σij is the pooled standard deviation of log10(LR) across populations i and j.
We applied false discovery rate (FDR < 0.05) multiple testing correction to all statistical tests to account for the potential lack of independence of each statistic (e.g., comparing population N LRs to Novroski et al. population LRs) (15).
Results
Forensim population genetic modeling
Across 200 simulated populations, population genetic parameters modestly varied relative to those reported by Novroski et al. (15). Allele frequencies for all 27 STRs of interest fell within the 95% confidence intervals of the Novroski et al. estimate (Figure S1; Tables S1–S2) (15).
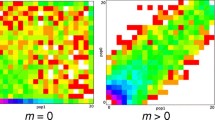
We observed a total of 289 deviations from HWE expectations (p < 0.05) where every STR evaluated had at least one deviation from expectations in at least one simulated population (Fig. 1; Table S3). Overall, 32% (ASA) to 50% (HIS) of the simulated population data had more HWE deviations than expected by chance alone (~ two observations in population data for 27 loci). There was no single locus driving the observed deviations from HWE, but after multiple testing correction per profile (p < 0.0018), we identified ten loci violating HWE expectations in at least one simulated population dataset.

Hardy–Weinberg equilibrium (HWE) results. A Density plots of HWE p-values from 50 simulated populations per “truth” population (AFA, African-American; ASA, Asian-American; EUA, European-American; HIS, Hispanic). Vertical dashed lines mark the nominal significance p-value (p = 0.05). (B) Locus-specific HWE information (simulated population unadjusted p-values in left panel and multiple-testing corrected p-values in middle panel) compared to the loci deviating from HWE expectations in Novroski et al. (15) (right panel, in black)
Single ancestry mixtures
A total of 2400 mixed DNA profiles (800 each of two-, three-, and four-person mixtures; 200 mixtures per ancestry group) were generated. Consistent with other simulation studies, based on the number of alleles observed in each mixture, higher contributor mixtures were more frequently predicted to be the result of an incorrect number of contributors (Fig. 2) (14, 17, 18). LRs for all 2400 mixtures were calculated relative to allele frequencies of the true contributor population and all three non-contributor populations. With few exceptions, the true contributor population log10(LR) was consistently less prejudicial than those estimated using non-contributor population allele frequencies (Fig. 3). Taken together, the LR observations indicate that the simulated DNA mixtures from forensim perform as expected.

Estimated contributors per mixture type. True versus estimated contributor N based on forensim allele-frequency aware contributor prediction algorithm for African-American (AFA), Asian-American (ASA), European-American (EUA), and Hispanic (HIS) same-ancestry DNA mixtures created using allele frequency population data from Novroski et al. (15). The true contributor (N) varied from two to four component contributors in a total of 200 simulated mixtures per mixture type per population (total N = 2400)

Two-, three-, and four-person mixture likelihood ratios (LRs) derived from Novroski et al. “truth” data. LRs for three types of same-population mixtures (N = 2400 mixtures; 200 per population per mixture type) derived from the Novroski et al. (15) population allele frequencies demonstrating suitability of simulated mixture data to appropriately model mixture statistics
Using simulated mixtures, we assessed how LRs change in the context of the observed within-ancestry allele frequency spectra (e.g., those observed in Figure S1). LRs were calculated for each mixture using allele frequencies from 50 simulated versions of the contributors’ origin population and compared to LRs generated from frequency data reported in Novroski et al. (15). The mean log10(LR) across two-person mixtures significantly differed from expected values in all four population groups with 34/50 AFA, 18/50 ASA, 26/50 EUA, and 41/50 HIS populations exhibiting significantly different mean LRs relative to Novroski et al. (Fig. 4) (15). Among two-person mixtures, log10(LRs) tended to be larger than expected as evidenced by negative standardized mean differences relative to Novroski et al. (15). In three outlier EUA populations, log10(LR) was 1.14- to 1.16-fold greater than expected.

Significant effects of population replicate selection of two-person mixture likelihood ratios (LR) across populations. Each data point represents the standardized mean difference (SMD) of LRs (N = 200 mixtures per data point) in each simulated population (African-American (AFA); Asian-American (ASA); European-American (EUA); and Hispanic (HIS)) relative to that of the corresponding LR derived from Novroski et al. truth data (15). The EUA subplot highlights three EUA populations with large changes in log10(LR). Statistical weight of SMD estimates was determined using two-sided paired T-tests
Among three- and four-person mixtures, the same effect was observed. The log10(LR) distributions of three- and four-person mixtures were significantly different from those derived from Novroski et al. population data for 26 and one AFA simulated populations, respectively; 27 and 44 ASA simulated populations respectively; 23 and 13 EUA simulated populations, respectively; and 24 and seven HIS simulated populations, respectively (15). The log10(LRs) of three-person mixtures derived from replicate population allele frequencies were up to 1.10-fold (AFA), 1.05-fold (ASA), 1.22-fold (EUA), and 1.06-fold (HIS) greater than those calculated using Novroski et al. allele frequencies. The log10(LRs) of four-person mixtures derived from replicate population allele frequencies were up to 1.04-fold (AFA), 1.04-fold (ASA), 1.27-fold (EUA), and 1.02-fold (HIS) greater than those calculated using Novroski et al. allele frequencies (15).
Mixture-specific observations
To test if population differences in LR per mixture category were due to mixture-specific features (i.e., an abundance of mixtures with low frequency alleles), we performed Z-tests between the distribution of mixture LRs across all 200 simulated population datasets and their matched LRs calculated with Novroski et al. allele frequencies (15).
Observations among population replicates were not attributed to a select few mixtures. A total of 54/200 AFA, 46/200 ASA, 36/200 EUA, and 56/200 HIS log10(LRs) from two-person mixtures were significantly (FDR < 5%; Table S4) different from the corresponding expected estimates using Novroski et al. population data (15). In three-person mixtures, there were 62/200 AFA, 64/200 ASA, 28/200 EUA, and 45/200 HIS significant differences, and in four-person mixtures, there were 26/200 AFA, 77/200 ASA, 32/200 EUA, and 30/200 HIS significant differences. These deviations are substantially larger than expected by chance (~ 10/200 significant differences per mixture per population). Among the significant differences from Novroski et al. population data (15), 100% of two-person, 73.4% of three-person, and 79.5% of four-person mixture log10(LRs) were overestimated in the simulated population data.
Replicate count required to converge match statistics
Given the wide distribution of allele frequencies observed across STR loci, we tested how many replicate populations were required to converge on the “truth” match statistics derived from Novroski et al. For each mixture, we calculated the mean log10(LR) difference between Novroski et al. (15) with sequentially increasing and randomly selecting numbers of population replicates. Among two-person same-ancestry mixtures (N = 200 per population) of AFA (p = 1.09 × 10−15), EUA (p = 4.98 × 10−50), and HIS (p = 0.003) ancestries, we detected a significant effect of number of replicates on producing log10(LRs) more similar to those derived from Novroski et al. (15). There was no significant difference in mean log10(LR) among ASA population replicates (p = 0.370).
Post hoc pairwise comparisons of each number of population replicates (Fig. 5; Figure S4) support the use of a larger number of replicates both statistically and visually presented by narrower distribution of mean difference in log10(LR) per mixture. For AFA and EUA two-person mixtures, the sequential increase in replicates significantly reduced the difference in log10(LR) relative to the Novroski et al. estimate (15). After multiple testing correction, the data support the use of at least 10 replicates of AFA-AFA two-person mixtures (1 replicate versus 10 replicates Wilcox p = 0.0073; F-test F = 4.69, df = 199, p < 2.2 × 10−16) and EUA-EUA two-person mixtures (1 replicate versus 10 replicates Wilcox p = 9.70 × 10−4; F-test F = 4.40, df = 199, p < 2.2 × 10−16). There was no significant effect of replicate count with respect to mean difference in log10(LR) for HIS-HIS mixtures; however, 30 population replicates produced the most significant reduction in log10(LR) variance relative to a single replicate (1 replicate versus 30 replicates F-test F = 9.65, df = 199, p < 2.2 × 10−16). Finally, among ASA-ASA mixtures, no differences in mean difference in log10(LR) were observed; however, 50 population replicates produced the most significant reduction in log10(LR) variance relative to a single replicate (1 replicate versus 50 replicates F-test F = 13.84, df = 199, p < 2.2 × 10−16).

Estimated appropriate number of population replicates to minimize noise in two-person mixture likelihood ratio (LR) estimates. Violin halves (right) represent data distribution while dot-plot halves (left) represent individual observations. Each data point in the dot plots represents a single mixture assayed in N population replicates (x-axis) for four population groups (African-American (AFA); Asian-American (ASA); European-American (EUA); and Hispanic (HIS)). The position on the y-axis corresponds to the difference between mean log10(LR) and Novroski et al. log10(LR) (15). Significant differences were observed among AFA (p = 1.09 × 10−15), EUA (p = 4.98 × 10−50), and HIS (p = 0.003) populations but not ASA (p = 0.37); pairwise significant differences are shown in supplementary material. Figure S4 shows replicate pairwise comparison p-values
In three-person mixtures from AFA (p = 1.10 × 10−15), EUA (p = 2.20 × 10−16), and HIS (p = 0.003) populations, we detected a significant effect of replicate number on mean log10(LR) difference relative to Novroski et al. (15). In AFA and EUA populations, we recapitulate the requirement for a minimum of 10 replicate populations (Table S5; Figure S5). Though a significant effect was detected in the HIS population, we could not verify an appropriate number of replicates using the data presented here. In four-person mixtures from AFA (p = 8.30 × 10−5), EUA (p = 2.20 × 10−16), and HIS (p = 0.007) populations, we detected a significant effect of replicate number on mean log10(LR) difference relative to Novroski et al. (15). In four-person mixtures, between 10 (EUA) and 20 (AFA and HIS) replicates significantly reduced the differences in log10(LR) estimates (Table S5; Figure S6).
Discussion and conclusions
It is well understood that DNA match statistics, which rely on allele frequencies, are affected by sampling variation of those frequencies from their respective populations (19). As published allele frequencies reflect relatively small samplings of large populations, we hypothesized that reports of minor contributor matches from low genetic diversity populations may be biased by the sampling of those populations. We used major US population groups to quantify how variable these match statistics may be when the population is randomly sampled many times. Our findings confirm the wide range of allele frequencies captured by population samplings and translate this into match statistics distribution across samplings. We demonstrated that this variation in allele frequency may bias the most conservative match statistic for a mixture away from its true population origin.
Forensim is a fast and efficient way to simulate mock forensic genotypes and DNA mixtures, and the data generated here demonstrates a broad range of allele frequencies across simulated populations and a relatively high chance of generating population data with at least one significant HWE event. Because populations are generated in forensim using a random sampling of alleles with replacement, these observations are most likely attributed to noise; however, their detection is pertinent to the use of each population for statistical considerations. Because of the random sampling of alleles, it is less likely that these HWE deviations represent the induction of residual population substructure in the simulated populations. It is, of course, important to understand when these events occur in the context of relatively admixed populations such as those from low diversity populations.
In the context of variable allele frequencies across simulated populations, we tested the extent to which match statistics varied. While allele frequencies generally fell within the 95% confidence interval of the Novroski et al. reported allele frequency, LRs were highly variable across population replicate indicating that small, largely insignificant, changes in allele frequency can have drastic effects on match statistics. In two-, three-, and four-person mixtures, we identified a significant effect of replicate number on reducing the variance of STR match statistics. Importantly, these effects were not driven but a single population nor a single profile and recapitulate more systemic variability of allele frequencies across population replicates. In nearly all cases, the LR from a sampled population was stronger than that from the truth population. This finding highlights how variability across populations, even those sampled from the same reference, can influence conclusions about minor contributors to a mixture. Our data suggest a different minimum number of replicates based on population but clearly highlighted a necessity for at least 10 population replicates (e.g., for questions related to two-person mixtures of contributors with matched ancestry) regardless of population. It is important to note that the observed effects reflect same-ancestry mixtures only, and we hypothesize that these consequences may be more profound when the major and minor contributor represent discordant prominent ancestry proportions. In other words, observations from Ceberio et al. (8) are likely real but the magnitude of this effect requires careful consideration for the genetic ancestry proportion of each contributor to a given mixture.
While contextualizing the effects of allele frequency variation on mixture match statistics in a novel manner, our study has three limitations to consider. First, we assumed optimal scenarios for all mixtures: no stutter events, no drop-in or drop-out events, and simple LR hypotheses. We recognize that these assumptions do not reflect the reality of DNA casework, and future research will build upon our results to systematically assess the nuances of each parameter in the context of within- and between-population mixtures. Second, we exclusively consider length-based STR data with no consideration for capillary electrophoretic peak heights or sequence-based read depths. We expect that sequence-based STR data will demonstrate similar observations as length-based tests but may permit better resolution of mixture events in the presence of loci with greater sequence-based heterozygosity (15, 20, 21) and/or the presence of STR sequence level diversity that sheds light on potential population-specific origins of an allele (22). Third, this study only considered within-population mixtures. Mixed-ancestry profiles have demonstrated that populations with lower genetic diversity can be overrepresented in mixture deconvolution match statistics (23). In the context of the findings presented herein, future studies with appropriate replicates must focus on how genetic diversity among mixture contributors exacerbates social and legal injustices associated with race, ethnicity, and genetically determined ancestry.
The data presented herein highlight the feasibility of studying mixtures with freely available resources and published STR allele frequency data. We described how variation in random sampling from a reference population contributes to variation in DNA match statistics. Depending on the pattern of random sampling, this match statistic variation may have considerable societal implications in populations with low genetic diversity and/or scenarios of partial DNA matches [20]. These data form the groundwork for future controlled mixture simulation studies, including multi-ancestry mixtures, and contribute suggested best practices for simulated data reliability and reproducibility.
References
Haned H (2011) Forensim: an open-source initiative for the evaluation of statistical methods in forensic genetics. Forensic Sci Int Genet 5:265–268
Haned H, Benschop CCG, Gill PD, Sijen T (2015) Complex DNA mixture analysis in a forensic context: evaluating the probative value using a likelihood ratio model. Forensic Sci Int Genet 16:17–25
Curran JM (2008) A MCMC method for resolving two person mixtures. Sci Justice 48:168–177
Haned H, Gill P, Lohmueller K, Inman K, Rudin N (2016) Validation of probabilistic genotyping software for use in forensic DNA casework: definitions and illustrations. Sci Justice 56:104–108
Kelly H, Bright JA, Buckleton JS, Curran JM (2014) A comparison of statistical models for the analysis of complex forensic DNA profiles. Sci Justice 54:66–70
Manabe S, Morimoto C, Hamano Y, Fujimoto S, Tamaki K (2017): Development and validation of open-source software for DNA mixture interpretation based on a quantitative continuous model. PLoS One. 12:e0188183.
Marciano MA, Adelman JD (2017) PACE: probabilistic assessment for contributor estimation- a machine learning-based assessment of the number of contributors in DNA mixtures. Forensic Sci Int Genet 27:82–91
Caberio N, Flores M, Kalaydjian C, Paunovich M, Lobpez E, Reyes R, et al. (2019): Are individuals from populations with low genetic diversity more likely to be wrongly identified as contributors to DNA mixtures? . 30th International Symposium on Human Identification. Palm Springs, CA, USA: Promega Corporation.
Bright JA, Curran JM, Buckleton JS (2014) The effect of the uncertainty in the number of contributors to mixed DNA profiles on profile interpretation. Forensic Sci Int Genet 12:208–214
Haned H, Egeland T, Pontier D, Pène L, Gill P (2011) Estimating drop-out probabilities in forensic DNA samples: a simulation approach to evaluate different models. Forensic Sci Int Genet 5:525–531
Haned H, Slooten K, Gill P (2012) Exploratory data analysis for the interpretation of low template DNA mixtures. Forensic Sci Int Genet 6:762–774
Prieto L, Haned H, Mosquera A, Crespillo M, Alemañ M, Aler M et al (2014) Euroforgen-NoE collaborative exercise on LRmix to demonstrate standardization of the interpretation of complex DNA profiles. Forensic Sci Int Genet 9:47–54
Smart U, Cihlar JC, Mandape SN, Muenzler M, King JL, Budowle B, et al. (2021): A continuous statistical phasing framework for the analysis of forensic mitochondrial DNA mixtures. Genes (Basel). 12.
Coble MD, Bright JA, Buckleton JS, Curran JM (2015) Uncertainty in the number of contributors in the proposed new CODIS set. Forensic Sci Int Genet 19:207–211
Novroski NMM, King JL, Churchill JD, Seah LH, Budowle B (2016) Characterization of genetic sequence variation of 58 STR loci in four major population groups. Forensic Sci Int Genet 25:214–226
Ye Z, Wang Z, Hou Y (2020): Does Bonferroni correction “rescue” the deviation from Hardy-Weinberg equilibrium? Forensic Sci Int Genet. 46:102254.
Chong KWY, Syn CK (2021) Uncertainty in estimating the number of contributors from simulated DNA mixture profiles, with and without allele dropout, from Chinese, Malay, Indian, and Caucasian ethnic populations. Sci Rep 11:5249
Norsworthy S, Lun DS, Grgicak CM (2018) Determining the number of contributors to DNA mixtures in the low-template regime: exploring the impacts of sampling and detection effects. Leg Med (Tokyo) 32:1–8
Rosenberg NA, Kang JT (2015) Genetic diversity and societally important disparities. Genetics 201:1–12
Gettings KB, Borsuk LA, Steffen CR, Kiesler KM, Vallone PM (2018) Sequence-based U.S. population data for 27 autosomal STR loci. Forensic Sci Int Genet 37:106–115
Young BA, Gettings KB, McCord B, Vallone PM (2019) Estimating number of contributors in massively parallel sequencing data of STR loci. Forensic Sci Int Genet 38:15–22
Devesse L, Davenport L, Borsuk L, Gettings K, Mason-Buck G, Vallone PM, et al. (2020): Classification of STR allelic variation using massively parallel sequencing and assessment of flanking region power. Forensic Sci Int Genet. 48:102356.
Rohlfs RV, Fullerton SM, Weir BS (2012): Familial identification: population structure and relationship distinguishability. PLoS Genet. 8:e1002469.
Acknowledgements
Points of view in this document are those of the authors and do not necessarily represent the official position or policies of their organizations. The data generation from Novroski et al., were supported in part by award no. 2015-DN-BX-K067, awarded by the National Institute of Justice, Office of Justice Programs, US Department of Justice.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Research involving human participants and/or animals
The data used in this study are freely available datasets, where no Institutional Review Board/Research Ethics Board permissions were required.
Informed consent
Not applicable.
Conflict of interest
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Nicole MM Novroski and Frank R Wendt equally contributed to this paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Novroski, N.M.M., Moo-Choy, A. & Wendt, F.R. Allele frequencies and minor contributor match statistic convergence using simulated population replicates. Int J Legal Med 136, 1227–1235 (2022). https://doi.org/10.1007/s00414-022-02822-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-022-02822-0