
Abstract
The HomyGene19+14Y System (HG19+14Y) is a PCR-based amplification kit that enables typing of 18 autosomal short tandem repeat (STR) loci (i.e., CSF1PO, D2S1338, D3S1358, D5S818, D6S1043, D7S820, D8S1179, D12S391, D13S317, D16S539, D18S51, D19S433, D21S11, FGA, Penta E, TPOX, TH01, vWA), 14 widely used Y chromosome STR (Y-STR) loci (Y_GATA_H4, DYS385a/b, DYS389I/II, DYS390, DYS391, DYS392, DYS393, DYS438, DYS439, DYS456, DYS458, DYS635), and amelogenin. This multiplex system was designed for the simultaneous analysis of amelogenin-Y allele mutation, single-source searches, kinship (including familial searching), mixture profiles, international data sharing, and other forensic applications. In this study, the multiplex system was validated for sensitivity, specificity, DNA mixtures, stability, precision, stutter, reproducibility, parallel tests, PCR-based conditions, and population analysis according to the Scientific Working Group on DNA Analysis Methods (SWGDAM) developmental validation guidelines. A total of 212 alleles were detected for the 18 autosomal STR loci among 528 Guangdong Han individuals, and 431 haplotypes were found for 14 Y-STRs among 452 unrelated males. The combined match probability (CMP) of the HG19+14Y System was calculated as 2.39 × 10−29. All the validation results showed that the HG19+14Y System would be a robust, reliable, highly polymorphic, and informative forensic kit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autosomal short tandem repeats (STRs) and Y chromosome STRs (Y-STRs) have been widely applied in forensic DNA analyses for many years [1–3]. In the past decade, a variety of commercially available STRs and Y-STR kits with different loci combination has been developed [4–7]. However, the rapid growth of DNA databases requires more genetic markers included in one multiplex system to meet the challenges of adventitious hits, missing person identification, international data sharing, etc. [8, 9]. Moreover, the challenges of mixture, degraded, and low copy number (LCN) DNA analysis are considered to be routine and need more new strategies to solve [10–14].
With development of the STR-PCR and capillary electrophoresis (CE) technology, more and more multiplex systems were designed up to 24–27 STR loci which were the principle DNA databases of the combined DNA index system (expanded CODIS), extended European standard set (ESS-extended), interpol standard set of loci (ISSL) German core loci (GCL), and UK core loci (UKL) [15–18]. In recent 2 years, a novel multiplex approach with combination of STRs and several Y-STRs has been presented to provide a higher kinship index with sufficient discriminating power for single source and mixture comparisons compared with a similar number of only autosomal STR loci [19, 20]. In addition, the technology of mini-STRs by using reduced-size STR amplicons and direct PCR without template extraction by promoting the inhibitors’ tolerance of PCR buffer can improve the detection rate of LCN and high inhibitive samples [21–23].
However, the utility of several Y-STR loci in one multiplex system is insufficiently powerful for kinship analyses and determining potential contributors of mixtures. More Y-STR loci are very useful to resolve amelogenin-Y allele dropout, deconvolve male and female mixtures, and trace paternal lineage. It is necessary to design a new multiplex system which combines a dozer Y-STRs with available autosomol STRs.
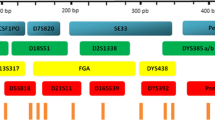
In this study, the HG19+14Y System which contains 18 STRs and 14 Y-STRs along with sex determination marker amelogenin was developed and validated. The multiplex is a 6-dye system that co-amplifies nine mini-STRs (D3S1358, D13S317, D7S820, TPOX, TH01, D19S433, vWA, D8S1179, D5S818), four mini Y-STRs (DYS456, DYS391, DYS393, DYS389I), and other highly polymorphic nine STRs (D16S539, Penta E, D2S1338, CSF1PO, D21S11, D18S51, D6S1043, D12S391, FGA) and ten Y-STRs (DYS635, DYS385a/b, DYS458, DYS428, DYS439, DYS389II, DYS392, Y_GATA_H4, DYS390) markers. The configuration of HG19+14Y System and the information of each locus are shown in Fig. 1 and Table 1.

Configuration of the HG19+14Y System
The HG19+14Y System was performed to evaluate sensitivity, specificity, DNA mixtures, stability, precision, stutter, reproducibility, parallel tests, and PCR-based conditions using control DNA 9948 and 2800M (Promega, WI, USA) according to FBI QAS (2009/2011) [24], Scientific Working Group on DNA Analysis Methods (SWGDAM) developmental validation guidelines [25], and Chinese criteria [26]. The 528 unrelated Guangdong Han individuals (including 452 males) were conducted to analyze forensic parameters.
Materials and methods
Samples
In the sensitivity study, both control DNA 9948 and 2800M were amplified, respectively, in a series of dilutions with the following template quantities: 1.0, 0.5, 0.25, 0.125, 0.0625, and 0.03125 ng. The species specificity studies were conducted using 1.0 ng each of extracted DNA from human, chimpanzees, and macaques; 10 ng each of purified DNA from common domestic animals (cat, dog, sheep, bull, and fish); 10 ng of microbial DNA pool from Escherichia coli, Lactobacillus acidophilus, Streptococcus salivarius, Saccharomyces cerevisiae, and Enterococcus faecalis; and a negative control. These samples were kindly donated from Guangzhou Zoo (Guangzhou, China) and AGCU ScienTech Incorporation (Wuxi, China). The quantities were determined using a Qubit® ssDNA Assay Kit with a Qubit® 3.0 Fluorometer (Life Technologies, CA, USA).
Forensic casework analysis usually involves mixture DNA samples from more than one individual. Therefore, it is very important to reliably interpret results from mixed-source samples. In this validation, mixture studies were performed with mixed DNA samples at known ratios. Male/male mixtures were prepared using 9948 and 2800M human genomic DNA with mixture ratios of 1:1, 1:4, 1:9, or 1:19, respectively. Male/female mixtures were prepared using 9948 and 9947A (Promega, WI, USA) human genomic DNA with mixture ratios of 1:1, 1:4, 1:9, or 1:19, respectively. Each mixture was tested in triplicate. These mixed samples were formulated at a total concentration of 1 ng in 25 μL reaction volume.
Several common forensic inhibitors were tested in this studies, including hematin, hemoglobin, humic acid, indigo, calcium ion, and EDTA. The quantity of 9948 control DNA was constant at 0.5 ng, while the inhibitor concentrations varied: 20, 50, 80, 100, or 150 μmol/L of hematin; 50, 80, 100, 150, or 200 μmol/L of hemoglobin; 20, 50, 80, 100, or 150 ng/μL of humic acid; 4, 5, 8, 10, or 12 mmol/L of indigo; 0.1, 0.2, 0.5, 0.7, or 1.0 mmol/L of calcium ions; and 0.1, 0.5, 0.8, 1.0, or 1.2 mmol/L of EDTA.
Parallel test was conducted using case samples collected in daily work at Guangzhou Forensic Science Institute. These samples included two blood FTA® cards, two buccal swabs, three cigarette butts, five nails, and three hair roots. The samples on blood FTA® cards were directly amplified (1.2-mm punch), while the others were extracted using magnetic beads. These results were compared with PowerPlex 21 System and PowerPlex Y23 System.
To calculate the stutter of each locus and to study the population analysis, 528 samples (452 males and 76 females) prepared by spotting 50 μL whole blood onto FTA® cards (GE Healthcare/Whatman, Buckinghamshire, UK) were directly amplified (1.2-mm punch) and analyzed. All samples were collected from 528 unrelated healthy individuals of Guangdong Han population with informed consent approved by Nanfang Hospital medical ethics committee of Southern Medical University, Guangzhou, China.
Reproducibility study was conducted by randomly choosing 90 samples collected above at three laboratories (Guangzhou Forensic Science Institute and two of its sub-institutes).
The PCR condition studies included the tests of cycle number, annealing temperature, final extension time, concentrations of PCR Master Mix, Taq DNA polymerase, and primer set.
DNA amplification
DNA amplification reaction was carried out on GeneAmp® PCR System 9700 thermal cycler (Life Technologies, CA, USA) using the 9600 mode on a gold-plated silver block in a total volume of 25 μL including 10 μL of 2.5× PCR Master Mix, 5 μL of 5× HG19+14Y primer set, 1 μL of 5 U/μL Taq DNA polymerase (AGCU, Wuxi, China), and 0.5–2 ng of template DNA. According to the manufacturer’s technical manual, the following amplification setup and cycling parameters were used: initial denaturation of 95 °C for 2 min, 15 cycles of 94 °C for 30 s, 60 °C for 1 min, and 72 °C for 1 min, 15 cycles of 90 °C for 30 s, 59 °C for 1 min, and 72 °C for 1 min, a final extension at 60 °C for 30 min, and a 4 °C soak.
Electrophoresis and analysis
Electrophoresis was run on the Applied Biosystems 3500xl Genetic Analyzer using 36-cm capillary arrays with POP-4® Polymer (All from Life Technologies, CA, USA). Spectral calibration was performed using J6 Dyeset with the 6-Dye Matrix Standards (containing six DNA fragments labeled FAM, HEX, TAMRA, ROX, ATTO590, SIZ, respectively). Samples were prepared in MicroAmp Optical 96-Well Plate (Life Technologies, CA, USA) for CE by adding 1.0 μL of the PCR product or HG19+14Y allelic ladder to 10.0 μL of formamide/SIZ mixture (0.5 μL of Marker SIZ-500 size standard and 9.5 μL Hi-Di™ Formamide). The prepared samples were denatured for 3 min at 95 °C and then immediately chilled on ice or in an ice-water bath. Standard run conditions were sample injection for 10 s at 2 kV and electrophoresis at 15 kV for 1500 s with the indicated run temperature of 60 °C. GeneMapper ID-X v1.3 (Life Technologies, USA) was used to determine fragment size and genotyping. Allele peak of 100 relative fluorescence unit (RFU) was implemented as the peak detection threshold when analyzing data from CE instruments.
Statistical analysis
Sizing precision is critical for accurate genotyping. Sizing precision of the HG19+14Y System was evaluated by comparing each allele size of the HG19+14Y allelic ladder calculated with the supplied internal lane standard (Marker SIZ-500) from 24 lanes of a single injection on 3500xl Genetic Analyzer. The size of each allele was determined with GeneMapper ID-X software using the local southern method. The sizes were averaged, and a standard deviation of the mean size for each allele was calculated and plotted against the mean allele size.
Stutter products which are caused by slippage of the Taq DNA polymerase during the elongation step are common artifacts observed during the DNA-STR amplification [27, 28]. Peaks of one repeat smaller or larger than the true allele were the most common stutter product. Proportion of the stutter peaks relative to the true allele was measured by dividing the stutter peaks by the true peak height. Five hundred twenty-eight samples were used for the stutter ratio calculation.
Population statistical parameters of autosomal STR loci such as heterozygosity (H), matching probability (MP), polymorphism information content (PIC), discrimination power (DP), and power of exclusion (PE) were calculated using PowerStats v1.2 software (Promega, WI, USA) [29]. The gene diversity (GD) and haplotype diversities (HD) for Y-STRs were executed using the PowerMarker v3.25 [30]. The combined match probability (CMP) value of 18 STRs and 14 Y-STR combination was calculated using previously described methods [9, 31–33].
Quality control measures
All methods were carried out following ISFG recommendation as described by Schneider on the analysis of DNA polymorphisms [34].
Results and discussion
Sensitivity
Sensitivity studies were performed to assess the capability of the HG19+14Y System to amplify efficiently at a range of template concentrations. In this study, DNA templates at different concentrations were amplified and run in triplicate. Figure 2 summarized the results of the sensitivity study. Full profiles were obtained with DNA amount from 1 ng down to 0.125 ng, and perfect profiles were observed at 0.5 ng DNA amount. The alleles of D12S391, DYS635, DYS389II, DYS392, Y_GTAT_H4, and DYS390 were less than 100 RFU at the amount of 0.0625 ng. The haplotype of Y chromosome and the lower fluorescent efficient of ATTO may cause Y-STR loci with longer fragments to be lost easily at low amount of DNA template. When the amount of template DNA reduced to 0.03125 ng, almost 80% alleles observed were dropouts. Therefore, the HG19+14Y System can obtain reliable profiles at threshold 100 RFU above the DNA concentration of 0.125 ng, and the sensitivity is determined to be 0.125 ng/25 μL.

Heat maps summarizing the sensitivity results of the last three serial dilutions (from 0.125 to 0.03125 ng) of control DNA 9948 and 2800M (stochastic threshold was 100 RFU). Allele and locus dropping out was shown in yellow and red, as well as the heterozygous balance in green and light green
Species specificity
Non-human genomic DNA samples were tested for cross-reaction with the HG19+14Y System. As seen with Fig. 3, some squatty peaks between 90 and 300 bp were observed in the profiles of primate DNA (chimpanzee and macaque) which were distinguishable from human genomic DNA profiles by their allele number of off-ladder. No peak was observed at 10 ng DNA per PCR for other common animals and the microbial pool.

Representative electropherograms from species specificity studies: The templates were 1 ng of human DNA, 1 ng each of chimpanzee and macaque DNA, 10 ng each of dog, cat, sheep, bull, and fish DNA, a microbial pool (10 ng each purified DNA from Escherichia coli, Lactobacillus acidophilus, Streptococcus salivarius, Saccharomyces cervisiae, and Enterococcus faecalis mixed together), and a negative control. All samples were amplified for 30 cycles and analyzed on an Applied Biosystems 3500xl Genetic Analyzer. The Y-axis was adjusted to 5000 RFU
Mixture
Male DNA 9948/male DNA 2800M mixtures and male DNA 9948/female DNA 9947A mixtures were tested and summarized as listed in Table 2. All alleles of both mixtures were identified at the ratio of 1:1, 1:4, and 1:9. When the mixture ratio was increased to 1:19, the alleles of the eight STR loci including six Y-STRs dropped out (except D12S391, DYS635, DYS389II, DYS392, Y_GTAT_H4, and DYS390, plus DYS458 and CSF1PO), but no allele drop-in was observed. The mixture studies show that this system can meet the requirement of the analysis for mix samples and Chinese criteria for the human fluorescent STR multiplex PCR reagent, which requires that a valid kit should be able to detect all the alleles for 1:4 mixtures.
Stability
Six common inhibitors (hematin, hemoglobin, humic acid, indigo, calcium ion, and EDTA) were tested to evaluate inhibition tolerance of the HG19+14Y System. Complete profiles were obtained below the concentrations at 50 μmol/L of hematin, 100 μmol/L of hemoglobin, 20 ng/μL of humic acid, 12 mmol/L of indigo, 0.7 mmol/L of calcium ion, and 1.0 mmol/L of EDTA. When the concentration of hematin increased to 80 μmol/L and calcium ion increased to 1.0 mmol/L, the DYS392 locus dropped out. And when the concentration of hemoglobin increased to 150 μmol/L and EDTA increased to 1.2 mmol/L, the DYS635 locus dropped out. As the results seen in the Table 3, the HG19+14Y System had poor performance of the humic acid tolerance. There were only 33% alleles called if the concentration of humic acid increased to 80 ng/μL, while no profile was obtained in the presence of 150 ng/μL.
Size precision
Size precision of the HG19+14Y System was evaluated on 3500xl Genetic Analyzer by running three full injection of allelic ladder (Fig. 4). Size mean variability was determined by calculating the standard deviation for each allele. Very little variation at each locus was seen in the size of the HG19+14Y allelic ladder mix across the CE tested, and most allele deviations were nearly 0.06 base (Fig. 5). As expected, there was a slight trend of increasing standard deviation with increased fragment size, and the maximum standard deviation of an allele was close to 0.1 base at DYS385a/b and DYS390. Therefore, these results demonstrate that migration and size precision of the system can ensure proper allele to be consistent and within the bin window.

The profile of the HG19+14 allelic ladder on a 3500xl Genetic Analyzer

Precision across 24 CE lanes of the HG19+14Y allelic ladder on a 3500 Genetic Analyzer
Stutter
Stutter peaks are caused by PCR slippage events that are known to occur, and they are most notably found one repeat unit shorter in length than true allelic peaks. As stutter peaks are inevitable with any STR loci, it is better to do a series of experiments and statistics to ensure that stutter peaks in the forensic STR detection system would not affect the genotyping interpretation and also provide an effective reference to set the recommended filter values of each STR locus of the HG19+14Y System. The stutter average, range, and standard deviation (SD) are shown in Table 4. The average stutter ratio plus three standard deviations were used to set the stutter filter threshold. The meaningful data were selected from all loci profiles of 528 sample PCR products that had peak heights in the range varied from 400 to 20,000 RFU. The mean stutter ratios of all loci were less than 15% except D21S11 (15.32%) as shown in Table 4.
Parallel tests
In order to share the data among laboratories, the concordance between different kits provided by different companies is crucial. Therefore, parallel testing was performed using the HG19+14Y System, PowerPlex 21 System, and PowerPlex Y23 System to amplify 9948, 9947A, and some case samples, respectively. All DNA samples were obtained full profiles, and the same loci of the same sample had the concordant genotypes. As seen in Table 5, the results listed the genotypes of 9948 and one blood FTA® card.
Reproducibility
A reproducibility study was conducted to determine if the profiles were reliable and suitable for comparison between or among laboratories using the same DNA samples. To demonstrate compatibility on different capillary electrophoresis platforms, both the Applied Biosystems 3130xl (upgraded to 6-dye module) and 3500xl were used to run the HG19+14Y allelic ladder. The same locus detected was observed to be a consistent genotype, for example, the electropherograms of D16S539 allelic ladder were concordant between the 3500xl and 3130xl Genetic Analyzer (Fig. 6). Amplification reactions of 90 samples were performed by three different laboratories, and all genotypes of the same samples were consistent with known profiles (data not shown).

Concordant calls in ladder labeled with HEX and ATTO between 3500xl and 3130xl Genetic Analyzer
PCR-based conditions
The PCR-based condition studies included the following test of PCR Master Mix, primer set, Taq DNA polymerase, cycling number, and annealing temperature. All experiments were conducted using 0.5 ng 9948 DNA in 25 μL reaction volume.
Reaction components
The PCR Master Mix, primer set, and Taq DNA polymerase concentrations are important factors for consistent and robust results. It is necessary to evaluate whether the reaction component changes will affect the PCR performance of HG19+14Y System. The experiments studied the amplification of a series of concentration (0.5×, 0.75×, 1.0×, 1.25×, 1.5×) of PCR Master Mix, primer set, and Taq DNA polymerase, respectively. As Figs. 7, 8, and 9 shows, full profiles were obtained except at 0.5× concentration of PCR Master Mix and primer set, and the peak heights ascended with the concentration increasing. TH01 locus dropped out at 0.5× primer set concentration, while the alleles of D13S317, D7S820, TPOX, and TH01 were not detected at 0.5× PCR Master Mix. Moreover, a 436-base nonspecific peak was observed at 1.5× PCR Master Mix concentration. Variable concentration of Taq DNA polymerase could not impact the integral results of the HG19+14Y System significantly.

Effect of various reaction mix concentrations on the HG19+14Y System. Five concentrations were tested: 0.5×, 0.75×, 1.0× (standard), 1.25×, and 1.5×

Effect of various primer mix concentrations on the HG19+14Y System. Five concentrations were tested: 0.5×, 0.75×, 1.0× (standard), 1.25×, and 1.5×

Effect of various Taq DNA polymerase concentrations on the HG19+14Y System. Five concentrations were tested: 0.5×, 0.75×, 1.0× (standard), 1.25×, and 1.5×
Cycle number
The recommended optimal cycling number for the HG19+14Y System is 30 cycles. Of 9948 DNA, 0.5 ng was amplified at 28, 29, 30, 31, and 32 cycles, respectively. Full profiles were reliably generated at 28 cycles or more. For all tested samples, the overall balance had no effect basically by a variation in the cycle number. As expected, the peak heights increased with additional cycles. Figure 10 showed the mean peak heights from 500 RFU to 3000 RFU for the control DNA 9948 amplified for different cycle number. So, certain situations require the increased sensitivity afforded by additional cycles.

Effect of various cycle number on the HG19+14Y System. Five different cycles were tested: 28, 29, 30 (standard), 31, and 32
Annealing temperature
Temperature of different PCR thermocycler instruments among laboratories may slightly vary. The appropriate annealing temperature was conducted between 58 and 62 °C to ensure the stability and accuracy of genotyping results in this study. As seen at Fig. 11, the precise and accurate profiles could be observed at these annealing temperature conditions. The peak heights of the HG19+14Y System had slight impact with 2000 RFU at 60 °C and a little decreased to 1500 RFU at 62 °C. When the annealing temperature rose to 64 °C, a lot of loci dropped out (data not shown).

Electropherograms from the PCR anneal temperature study
Final extension time
The final PCR extension time was tested with 10, 20, 30 (recommended), 40, and 50 min. As Taq DNA polymerase has the tendency of adding an extra non-template adenosine nucleotide at the 3′ ends of DNA strands during thermal cycling [35]. Therefore, sufficient final extension time was needed to adenylate all amplified fragments. 9948 DNA at the quantity of 0.5 ng performed well above 30-min extension times, but the D7S820 locus showed small minus A(−A) peaks under the condition of 10 and 20 min and the DYS390 locus showed small plus A(+A) peaks under the condition of 40 and 50 min. As these results, 30 min was recommended to be the best final extension time (Fig. 12a, b).

a Effects of shortening the final extension time after normal thermal cycling for 0.5 ng 9948 DNA. The red arrows show the minus-A shoulder peaks for the D7S820. b Effects of lengthening the final extension time after normal thermal cycling for 0.5 ng 9948 DNA. The red arrows show the plus-A shoulder peaks for the DYS390
Population analysis
Relevant forensic parameters were investigated by genotyping 528 unrelated individuals (452 males, 76 females) from Guangdong Han population. As seen in supplementary Tables S1 and S2, a total of 212 alleles were detected for the 18 autosomal STR loci among 528 individuals, and 431 haplotypes were found for 14 Y-STRs among 452 unrelated males including 15 repeated twice haplotypes and 3 triplicate haplotypes. The allele number 8 of TPOX was the most frequent with frequency of 0.5227, while the allele number 11 of DYS391 was found with maximum frequency of 0.7102. The H, PM, PIC, DP, PE, and p value of autosomal STRs and the GD of Y-STRs were showed at Table 6, and there were no significant differences compared with previous data [36, 37]. The combined power of discrimination and combined probability of matching were calculated as 0.9999999999999999999998026 and 1.97 × 10−22, respectively, while the haplotype diversities were 0.999764535. The CMP of 18 STRs and 14 Y-STR combination was calculated as 2.39 × 10−29.
Conclusions
The HG19+14Y System was designed to amplify 18 autosomal STR loci and 14 Y-STR loci along with amelogenin simultaneously, which could be capable to obtain DNA genotype from those degraded biological samples as the result of 13 mini-STRs’ presence and determine the problem of amelgenin-Y allele loss [38] and male contributors in mixtures, meanwhile to provide Y-STR haplotypes in sexual assault cases. The 18 autosomal STRs selected in the system are the main principle loci of Chinese DNA database which can be compared with other commercial multiplex systems, while the 14 Y-STRs include the loci recommended in the European minimal haplotype (DYS385a/b, DYS389I/II, DYS390, DYS391, DYS392, DYS393, except DYS19) and two recommended loci by SWGDAM (DYS438 and DYS439), as well as four additional loci (DYS456, DYS458, DYS635, and Y_GATA_H4). This article described the developmental validation of the HG19+14Y System and examined the performance of the system by the PCR parameters established by its protocol, as well as examined factors related to the use of the HG19+14Y System. The validation studies demonstrate that the HG19+14Y System is a robust, accurate, stable, sensitive, and compatible multiplex PCR system which combines autosomal and Y chromosome STR loci making the system suitable for individual identification, paternity testing, paternal lineage testing, and mixture analysis.
References
Budowle B (1997) Studies for selecting core STR loci for CODIS in presented at DNA forensics science, evidence, and future prospects. November 17–18, McLean, VA
Butler JM (2006) Genetics and genomics of core short tandem repeat loci used in human identity testing. J Forensic Sci 51:253–265
Jiang XH (2011) Role of technologies in national DNA database in China: past, present and future trends. Chin. J Forensic Med 26:383–386
Ensenberger MG, Lenz KA, Matthies LK, Hadinoto GM et al (2016) Developmental validation of the PowerPlex Fusion 6C System. Forensic Sci Int Genet 21:134–144
Wang DY, Gopinath S, Lagace RE, Norona W, Hennessy LK, Short ML, Mulero JJ (2015) Developmental validation of the GlobalFiler Express PCR Amplification Kit: a 6-dye multiplex assay for the direct amplification of reference samples. Forensic Sci Int Genet 19:148–155
Thompson JM, Ewing MM, Frank WE, Pogemiller JJ et al (2013) Developmental validation of the PowerPlex Y23 System: a single multiplex Y-STR analysis system for casework and database samples. Forensic Sci Int Genet 7:240–250
Gopinath S, Zhong C, Nguyen V, Ge JY, Lagace RE, Short ML, Mulero JJ (2016) Developmental validation of the Yfiler Plus PCR Ampli fication Kit: an enhanced Y-STR multiplex for casework and database applications. Forensic Sci Int Genet 24:164–175
Hares DR (2012) Expanding the CODIS core loci in the United States. Forensic Sci Int Genet 6(1):52–54
Ge JY, Eisenberg A, Budowle B (2012) Developing criteria and data to determine best options for expanding the core CODIS loci. Invest Genet 3:1–48
Gill P, Haned H, Bleka O, Hansson O, Dorum G, Egeland T (2015) Genotyping and interpretation of STR-DNA: low-template, mixtures and database matches—twenty years of research and development. Forensic Sci Int Genet 18:100–117
Butler JM, Shen Y, McCord BR (2003) The development of reduced size STR amplicons as tools for analysis of degraded DNA. J Forensic Sci 48(5):1054–1064
Ballantyne KN, Oorschot RA, Mitchell RJ (2007) Comparison of two whole genome amplification methods for STR genotyping of LCN and degraded DNA samples. Forensic Sci Int 166:35–41
Lopes V, Andrade L, Carvalho M, Serra A, Balsa F, Bento AM et al (2009) Mini-STRs a powerful tool to identify genetic profiles in samples with small amounts of DNA. Forensic Sci Int Genet 2:121–122
Guo F, Zhou YS, Jiang XH et al (2016) Evaluation of the Early Access STR Kit v1 on the Ion Torrent PGM™ platform. Forensic Sci Int Genet 23:111–120
Martin PD, Schmitter H, Schneider PM (2001) A brief history of the formation of DNA databases in forensic science within Europe. Forensic Sci Int 119(2):225–231
Schneider PM (2009) Expansion of the European standard set of DNA database loci-the current situation. Profile in DNA 2009, 12(1): 6–7. At http://www.promega.com/profiles/1201/1201_06.html
Hares DR (2011) Expanding the CODIS core loci in the United States. Forensic Sci Int Genet 6(1):52–54
Butler JM, Hill CR (2012) Biology and genetics of new autosomal STR loci useful for forensic DNA analysis. Forensic Sci Rev 24:15
Li SL, Liu C, Liu H, Ge JY, Budowle B, Liu CH, Zheng WG, Li FY, Ge BW (2014) Developmental validation of the EX20+4 system. Forensic Sci Int Genet 11:207–213
Shafique M, Shahzad MS, Rahman Z, Shan MA, Perveen R, Shahzad M, Hussain M, Shahid AA, Husnain T (2016) Development of new PCR multiplex system by the simultaneous detection of 10 miniSTRs, SE33, Penta E, Penta D, and four Y-STRs. Int J Legal Med. doi:10.1007/s00414-016-1372-x
Lopes V, Andrade L, Carvalho M, Serra A, Bals F, Bento AM, Batista L, Oliceira C, Corte-Real F, Anjos MJ (2009) Mini-STRs: a powerful tool to identify genetic profiles in samples with small amounts of DNA. Forensic Sci Int Genet Supp S2:121–122
Tenpleton J, Ottens R, Paradiso V, Handt O, Taylor D, Linacre A (2013) Genetic profiling from challenging samples: direct PCR of touch DNA. Forensic Sci Int Genet Supp S4(1):224–225
Zhao G, Zhai H, Yuan Q, Sun S, Liu T, Xie L (2014) Rapid and sensitive diagnosis of fungal keratitis with direct PCR without template DNA extraction. Clin Microbiol Infect 20(10):776–782
Quality Assurance Standards (QAS) for DNA Databasing Laboratories. (2009). Available at: http://www.cstl.nist.gov/strbase/QAS/Final-FBI-Director-Databasing-Standards.pdf
Revised validation guidelines-scientific working group on DNA analysis methods (SWGDAM), Forensic Sci. Commun. 6 (3) (2004), Available at: http://swgdam.org/SWGDAM_Validation_ Guidelines_ APPROVED_Dec_2012.pdf
The implementation of voluntary certification of forensic science product rules—DNA kits, Available at: http://www.csp.gov.cn/UploadFiles/P-V03-005.pdf
Walsh PS, Fildes NJ, Reynolds R (1996) Sequence analysis and characterization of stutter products at the tetranucleotide repeat locus vWA. Nucleic Acids Res 24:2807–2812
Leclair B, Frégeau CJ, Bowen KL, Fourney RM (2004) Systematic analysis of stutter percentages and allele peak height and peak area ratios at heterozygous STR loci for forensic casework and database samples. J Forensic Sci 49(5):968–980
Tereba A (1999) Tools for analysis of population statistics. Profiles DNA 2(3):14–16
Lui K, Muse SV (2005) PowerMarker: integrated analysis environment for genetic marker data. Bioinformatics 21(9):2128–2129
Weir BS (2007) Matching and partially-matching DNA profiles. Ann Appl Stal 1(2):358–370
Ge JY, Chakraborly R, Eisenberg AJ, Budowle B (2011) Comparisons of familial DNA database searching strategied. J Forensic Sci 56(6):1448–1456
Ge JY, Budowle B, Chakraborly R (2011) Choosing relatives for DNA identification of missing persons. J Forensic Sci 56(s1):23–28
Schneider PM (2007) Scientific standards for studies in forensic genetics. Forensic Sci Int 165(2–3):238–243
Magnuson VL, Ally DS, Nylund SJ, Karanjawala ZE, Rayman JB, Knapp JI, Lowe AL, Ghosh S, Collins FS (1996) Substrate nucleotide-determined non-templated addition of adenine by Taq DNA polymerase: implications for PCR-based genotyping and cloning. BioTechniques 21:700–709
Liu QL, Chen YF, Huang XL, Liu KY, Zhao H, Lu DJ (2016) Population data and mutation rates of 19 STR loci in seven provinces from China based on Goldeneye™ DNA ID System 20A. Int J Legal Med Available at. doi:10.1007/s00414-016-1441-1
Wang Y, Liu C, Zhang CC, Li R, Liu H, Qu XL, Li HX, Sun HY (2016) Analysis of 24 Y-STR haplotype data in a Chinese Han population from Guangdong Province. Int J Legal Med 130(3):689–691
Tozzo P, Giuliodori A, Corato S, Ponzano E, Rodriguez D, Caenazzo L (2013) Deletion of Amelogenin Y-locus in forensics: literature revision and description of a novel method for sex confirmation. J Forensic Legal Med 20(5):387–391
Acknowledgements
This work was supported by a grant from Science and Technology Planning Project of Guangdong Province, China (Grant No. 2013B021500010), Natural Science Foundation of Guangdong Province (Grant No. 2014A030310025), and Medical Science and Technology Research Foundation of Guangdong Province (A2015043).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Weian Du and Ling Chen contributed equally to this work
Rights and permissions
About this article

Cite this article
Du, W., Chen, L., Liu, H. et al. Developmental validation of the HomyGene19+14Y System. Int J Legal Med 131, 605–620 (2017). https://doi.org/10.1007/s00414-016-1505-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-016-1505-2