
Abstract
The genome of proliferating cells must be precisely duplicated in each cell division cycle. Chromosomal replication entails risks such as the possibility of introducing breaks and/or mutations in the genome. Hence, DNA replication requires the coordinated action of multiple proteins and regulatory factors, whose deregulation causes severe developmental diseases and predisposes to cancer. In recent years, the concept of “replicative stress” (RS) has attracted much attention as it impinges directly on genomic stability and offers a promising new avenue to design anticancer therapies. In this review, we summarize recent progress in three areas: (1) endogenous and exogenous factors that contribute to RS, (2) molecular mechanisms that mediate the cellular responses to RS, and (3) the large list of diseases that are directly or indirectly linked to RS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Fundamental aspects of eukaryotic DNA replication
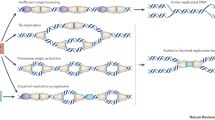
The first steps toward genome duplication occur before any new DNA is synthesized. Starting in late telophase and during G1, prereplicative complexes (pre-RCs) are assembled at genomic points called origins, whose number fluctuates between several hundreds in unicellular yeasts and tens of thousands in mammalian cells. Pre-RC formation, also called origin “licensing,” involves the origin recognition complex (ORC), CDC6 and CDT1 proteins, which attract and engage the ring-shaped minichromosome maintenance (MCM) DNA helicase with the DNA. Then, the coordinated action of two kinases (CDK and DBF4-CDC7) promotes the recruitment of additional factors including Sld2 (yeast)/RecQL4 (mammalian), Sld3/Treslin, Dbp11/TopBP1, Cdc45, GINS, and DNA polymerases along with their accessory factors. Each origin gives rise to two replication forks that synthesize new DNA while moving in opposite directions (Fig. 1; reviewed by Masai et al. 2010). The past year has seen remarkable advances in the elucidation of the origin activation pathway, including three-dimensional structures of ORC (Bleichert et al. 2015) and MCM (Li et al. 2015), as well as the full reconstitution of plasmid replication in vitro with purified yeast components (Yeeles et al. 2015).

Replication origin activation pathway. Schematic of the different proteins that license replication origins in G1 and activate them in S phase, giving rise to two replication forks from each origin. See text for details
The protein complexes responsible for DNA synthesis at the forks are called “replisomes” and include a DNA helicase that separates the parental DNA, at least three DNA polymerases (α, δ, and ε), a DNA polymerase processivity factor called proliferating cell nuclear antigen (PCNA), single-stranded DNA (ssDNA) binding replication protein A (RPA), and proteins that tether the polymerases to the helicase such as Tipin, Tim1, And1, and Claspin (Gambus et al. 2006, 2009; Errico et al. 2009). Several proteomic studies have identified many other replisome components in mammalian cells, whose functions are related to chromatin remodeling or remain to be elucidated (Alabert et al. 2014; Lopez-Contreras et al. 2013; Sirbu et al. 2013).
Importantly, replication forks slow down or completely pause when they encounter “blocks” in the template or when the concentration of free dNTPs is limiting. Under this circumstances, replisomes that stay stably associated with the DNA may restart DNA synthesis after the problem has been solved. However, when the arrest persists for a long period of time, replisomes collapse and the aborted forks are processed into DNA double-strand breaks (DSBs).
An evolving definition of replicative stress
Despite its interest and implications, the DNA replication and DNA damage fields are yet to reach a consensus definition for replicative stress (RS). Early usage of the term referred to aberrant events in cells undergoing rapid proliferation, such as uridine incorporation into DNA by Ehrlich ascites tumor cells (Yost et al. 1976). Later on, RS was used to describe “the vulnerability to genotoxic insults and other stochastic events that impede the proper replication and segregation of their genomes to daughter cells,” and it was established that it triggered specific cellular responses (Osborn et al. 2002). In recent years, RS has referred specifically to DNA replication disturbances that generate stretches of ssDNA that is covered by RPA (Zou and Elledge 2003). The strength of the cellular response is influenced by the amount of ssDNA-bound RPA (MacDougall et al. 2007). A rather minimalistic definition of RS has been recently put forward by Zeman and Cimprich (2014): “the slowing or stalling of replication fork progression and/or DNA synthesis”. To this elegant description, we would only add that, depending on the extent and duration of fork slowing or stalling, a specific checkpoint response is activated.
Causes of RS
Lessons from chromosome fragile sites
Common fragile sites (CFSs) are chromosomal points prone to break in the presence of DNA replication inhibitors such as aphidicolin. CFSs facilitate gross chromosomal rearrangements and may contribute to cancer development (reviewed by Durkin and Glover 2007). While CFSs were originally identified in blood cells (Glover et al. 1984), they have also been detected in fibroblasts and epithelial cells (Le Tallec et al. 2011, 2013). Interestingly, CFSs are largely tissue-specific; a similar DNA sequence can be stable in one cell type and fragile in another (Letessier et al. 2011).
The genomic features of CFSs provide clues into the elements that pose a risk for the DNA replication machinery. For instance, AT-rich regions found at some CFSs can form secondary structures that stall replication forks (Zlotorynski et al. 2003). Some CFSs are located in very long (>800 kb) genes in which collisions between the replication and transcription machineries are unavoidable (Helmrich et al. 2011; Le Tallec et al. 2013). However, not all CFSs contain AT-rich regions, and long genes are stable in some cell types. The specific replication program of each chromosomal domain is a major determinant of the “expression” of CFSs. Late-replicating domains with a low density of origins are prone to fragility because forks cover long distances, and in the event of collapse, the chances of activation of a nearby origins are minimal (Letessier et al. 2011; Ozeri-Galai et al. 2011). In contrast, the recently discovered early-replicating fragile sites (ERFS), responsible for >50 % of the translocations observed in B-cell lymphomas, are located in early-replicating, highly expressed gene clusters. In these cases, frequent replication-transcription encounters may account for the fragility (Barlow et al. 2013).
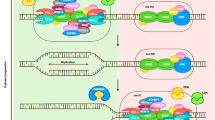
This large body of work with fragile sites point at special DNA structures, low origin density, and replication-transcription collisions as probable causes of RS. These causes, and others, are compiled in Fig. 2 and briefly described in the next sections.

Common causes of RS. Schematics show different DNA replication challenges that lead to RS. See text for details
Endogenous causes of RS
Special DNA structures
In addition to the AT-rich domains present in some CFSs, other DNA sequences can form special structures such as cruciforms, hairpins, G-quadruplexes, H-DNA, or Z-DNA, all of which are natural barriers for replication forks (reviewed in Mirkin and Mirkin 2007). Replication through G-quadruplexes, formed in quartets of guanines stabilized in the same plane by noncanonical Hoogsteen hydrogen bonds, is facilitated by Pif1, BLM, and WRN helicases (Kamath-Loeb et al. 2001; Huber et al. 2002; Paeschke et al. 2011). Short tandem nucleotide repeats (microsatellites) are not a physical barrier but favor the slippage of DNA polymerases, leading to high error rates. Defective replication of trinucleotide repeats has been linked to neurodegenerative diseases (reviewed by Kim and Mirkin 2013), and microsatellite instability is one of the causes of colon cancer (reviewed in Markowitz and Bertagnolli 2009).
Endogenous DNA damage
DNA can be damaged as a consequence of “natural” reactions such as depurination, base oxidation, or interstrand cross links (ICLs) caused by aldehydes (reviewed by Hoeijmakers 2009). Generally, damaged DNA bases and bulky adducts are not recognized as valid templates by replicative DNA polymerases, creating a classic source of RS: the uncoupling of DNA synthesis from DNA unwinding by helicases, which exposes stretches of ssDNA (Byun et al. 2005). ICLs are an exception because they do not create extensive areas of ssDNA (Huang et al. 2010). Cells use the Fanconi anemia (FA) pathway to signal and repair ICLs (Knipscheer et al. 2009; reviewed by Smogorzewska 2013). The majority of FA proteins form a core complex involved in monoubiquitylation of FANCD2 and FANCI (Smogorzewska et al. 2007; Taniguchi et al. 2002), which localize to damaged sites and coordinate the repair of ICLs using a combination of homologous recombination (HR), nucleotide excision repair (NER), and translesion synthesis (TLS; Budzowska et al. 2015; Klein Douwel et al. 2014; Long et al. 2011; Räschle et al. 2008, 2015).
Centromeres, telomeres, and DNA-bound nonhistone proteins
Replication forks also operate on genomic regions with special chromatin structures and encounter nonhistone proteins bound to the template DNA. Budding yeast forks slow down at some of these elements, e.g., centromeres (Greenfeder and Newlon 1992), telomeres (Ivessa et al. 2002), and the silent mating type locus (Ivessa et al. 2003). Yeast Rrm3 is an ancillary helicase that assists the replisome in progressing through such potential blocks (Ivessa et al. 2003). In mammalian cells, it is not yet clear whether these regions represent a challenge for the replisome.
Replication-transcription collisions: topological conflicts and R-loops
Collisions between replication and transcription forks can lead to transcription-associated recombination (TAR) and chromosomal rearrangements (reviewed by Aguilera and Gaillard 2014). Therefore, cells have evolved strategies to minimize these encounters, including temporal and spatial separation between both processes. Most transcription takes place in G1 while replication occurs in S phase, and genes transcribed during S phase may be physically separated from active replication forks (Wei et al. 1998; Helmrich et al. 2011). Actually, some highly transcribed regions are depleted of active origins (Martin et al. 2011). In the tandem repetitions or highly transcribed ribosomal DNA (rDNA) genes in Saccharomyces cerevisiae, collisions are prevented by the formation of a replication fork barrier after each copy of rDNA, leading to effective unidirectional replication (reviewed by Tsang and Carr 2008). In addition, DNA replication can be inhibited in special situations requiring increased transcription. For instance, S. cerevisiae triggers the activation of ~600 genes in response to osmotic stress, concomitant to the inactivation of Mrc1/claspin to minimize DNA replication (Duch et al. 2013).
A first problem that arises in head-on encounters between replication and transcription machineries is the positive supercoiling generated between them. For this reason, topoisomerases are essential to prevent instability at transcriptionally active sites in S phase (Bermejo et al. 2009). Topological constrains are also caused, at least in yeast, by the process of gene gating that brings transcribed genes to the nuclear pores. The Rad53 pathway is needed to release transcribed genes from the nuclear pore, preventing replication forks from stalling at supercoiled regions (Bermejo et al. 2011).
R-loops are three-stranded structures formed by a DNA-RNA duplex and an excluded ssDNA strand that arise during regular transcription and after replication-transcription collisions (reviewed by Hamperl and Cimprich 2014). G-rich areas and G-quadruplexes are prone to R-loop stabilization (Belotserkovskii et al. 2010). R-loops promote TAR and genome instability in mitotic and meiotic cell cycles (Huertas and Aguilera 2003; Castellano-Pozo et al. 2012). Besides the intrinsic vulnerability of the exposed ssDNA, R-loops can be processed into DSBs by the transcription-coupled nucleotide excision repair pathway (Sollier et al. 2014).
The THO/TREX and THSC/TREX-2 complexes that mediate the processing of messenger RNAs (mRNAs) and their export through the nuclear pore complex are required to avoid replication-transcription conflicts (reviewed by Aguilera and Gaillard 2014). THO/TREX may also prevent R-loop formation (Gómez-González et al. 2011). In its absence, the accumulation of R-loops leads to hyperrecombination that can be alleviated by RNAse H, which degrades the RNA component of R-loops (Huertas and Aguilera 2003). In addition to RNAse H, other proteins involved in mRNA processing and RNA biogenesis can minimize the RS associated to R-loops (Wahba et al. 2011; Stirling et al. 2012). Their protective function may rely on their ability to interact with mRNA and prevent its annealing with DNA. For instance, DNA topoisomerase I, splicing factor ASF, and DNA repair protein BRCA2 prevent R-loop formation (Tuduri et al. 2009; Bhatia et al. 2014). Other proteins may facilitate the resolution of R-loops, including helicases Aquarius and Senataxin that are capable of unwinding DNA-RNA structures and the FACT chromatin reorganizing complex (Alzu et al. 2012; Herrera-Moyano et al. 2014; Sollier et al. 2014). Finally, RecQL5 helicase prevents transcription-associated RS by controlling RNA polymerase II progression (Saponaro et al. 2014).
Exogenous causes of RS
DNA lesions caused by irradiation or cytotoxic agents
Ionizing radiation (IR) is a classic cause of DSBs, while ultraviolet (UV) irradiation leads to the formation of photoproducts such as cyclobutane pyrimidine dimers (CPD) and 6-4 pyrimidine pyrimidone ((6-4)pp) that cannot be replicated by DNA polymerases δ or ε, thus causing forks to stall.
Certain drugs used in chemotherapeutic regimes, such as cisplatin, etoposide, and derivatives of camptothecin, are very efficient in killing fast-proliferating tumor cells but also have secondary effects related to RS. Cisplatin is an alkylating agent that interacts with nucleophilic N7-sites of purinic bases to form ICLs and intrastrand cross links (Eastman 1987). As mentioned before, the FA pathway is largely responsible for the repair of ICLs. Camptothecin and etoposide inhibit topoisomerases I and II, respectively, by trapping them in an intermediate step between the DNA break and religation reactions (Hsiang et al. 1985; Osheroff 1989). In addition to chemotherapy drugs, it has been hypothesized that continued exposure to low doses of many chemicals present in modern life, including heavy metals and acrylamide, may contribute to genomic instability by interfering with DNA replication and repair (Langie et al. 2015).
Nucleotide attrition or imbalance
Hydroxyurea (HU), a drug used to treat resistant chronic myelocytic leukemias and other tumors, inhibits ribonucleotide reductase (RNR) and creates imbalances in the cellular pool of dNTPs that affect DNA polymerases and contribute to RS. Prolonged exposure to HU results in irreversible fork collapse and DSBs (Petermann et al. 2010). The pathways of dNTP biosynthesis are carefully controlled in vivo and may be affected by oncogenic activation (reviewed by Aye et al. 2014). In yeast, checkpoint kinase Dun1 activates RNR in response to DNA damage and increases the dNTP pool, improving cell survival (Chabes et al. 2003) but also increasing mutation rates (Davidson et al. 2012; Poli et al. 2012). Interestingly, a mouse model carrying an extra allele of RNR small subunit RRM2 reduces breakage at fragile sites, suggestive of enhanced resistance to RS (Lopez-Contreras et al. 2015).
Aberrant replication triggered by oncogenes
Strictly speaking, oncogene activation is not an exogenous event but is included in this section as a nonphysiological cause of RS. Oncogenic stress activates the DNA damage response (DDR) as a first line of defense against cell transformation (reviewed by Halazonetis et al. 2008). Oncogenes affect the activity of replication origins, either repressing or hyperactivating them, depending on the cellular context and experimental conditions. Premature expression of CycE during G1 restricts the licensing process and accelerates entry into S phase under suboptimal conditions, creating chromosomal instability (Ekholm-Reed et al. 2004). In this situation, cells enter mitosis with under-replicated DNA, promoting the loss of specific genomic regions (Teixeira et al. 2015). It has been hypothesized that cells have an “origin licensing checkpoint” that prevents entry into S phase until a sufficient number of origins have been licensed; the precise mechanism behind this checkpoint remains unknown, but logic dictates that it may be lost in many cancer cells (Shreeram et al. 2002; reviewed by Hills and Diffley 2014).
On the other hand, examples of origin overusage upon oncogene activation have also been reported, which may generate DNA damage in different ways: (1) origin refiring events leading to DSBs (Di Micco et al. 2006), (2) accumulation of active forks that rapidly exhaust dNTPs or other replisome components (Bester et al. 2011; Toledo et al. 2013), and (3) increased probability of collisions between replication and transcription machineries (Jones et al. 2013).
In summary, RS can arise as a consequence of normal cellular reactions involving DNA, upon exposure to external agents or after oncogene activation (Fig. 2). In the next section, we discuss how cells react to RS.
Cellular responses to RS
Some of the responses to RS do not require checkpoint activation, such as the bypass of DNA lesions by restarting DNA synthesis downstream of a damaged template, or the activation of “dormant” origins proximal to stalled forks. Above a certain threshold of RS, a systemic response is activated that shares components with other cellular checkpoints (MacDougall et al. 2007).
Repriming DNA synthesis ahead of a lesion
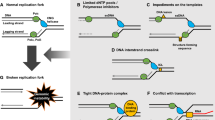
The persistence of unreplicated ssDNA gaps after S phase hints at the possibility that forks paused by different obstacles could bypass them by reinitiating DNA synthesis at a downstream position. Short ssDNA gaps have been observed by electron microscopy in NER-deficient yeast cells irradiated with UV (Lopes et al. 2006). Reinitiation events have also been described in bacteria (Heller and Marians 2006) and human cells after UV irradiation (Elvers et al. 2011). As DNA polymerases do not initiate DNA synthesis de novo, this mechanism depends on a primase activity (Fig. 3a). The canonical Polα/Primase complex initiates Okazaki fragments in the lagging strand, but whether it could perform the repriming task in the leading strand is unclear. The recently discovered PrimPol protein (a primase-polymerase) is rapidly recruited to chromatin upon UV irradiation to facilitate fork restart (Bianchi et al. 2013; García-Gómez et al. 2013; Mouron et al. 2013; Wan et al. 2013). Interestingly, a mutant version of PrimPol retaining TLS activity but lacking primase activity failed to recover fork progression, suggesting that its primase activity is fundamental (Mouron et al. 2013). PrimPol is an error-prone enzyme (Guilliam et al. 2015; Martínez-Jiménez et al. 2015); therefore, rapid fork restart may occur at the partial expense of replication fidelity. A role for PrimPol in reinitiating DNA synthesis downstream of G-quadruplexes has also recently been proposed in avian DT40 cells (Schiavone et al 2015). Interestingly, yeasts do not encode PrimPol homologues, so these reactions must depend on Polα/primase (Fumasoni et al. 2015).

Cellular mechanisms to counteract RS. Schematics summarize the different ways to restart DNA synthesis after an ongoing fork has stalled and/or collapsed. a Repriming ahead of a lesion. b Fork regression and eventual restart. c Translesion synthesis (TLS). d Template switch (TS). e Break-induced replication (BIR). The main proteins involved in each process are indicated in red. See text for details
Activation of dormant origins
As described above, origins are licensed during G1 and activated in the S phase. This temporal regulation avoids origin relicensing that could lead to rereplication events (reviewed by Arias and Walter 2007). However, in the event that two converging forks collapse, no new origins could be assembled in the unreplicated area. This limitation is overcome by the licensing of a “surplus” of origins in G1 (Fig. 4a). Most of these origins remain in a dormant state and provide a backup mechanism as they can be activated to replicate DNA between two collapsed forks (Fig. 4b). When the concentration of MCM complexes is reduced to limit the licensing of dormant origins, cells still replicate DNA in normal growth conditions but accumulate DNA damage in the presence of drugs that challenge fork progression (Ge et al. 2007; Ibarra et al. 2008). The contribution of dormant origins to the maintenance of genomic integrity has been validated in vivo even in an unperturbed S phase. Mouse models with hypomorphic expression of MCM are cancer-prone and their cells display genomic instability (Pruitt et al. 2007; Shima et al. 2007; Kawabata et al. 2011; Bagley et al. 2012; Alvarez et al. 2015).

Dormant origins as a safeguard of genomic integrity. a Schematic showing a region of DNA in which six potential origins have been licensed although only two of them (marked with green arrows) will be activated under normal growth conditions. The rest (red arrows) remain dormant during S phase. b “Emergency situation” in which two converging forks collapse after facing one of the challenges described in Fig. 2. In this setting, a dormant origin situated between the forks gets activated to rescue replication of this chromosome region
The mechanisms that regulate dormant origins are still under investigation. A model developed from biochemical work in Xenopus egg extracts invokes the action of Polo kinase 1 (PLK1) at stalled forks after MCM2 phosphorylation by ataxia-telangiectasia mutated and RAD3-related kinase (ATR). PLK1 could facilitate the loading of CDC45, a cofactor of MCM helicase, to proximal dormant origins to promote their activation (Trenz et al. 2008). Alternatively, PLK1 could restrict CHK1 activity (responsible for origin inhibition) by modulating Claspin levels (Mailand et al. 2006; Peschiaroli et al. 2006). FANCI protein, a member of the FA repair pathway, has also been linked to the regulation of dormant origins, although the precise mechanism remains unclear (Chen et al. 2015).
Checkpoint activation: cell cycle arrest and inhibition of late origins
The central regulator of the response to RS is ATR, a member of the phosphoinositide 3-kinase (PI3K) family of kinases. The canonical activation pathway starts with the recruitment of ATR and its interacting partner ATRIP to stretches of ssDNA covered by RPA. Once activated, ATR phosphorylates multiple proteins including the effector Chk1 kinase that blocks entry into mitosis and inhibits late origin firing. Mitotic entry is inhibited through phosphorylation of Cdc25 phosphatases, which prevents subsequent activation of mitotic CDKs. This mechanism is well studied and it is shared with other checkpoint pathways (reviewed by Cimprich and Cortez 2008).
Inhibition of origin activity by ATR-CHK1 activity (Maya-Mendoza et al. 2007; Syljuasen et al. 2005) could be mediated by the inhibition of Sld3/Treslin/Ticcr and/or the Dbf4 subunit of the DDK kinase (Costanzo et al. 2003; Lopez-Mosqueda et al. 2010; Zegerman and Diffley 2010; Guo et al. 2015). Checkpoint-mediated inhibition of late origins poses a paradox, as dormant origins need to be activated to rescue stalled forks (Fig. 4). The solution to this conflict resides in the capacity of Chk1 to inhibit origins “globally” while allowing the activation of those dormant origins in the proximity of stalled forks (Ge and Blow 2010). In this regard, the organization of clusters of adjacent origins in discrete “DNA replication factories” (Jackson and Pombo 1998; Guillou et al. 2010; Aparicio et al. 2012) contributes to separate local and global effects.
Stabilization of stalled forks
When forks stall upon stress, it is important to preserve the integrity of the replisome in order to facilitate fork restart. In yeast, checkpoint kinases Mec1/ATR and Rad53/CHK1 are needed to maintain fork integrity (Lopes et al. 2001; Tercero and Diffley 2001). Early studies suggested that Mec1 and Rad53 stabilized the replisome at stalled forks (Cobb et al. 2003, 2005; Katou et al. 2003; Lucca et al. 2004), but a later study showed replisome stability to be independent of the checkpoint kinases (De Piccoli et al. 2012). The role of checkpoint kinases may instead be directed toward controlling DNA processing at stalled forks by nucleases such as EXO1 (El-Shemerly et al. 2008; Morin et al. 2008). Loss of EXO1 prevents fork breakdown in Rad53 mutants treated with MMS or UV (Segurado and Diffley 2008). HR factor Rad51 also protects stalled forks from excessive Mre11 resection (Hashimoto et al. 2010). FA proteins participate in this protective response by cooperating with BRCA1/2 to stabilize RAD51 at stalled forks (Schlacher et al. 2011, 2012).
The detailed mechanisms underlying mammalian fork stabilization are complex and only partially understood, but the emerging view is that they involve DNA helicases, translocases, and FA proteins such as BLM, WRN, ZRANB3, SMARCAL1, and FANCM (Bansbach et al. 2009; Ciccia et al. 2012; Davies et al. 2007; Kim et al. 2008; Sidorova et al. 2008; Yuan et al. 2009). Some of these factors seem to regulate fork reversal, an intriguing remodeling mechanism that is discussed next.
Fork reversal: friend or foe?
Stalled forks can be reversed to form four-way “chicken foot” structures that resemble Holliday junctions (Fig. 3b). These structures were first identified in yeast checkpoint-deficient strains (Lopes et al. 2001; Sogo et al. 2002) and their biological significance has been debated (reviewed by Neelsen and Lopes 2015). Initially, they were considered aberrant recombinogenic structures that must be processed by exonucleases to maintain genome integrity (Cotta-Ramusino et al. 2005; Hu et al. 2012). However, recent studies in human cells suggest that reversal is a physiological way to protect fork integrity (Zellweger et al. 2015) and induce the checkpoint response (Fugger et al. 2015). Several factors that mediate fork reversal and/or stabilization of reversed intermediates have been identified in recent years, including PARP1, RAD51, SMARCAL, and FBH1 (Berti et al. 2013; Couch et al. 2013; Fugger et al. 2015; Zellweger et al. 2015). To resume DNA synthesis, reversed forks need to be remodeled again into forks that move in the forward direction. This remodeling may be mediated by RECQ1 DNA helicase (Berti et al. 2013) or by WRN helicase and DNA2 nuclease, which catalyze the nucleolytic degradation of reversed DNA arms (Thangavel et al. 2015). It is possible that other helicases as BLM and ZRANB3, which possess fork remodeling activities, also mediated fork reversal and/or fork restart (reviewed by Petermann and Helleday 2010).
Next, we present other mechanisms that allow the continuation of DNA synthesis upon fork stalling: TLS and template switch (TS)-based pathways. The choice between them is dictated by posttranslational modifications in PCNA (Hoege et al. 2002).
Translesion synthesis
TLS, favored by PCNA monoubiquitylation at Lys164 by RAD6 and RAD18 ubiquitin ligase (Kannouche et al. 2004; Stelter and Ulrich 2003), is carried out by specialized DNA polymerases whose flexible active sites allow the insertion of nucleotides opposite damaged templates at the expense of fidelity (Fig. 3c). Each one of the TLS polymerases identified to date (Rev1, Polη, Polι, Polκ, Polζ, and PrimPol) has evolved to bypass specific lesions, including thymidine adducts, 8-oxoguanine, (6,4)pp, CPD, or apurinic sites (reviewed by Sale et al. 2012). The proposed mechanism for TLS implicates two steps. First, the replicative polymerase is replaced by a TLS polymerase that incorporates a nucleotide opposite the damaged template. Then, the same or a different polymerase, usually Polζ, elongates the TLS product until the replicative polymerase can be engaged again (Shachar et al. 2009). It is worth noting that Polη catalyzes error-free synthesis opposite CPD lesions (Masutani et al. 1999; Johnson et al. 2000), so it is conceivable that each TLS polymerase has the capacity to deal with a specific lesion in an essentially error-free manner but introduces errors when replicating different lesions or an intact template.
A particularity of Rev1 is its capacity to bind to PCNA and Polη, Polι, Polκ, and Polζ (Guo et al. 2003, 2006). Hence, Rev1 could provide an “exchange platform” for TLS polymerases at stalled forks. This mechanism is partially independent of PCNA ubiquitylation and suggests an interesting model for the regulation of TLS polymerases: their recruitment to stalled forks during S phase could be mediated by Rev1, while PCNA ubiquitylation would be a signal to recruit them during postreplicative gap filling (Edmunds et al. 2008; reviewed by Sale 2012).
Within TLS enzymes, PrimPol is unusual in the sense that it belongs to the family of archeal eukaryotic primases (AEP) and contains both primase and TLS polymerase activities. Besides its function in repriming DNA synthesis ahead of a lesion (described above), as a TLS polymerase, it has the potential to replicate through oxidative lesions (8-oxo-G) and UV-induced CPD and 6,4pp (Bianchi et al. 2013; García-Gómez et al. 2013; Mourón et al. 2013).
Template switch
PCNA polyubiquitylation by Rad18, Rad5, and Ubc13-Mms2 and PCNA sumoylation by Ubc9 and Siz1 tip the balance toward TS mechanisms (Hoege et al. 2002; Branzei et al. 2008). In TS, a stalled nascent DNA strand invades the sister chromatid and is used as a primer to continue replication using the newly synthesized undamaged strand as template (Fig. 3d). TS intermediates have been recently characterized by a combination of two-dimensional gel electrophoresis and electron microscopy techniques (Giannattasio et al. 2014). TS is considered an error-free mechanism because the emerging X-shaped structures can be processed by BLM-TOPIIIa-RMI2 (Sgs1/Top3/Rmi1 in yeast) without generating crossovers (Liberi et al. 2005; Mankouri and Hickson 2006; Raynard et al. 2006).
The current view is that HR and TS factors are coordinated to promote fork restart. For instance, Rad51 mediates strand invasion in TS (Vanoli et al. 2010; Petermann et al. 2010) but its recombinogenic activity must be controlled. In cells exposed to a short HU treatment, TS is favored over HR (Petermann et al. 2010). In this case, Fbh1 helicase displaces Rad51 filaments from ssDNA (Simandlova et al. 2013; Chu et al. 2015), while its ubiquitin ligase activity monoubiquitylates Rad51 to export it from the nucleus (Chu et al. 2015). Other antirecombinogenic factors include BLM, RECQ1, RECQ5, FANCJ, and FANCM (Bugreev et al. 2007, 2008; Hu et al. 2007; Rosado et al. 2009; Sommers et al. 2009; Wu et al. 2001).
TLS and TS mediate fork restart during S phase but they can also operate postreplicatively. In yeast, both TLS and TS-based pathways can be restricted to G2 without compromising viability (Daigaku et al. 2010; Karras and Jentsch 2010). In these situations, after the bulk of DNA synthesis is completed, any remaining gaps are filled by TLS and/or TS, while lesions in the template strand are eliminated by NER or base excision repair (BER) mechanisms. The molecular details of these repair pathways can be found in recent reviews (Marteijn et al. 2014; Parsons and Dianov 2013).
HR and break-induced replication
HR plays a fundamental role in DSB repair. An HR variant called break-induced replication (BIR) may reinitiate DNA synthesis when collapsed forks are processed by structure-specific endonucleases such as Mus81-Eme1 to generate one-ended DSBs (Hanada et al. 2007). BIR was initially described in bacteria and bacteriophage T4 and has been largely studied in yeast (reviewed by Anand et al. 2013). A recent report suggests that it is also conserved in mammalian cells (Costantino et al. 2014).
BIR starts like the classical HR pathway (Fig. 3e). The Mre11-Rad50-Nbs1 (MRN) complex is recruited to breaks, where Mre11 nuclease resects one of the DNA ends while Nbs1 activates ATM. Subsequently, Brca2 targets Rad51 to ssDNA and the Rad51-ssDNA filament invades the sister chromatid. BIR-mediated DNA synthesis then proceeds in a migrating bubble that requires a full replisome but not origin licensing factors (Lydeard et al. 2010). Of note, BIR results in a conservative inheritance of DNA (Donnianni and Symington 2013; Saini et al. 2013; Wilson et al. 2013). Synthesis of the leading and lagging strands are uncoupled and ssDNA accumulates behind the migrating bubble (Fig. 3e) making BIR a highly mutagenic process (Deem et al. 2011; Sakofsky et al. 2014). In fact, a variation called microhomology-mediated BIR may be responsible for complex genomic rearrangements similar to those described in chromothripsis (Liu et al. 2011). In addition, BIR may lead to telomere lengthening by telomerase-independent recombination (Lydeard et al. 2007) but also by promoting telomerase activity in yeast (Vasianovich et al. 2014).
RS and human disease
The importance of the cellular responses to RS for human health is highlighted by the array of genetic diseases, as well as increased cancer predisposition, associated with alterations in the genes that participate in these responses.
Rare genetic diseases associated with RS
Mutations in components of the checkpoint response such as the ATM, ATR, and the MRN complex cause severe diseases such as ataxia-telangiectasia, Seckel syndrome, ataxia-telangiectasia-like disease (A-TLD), Nijmegen breakage syndrome (NBS), and Nijmegen breakage syndrome-like disease (NBSLD). A-TLD is linked to mutations in Mre11 that disrupt ATM activation, and consequently, it is very similar to ataxia-telangiectasia caused by ATM mutations (reviewed by Biton et al. 2008). Loss of function of ATR or ATRIP causes Seckel syndrome, characterized by mental retardation, growth defects, and abnormal craniofacial features (reviewed by O’Driscoll and Jeggo 2008). NBS and NBSLD are the consequence of mutations in Nbs1 and Rad50 genes, respectively, which compromise their roles in ATR activation or HR. Patients display microcephaly, mental retardation, growth defects and, in the case of NBS, cancer susceptibility (Demuth and Digweed 2007; Waltes et al. 2009). Mutations in genes responsible for origin licensing (Orc1, Orc4, Orc6, Cdc6, and Cdt1) are linked to a related genetic condition called Meier-Gorlin syndrome, whose patients display growth impairment, microcephaly, and lack of patellae in many cases (Guernsey et al. 2011; Bicknell et al. 2011a, b).
Mutations in the helicases that mediate fork remodeling and restart, BLM, WRN, and RecQ4L, cause Bloom (BS), Werner (WS), and Rothmund-Thomson (RTS) syndromes, respectively. These diseases are not only caused by RS but also by the participation of these proteins in classical DNA repair pathways, e.g., BLM is required for the dissolution of double Holliday junctions (reviewed by Bizard and Hickson 2014). BS patients show cancer predisposition, microcephaly, mental retardation, infertility, growth defects, and premature aging. WS patients suffer from age-related symptoms including atherosclerosis, cataracts, osteoporosis, and diabetes. Its morbidity is frequently caused by cancer or vascular disease. RTS also causes aging phenotypes, growth defects, and skin and skeletal abnormalities. Mutations in RecQ4L cause two additional independent diseases, RAPADILINO syndrome and Baller-Gelrold syndrome (reviewed by Chu and Hickson 2009; Croteau et al. 2014). Loss of SMARCAL1 function is responsible of Schimke immuno-oseo dysplasia (SIOD), whose patients suffer from immunodeficiency, renal defects, skeletal abnormalities, and dwarfism (Boerkoel et al. 2002).
Fanconi anemia, a disease that can be autosomal recessive or X-linked, is characterized by skeletal defects, hypopigmentation, progressive bone marrow failure, and cancer predisposition (Fanconi 1967). It is caused by mutations in any of the 18 identified genes of the FA pathway (Rickman et al. 2015; reviewed by Wang and Smogorzewska 2015). FA cells fail to repair ICLs, which elevates their genome instability.
Finally, mutations in TLS polymerase Polη are linked to Xeroderma pigmentosum (XP), which increases photosensitivity and skin cancer predisposition (Masutani et al. 1999). Whether the loss of other TLS polymerases results in related diseases remains to be elucidated.
The phenotypes of most of these genetic diseases have a common theme, i.e., incomplete development that can be linked to deficiency in stem cell abundance and/or functionality. In this regard, a mouse model hypomorphic for Mcm2 shows deficiency in several stem cell niches (Pruitt et al. 2007), and mouse models defective in DDR components display depletion of hematopoietic stem cells (HSCs; reviewed by Rossi et al. 2008). RS associated to low MCM expression also underlies the progressive loss of HSC regenerative capacity with age (Flach et al. 2014) and impairs fetal erythropoiesis (Alvarez et al. 2015). Of note, FA patients also display a defect in hematopoietic stem and progenitors cells due to an exacerbated response to unrepaired DNA damage (Ceccaldi et al. 2012).
Cancer predisposition
Besides the genetic conditions described in the previous section, RS contributes to cell transformation and cancer predisposition. Recent work has unveiled a positive correlation between cancer risk and the number of stem cell divisions in different tissues, due to stochastic problems that arise during DNA replication (Tomasetti and Vogelstein 2015). In addition, fragile sites have been associated to tumor development (Pelliccia et al. 2010; Blumrich et al. 2011; Barlow et al. 2013). Loss or hypomorphic mutation of genes involved in origin licensing, fork stability and restart, TLS, and DNA repair are related to cancer predisposition in patients and/or animal models. These include components of the MCM helicase, four of the five RecQ helicases (BLM, WNR, RECQ4L, and RECQ5L), Polη and Polζ, BRCA1 and BRCA2, endonucleases Mus81 and Slx4, and FA members (reviewed by Gaillard et al. 2015).
Upon oncogene expression, the DDR is activated in precancerous lesions, inducing senescence or apoptosis programs in damaged cells. Some cells may override this barrier and facilitate cancer development (Halazonetis et al. 2008). Interestingly, extensive sequencing of cancer genomes has revealed a low incidence of mutations, and the absence of deletions, in ATR or Chk1. On the contrary, the expression of these genes is frequently upregulated in cancer cells, probably because they need to deal with high levels of RS to survive (reviewed by Lecona and Fernández-Capetillo 2014). Chk1 overexpression increases the transformation capacity of Ras-E1A and c-fos oncogenes (Lopez-Contreras et al. 2012; Schulze et al. 2014) by shielding transformed cells from excessive RS. Mutations arising in cancer development could enhance RS (reviewed by Hills and Diffley 2014), making tumor cells hypersensitive to the inhibition of RS signaling. For instance, ATR inhibition in cancer cells driven by c-myc, which display high levels or RS, results in cell death and tumor regression (Murga et al. 2011). Several inhibitors of ATR, CHK1, or CDC7 are under clinical trials (for a recent review, see Dobbelstein and Sorensen 2015).
Because cancer mutations can inactivate specific DNA repair pathways, other therapeutic strategies exploit synthetic lethality effects caused by the loss of two modes of DNA repair, e.g., the use of PARP inhibitors in breast cancer cells with mutations in BRCA1 and BRCA2 (Farmer et al. 2005; Bryant et al. 2005), which targets the BER pathway in cells already deficient in HR.
Concluding remarks
DNA replication is a central process for tissue homeostasis and genetic inheritance. As described in this review, faulty replication, particularly when it occurs in stem cells during embryonic development, can lead to multiple diseases with severe phenotypes and increase the probability of developing cancer. Multiple mechanisms have evolved to counteract RS, and in the future, it will be very interesting to ascertain the relative contribution of each one of them in different cell types and growth conditions. In any case, as shown in this review, none of them seems to provide a bulletproof defense and many of them carry a significant risk of mutation or chromosomal instability.
Therapies directed to modulate RS have shown promising results in mouse models and need to be further developed to offer therapeutic alternatives for cancer patients. In addition, they could serve to counteract aging, which is possibly influenced by the levels of DNA damage suffered by fetal tissues during embryonic development (Fernandez-Capetillo 2010). Improving the resistance to RS extends the lifespan of mice suffering from premature aging (Lopez-Contreras et al. 2015) and alleviates the embryonic lethality associated to MCM hypomorphic expression (Alvarez et al. 2015). Transgenic mice with enhanced CDC6 expression in keratinocytes displayed more licensed origins and reached old age with remarkable preservation of hair density and color (Búa et al. 2015). Therefore, future anti-RS therapies may counterbalance the functional decline of tissues that today seems inevitable as we age.
References
Aguilera A, Gaillard H (2014) Transcription and recombination: when RNA meets DNA. Cold Spring Harb Perspect Biol 6. doi:10.1101/cshperspect.a016543
Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K et al (2014) Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16:281–293
Alvarez S, Díaz M, Flach J, Rodriguez-Acebes S, López-Contreras AJ, Martínez D, Cañamero M, Fernández-Capetillo O, Isern J, Passegué E, Méndez J (2015) Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun 6:8548
Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D et al (2012) Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151:835–846
Anand RP, Lovett ST, Haber JE (2013) Break-induced DNA replication. Cold Spring Harb Perspect Biol 5. doi:10.1101/cshperspect.a010397
Aparicio T, Megías D, Méndez J (2012) Visualization of the MCM DNA helicase at replication factories before the onset of DNA synthesis. Chromosoma 121:499–507
Arias EE, Walter JC (2007) Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev 21:497–518
Aye Y, Li M, Long MJ, Weiss RS (2014) Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34:2011–2021
Bagley BN, Keane TM, Maklakova VI, Marshall JG, Lester RA et al (2012) A dominantly acting murine allele of Mcm4 causes chromosomal abnormalities and promotes tumorigenesis. PLoS Genet 8, e1003034
Bansbach CE, Bétous R, Lovejoy CA, Glick GG, Cortez D (2009) The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23:2405–2414
Barlow JH, Faryabi RB, Callén E, Wong N, Malhowski A et al (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152:620–632
Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC (2010) Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci U S A 107:12816–12821
Bermejo R, Capra T, Gonzalez-Huici V, Fachinetti D, Cocito A et al (2009) Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 138:870–884
Bermejo R, Capra T, Jossen R, Colosio A, Frattini C et al (2011) The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 146:233–246
Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S et al (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20:347–354
Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446
Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A (2014) BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511:362–365
Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V et al (2013) PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52:566–573
Bicknell LS, Walker S, Klingseisen A, Stiff T, Leitch A et al (2011a) Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet 43:350–355
Bicknell LS, Bongers EM, Leitch A, Brown S, Schoots J et al (2011b) Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet 43:356–359
Biton S, Barzilai A, Shiloh Y (2008) The neurological phenotype of ataxia-telangiectasia: solving a persistent puzzle. DNA Repair (Amst) 7:1028–1038
Bizard AH, Hickson ID (2014) The dissolution of double Holliday junctions. Cold Spring Harb Perspect Biol 6(7):a016477. doi:10.1101/cshperspect.a016477
Bleichert F, Botchan MR, Berger JM (2015) Crystal structure of the eukaryotic origin recognition complex. Nature 519:321–326
Blumrich A, Zapatka M, Brueckner LM, Zheglo D, Schwab M, Savelyeva L (2011) The FRA2C common fragile site maps to the borders of MYCN amplicons in neuroblastoma and is associated with gross chromosomal rearrangements in different cancers. Hum Mol Genet 20:1488–1501
Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P et al (2002) Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet 30:215–220
Branzei D, Vanoli F, Foiani M (2008) SUMOylation regulates Rad18-mediated template switch. Nature 456:915–920
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–917
Búa S, Sotiropoulou P, Sgarlata C, Borlado LR, Eguren M, Domínguez O, Ortega S, Malumbres M, Blanpain C, Méndez J (2015) Deregulated expression of Cdc6 in the skin facilitates papilloma formation and affects the hair growth cycle. Cell Cycle 14:3897–3907
Budzowska M, Graham TG, Sobeck A, Waga S, Walter JC (2015) Regulation of the Rev1-polz complex during bypass of a DNA interstrand cross-link. EMBO J 34:1971–1985
Bugreev DV, Yu X, Egelman EH, Mazin AV (2007) Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev 21:3085–3094
Bugreev DV, Brosh RM Jr, Mazin AV (2008) RECQ1 possesses DNA branch migration activity. J Biol Chem 283:20231–20242
Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19:1040–1052
Castellano-Pozo M, García-Muse T, Aguilera A (2012) R-loops cause replication impairment and genome instability during meiosis. EMBO Rep 13:923–929
Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM et al (2012) Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 11:36–49
Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L (2003) Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112:391–401
Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, Rothenberg E, Jallepalli PV, Huang TT (2015) ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 58:323–338
Chu WK, Hickson ID (2009) RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer 9:644–654
Chu WK, Payne MJ, Beli P, Hanada K, Choudhary C, Hickson ID (2015) FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat Commun 6. doi:10.1038/ncomms6931
Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ et al (2012) Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47:396–409
Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9:616–627
Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM (2003) DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J 22:4325–4336
Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM (2005) Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19:3055–3069
Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD (2014) Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343:88–91
Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11:203–213
Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M (2005) Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 17:153–159
Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG et al (2013) ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27:1610–1623
Croteau DL, Popuri V, Opresko PL, Bohr VA (2014) Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem 83:519–552
Daigaku Y, Davies AA, Ulrich HD (2010) Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465:951–955
Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T et al (2012) Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J 31:895–907
Davies SL, North PS, Hickson ID (2007) Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol 14:677–679
De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, Labib K (2012) Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell 45:696–704
Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A (2011) Break-induced replication is highly inaccurate. PLoS Biol 9. doi:10.1371/journal.pbio.1000594
Demuth I, Digweed M (2007) The clinical manifestation of a defective response to DNA double-strand breaks as exemplified by Nijmegen breakage syndrome. Oncogene 26:7792–7798
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P et al (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642
Dobbelstein M, Sørensen CS (2015) Exploiting replicative stress to treat cancer. Nat Rev Drug Discov 14:405–423
Donnianni RA, Symington LS (2013) Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A 110:13475–13480
Duch A, Felipe-Abrio I, Barroso S, Yaakov G, García-Rubio M, Aguilera A, de Nadal E, Posas F (2013) Coordinated control of replication and transcription by a SAPK protects genomic integrity. Nature 493:116–119
Durkin SG, Glover TW (2007) Chromosome fragile sites. Annu Rev Genet 41:169–192
Eastman A (1987) The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol Ther 34:155–166
Edmunds CE, Simpson LJ, Sale JE (2008) PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30:519–529
Ekholm-Reed S, Méndez J, Tedesco D, Zetterberg A, Stillman B, Reed SI (2004) Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol 165:789–800
El-Shemerly M, Hess D, Pyakurel AK, Moselhy S, Ferrari S (2008) ATR-dependent pathways control hEXO1 stability in response to stalled forks. Nucleic Acids Res 36:511–519
Elvers I, Johansson F, Groth P, Erixon K, Helleday T (2011) UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res 39:7049–7057
Errico A, Cosentino C, Rivera T, Losada A, Schwob E, Hunt T, Costanzo V (2009) Tipin/Tim1/And1 protein complex promotes Pol alpha chromatin binding and sister chromatid cohesion. EMBO J 28:3681–3692
Fanconi G (1967) Familial constitutional panmyelocytopathy, Fanconi’s anemia (F. A.). I. Clinical aspects. Semin Hematol 4:233–240
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–921
Fernandez-Capetillo O (2010) Intrauterine programming of ageing. EMBO Rep 11:32–36
Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM et al (2014) Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512:198–202
Fugger K, Mistrik M, Neelsen KJ, Yao Q, Zellweger R et al (2015) FBH1 catalyzes regression of stalled replication forks. Cell Rep 10:1749–1757
Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D (2015) Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 complex. Mol Cell 57:812–823
Gaillard H, García-Muse T, Aguilera A (2015) Replication stress and cancer. Nat Rev Cancer 15:276–289
Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K (2006) GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol 8:358–366
Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, Labib K (2009) A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J 28:2992–3004
García-Gómez S, Reyes A, Martínez-Jiménez MI, Chocrón ES et al (2013) PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52:541–553
Ge XQ, Blow JJ (2010) Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 191:1285–1297
Ge XQ, Jackson DA, Blow JJ (2007) Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 21:3331–3341
Giannattasio M, Zwicky K, Follonier C, Foiani M, Lopes M, Branzei D (2014) Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Biol 21:884–892
Glover TW, Berger C, Coyle J, Echo B (1984) DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 67:136–142
Gómez-González B, García-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marín A, Foiani M, Aguilera A (2011) Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J 30:3106–3119
Greenfeder SA, Newlon CS (1992) Replication forks pause at yeast centromeres. Mol Cell Biol 12:4056–4066
Guernsey DL, Matsuoka M, Jiang H, Evans S, Macgillivray C et al (2011) Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat Genet 43:360–364
Guilliam TA, Jozwiakowski SK, Ehlinger A, Barnes RP, Rudd SG, Bailey LJ, Skehel JM, Eckert KA, Chazin WJ, Doherty AJ (2015) Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res 43:1056–1068
Guillou E, Ibarra A, Coulon V, Casado-Vela J, Rico D, Casal I, Schwob E, Losada A, Méndez J (2010) Cohesin organizes chromatin loops at DNA replication factories. Genes Dev 24:2812–2822
Guo C, Fischhaber PL, Luk-Paszyc MJ, Masuda Y, Zhou J, Kamiya K, Kisker C, Friedberg EC (2003) Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J 22:6621–6630
Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC (2006) REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell 23:265–271
Guo C, Kumagai A, Schlacher K, Shevchenko A, Shevchenko A, Dunphy WG (2015) Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol Cell 57:492–505
Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319:1352–1355
Hamperl S, Cimprich KA (2014) The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 19:84–94
Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R (2007) The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol 14:1096–1104
Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17:1305–1311
Heller RC, Marians KJ (2006) Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439:557–562
Helmrich A, Ballarino M, Tora L (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44:966–977
Herrera-Moyano E, Mergui X, García-Rubio ML, Barroso S, Aguilera A (2014) The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev 28:735–748
Hills SA, Diffley JF (2014) DNA replication and oncogene-induced replicative stress. Curr Biol 24:R435–R444
Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135–141
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361:1475–1485
Hsiang YH, Hertzberg R, Hecht S, Liu LF (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260:14873–14878
Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W et al (2007) RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21:3073–3084
Hu J, Sun L, Shen F, Chen Y, Hua Y et al (2012) The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149:1221–1232
Huang M, Kim JM, Shiotani B, Yang K, Zou L, D’Andrea AD (2010) The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell 39:259–268
Huber MD, Lee DC, Maizels N (2002) G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res 30:3954–3961
Huertas P, Aguilera A (2003) Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 12:711–721
Ibarra A, Schwob E, Méndez J (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci U S A 105:8956–8961
Ivessa AS, Zhou JQ, Schulz VP, Monson EK, Zakian VA (2002) Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev 16:1383–1396
Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein–DNA complexes. Mol Cell 12:1525–1536
Jackson DA, Pombo A (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140:1285–1295
Johnson RE, Washington MT, Prakash S, Prakash L (2000) Fidelity of human DNA polymerase eta. J Biol Chem 275:7447–7450
Jones RM, Mortusewicz O, Afzal I, Lorvellec M, García P, Helleday T, Petermann E (2013) Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 32:3744–3753
Kamath-Loeb AS, Loeb LA, Johansson E, Burgers PM, Fry M (2001) Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J Biol Chem 276:16439–16446
Kannouche PL, Wing J, Lehmann AR (2004) Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14:491–500
Karras GI, Jentsch S (2010) The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141:255–267
Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078–1083
Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N (2011) Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell 41:543–553
Kim JC, Mirkin SM (2013) The balancing act of DNA repeat expansions. Curr Opin Genet Dev 23:280–288
Kim JM, Kee Y, Gurtan A, D’Andrea AD (2008) Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 111:5215–5222
Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Räschle M, Walter JC, Knipscheer P (2014) XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54:460–471
Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC (2009) The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326:1698–1701
Kottemann MC, Smogorzewska A (2013) Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493:356–363
Langie SA, Koppen G, Desaulniers D, Al-Mulla F, Al-Temaimi R et al (2015) Causes of genome instability: the effect of low dose chemical exposures in modern society. Carcinogenesis 36:61–88
Le Tallec B, Dutrillaux B, Lachages AM, Millot GA, Brison O, Debatisse M (2011) Molecular profiling of common fragile sites in human fibroblasts. Nat Struct Mol Biol 18:1421–1423
Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M (2013) Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep 4:420–428
Lecona E, Fernández-Capetillo O (2014) Replication stress and cancer: it takes two to tango. Exp Cell Res 329:26–34
Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M (2011) Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470:120–123
Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, Tye BK, Gao N (2015) Structure of the eukaryotic MCM complex at 3.8 Å. Nature 524:186–191
Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19:339–350
Liu P, Erez A, Nagamani SC, Dhar SU, Kołodziejska KE et al (2011) Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146:889–903
Long DT, Räschle M, Joukov V, Walter JC (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333:84–87
Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412:557–561
Lopes M, Foiani M, Sogo JM (2006) Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21:15–27
López-Contreras AJ, Gutierrez-Martinez P, Specks J, Rodrigo-Perez S, Fernandez-Capetillo O (2012) An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J Exp Med 209:455–461
Lopez-Contreras AJ, Ruppen I, Nieto-Soler M, Murga M, Rodriguez-Acebes S, Remeseiro S, Rodrigo-Perez S, Rojas AM, Mendez J, Muñoz J, Fernandez-Capetillo O (2013) A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep 3:1105–1116
Lopez-Contreras AJ, Specks J, Barlow JH, Ambrogio C, Desler C et al (2015) Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev 29:690–695
Lopez-Mosqueda J, Maas NL, Jonsson ZO, Defazio-Eli LG, Wohlschlegel J, Toczyski DP (2010) Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 467:479–483
Lucca C, Vanoli F, Cotta-Ramusino C, Pellicioli A, Liberi G, Haber J, Foiani M (2004) Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene 23:1206–1213
Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448:820–823
Lydeard JR, Lipkin-Moore Z, Sheu YJ, Stillman B, Burgers PM, Haber JE (2010) Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 24:1133–1144
MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA (2007) The structural determinants of checkpoint activation. Genes Dev 21:898–903
Mailand N, Bekker-Jensen S, Bartek J, Lukas J (2006) Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 23:307–318
Mankouri HW, Hickson ID (2006) Top3 processes recombination intermediates and modulates checkpoint activity after DNA damage. Mol Biol Cell 17:4473–4483
Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361:2449–2460
Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15:465–481
Martin MM, Ryan M, Kim R, Zakas AL, Fu H et al (2011) Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res 21:1822–1832
Martínez-Jiménez MI, García-Gómez S, Bebenek K, Sastre-Moreno G, Calvo PA, Díaz-Talavera A, Kunkel TA, Blanco L (2015) Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair (Amst) 29:127–138
Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M (2010) Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem 79:89–130
Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399:700–704
Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA (2007) Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 26:2719–2731
Mirkin EV, Mirkin SM (2007) Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71:13–35
Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D (2008) Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J 27:2400–2410
Mourón S, Rodriguez-Acebes S, Martínez-Jiménez MI, García-Gómez S, Chocrón S, Blanco L, Méndez J (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20:1383–1389
Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R et al (2011) Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol 18:1331–1335
Neelsen KJ, Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16:207–220
O’Driscoll M, Jeggo PA (2008) The role of the DNA damage response pathways in brain development and microcephaly: insight from human disorders. DNA Repair (Amst) 7:1039–1050
Osborn AJ, Elledge SJ, Zou L (2002) Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol 12:509–516
Osheroff N (1989) Effect of antineoplastic agents on the DNA cleavage/religation reaction of eukaryotic topoisomerase II: inhibition of DNA religation by etoposide. Biochemistry 28:6157–6160
Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B (2011) Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 43:122–131
Paeschke K, Capra JA, Zakian VA (2011) DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145:678–691
Parsons JL, Dianov GL (2013) Co-ordination of base excision repair and genome stability. DNA Repair (Amst) 12:326–333
Pelliccia F, Bosco N, Rocchi A (2010) Breakages at common fragile sites set boundaries of amplified regions in two leukemia cell lines K562—molecular characterization of FRA2H and localization of a new CFS FRA2S. Cancer Lett 299:37–44
Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M (2006) SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 23:319–329
Petermann E, Helleday T (2010) Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11:683–687
Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010) Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37:492–502
Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P (2012) dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31:883–894
Pruitt SC, Bailey KJ, Freeland A (2007) Reduced Mcm2 expression results in severe stem/progenitor cell deficiency and cancer. Stem Cells 25:3121–3132
Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Schärer OD, Walter JC (2008) Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134:969–980
Räschle M, Smeenk G, Hansen RK, Temu T, Oka Y et al (2015) DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 348:1253671
Raynard S, Bussen W, Sung P (2006) A double Holliday junction dissolvasome comprising BLM, topoisomerase III alpha, and BLAP75. J Biol Chem 281:13861–13864
Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM et al (2015) Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Rep 12:35–41
Rosado IV, Niedzwiedz W, Alpi AF, Patel KJ (2009) The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res 37:4360–4370
Rossi DJ, Jamieson CH, Weissman IL (2008) Stems cells and the pathways to aging and cancer. Cell 132:681–696
Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A (2013) Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502:389–392
Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, Malkova A (2014) Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7:1640–1648
Sale JE (2012) Competition, collaboration and coordination—determining how cells bypass DNA damage. J Cell Sci 125:1633–1643
Sale JE, Lehmann AR, Woodgate R (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol 13:141–152
Saponaro M, Kantidakis T, Mitter R, Kelly GP, Heron M, Williams H, Söding J, Stewart A, Svejstrup JQ (2014) RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157:1037–1049
Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, Doherty AJ (2015) PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol Cell. S1097-2765(15)00829-1
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–542
Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22:106–116
Schulze J, Lopez-Contreras AJ, Uluçkan Ö, Graña-Castro O, Fernandez-Capetillo O, Wagner EF (2014) Fos-dependent induction of Chk1 protects osteoblasts from replication stress. Cell Cycle 13:1980–1986
Segurado M, Diffley JF (2008) Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev 22:1816–1827
Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J et al (2009) Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J 28:383–393
Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, Schimenti JC (2007) A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet 39:93–98
Shreeram S, Sparks A, Lane DP, Blow JJ (2002) Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene 21:6624–6632
Sidorova JM, Li N, Folch A, Monnat RJ Jr (2008) The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 7:796–807
Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I et al (2013) FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J Biol Chem 288:34168–34180
Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288:31458–31467
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE et al (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289–301
Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297:599–602
Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA (2014) Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56:777–785
Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, Brosh RM Jr (2009) FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem 284:7505–7517
Stelter P, Ulrich HD (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425:188–191
Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P (2012) R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26:163–175
Syljuåsen RG, Sørensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25:3553–3562
Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD (2002) S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100:2414–2420
Teixeira LK, Wang X, Li Y, Ekholm-Reed S, Wu X, Wang P, Reed SI (2015) Cyclin E deregulation promotes loss of specific genomic regions. Curr Biol 25:1327–1333
Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412:553–557
Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S et al (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208:545–562
Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155:1088–1103
Tomasetti C, Vogelstein B (2015) Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347:78–81
Trenz K, Errico A, Costanzo V (2008) Plx1 is required for chromosomal DNA replication under stressful conditions. EMBO J 27:876–885
Tsang E, Carr AM (2008) Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair (Amst) 7:1613–1623
Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve-Grez H et al (2009) Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11:1315–1324
Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D (2010) Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 6, e1001205. doi:10.1371/journal.pgen.1001205
Vasianovich Y, Harrington LA, Makovets S (2014) Break-induced replication requires DNA damage-induced phosphorylation of Pif1 and leads to telomere lengthening. PLoS Genet e1004679. doi:10.1371/journal.pgen.1004679.
Wahba L, Amon JD, Koshland D, Vuica-Ross M (2011) RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44:978–988
Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M et al (2009) Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet 84:605–616
Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, Huang J (2013) hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep 14:1104–1112
Wang AT, Smogorzewska A (2015) SnapShot: Fanconi anemia and associated proteins. Cell 160:354
Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R (1998) Segregation of transcription and replication sites into higher order domains. Science 281:1502–1506
Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P et al (2013) Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature 502:393–396
Wu L, Davies SL, Levitt NC, Hickson ID (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem 276:19375–19381
Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF (2015) Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519:431–435
Yost BK, Rosenberg MJ, Nishioka DJ (1976) Incorporation of tritiated uridine into DNA of Ehrlich ascites tumor cells. J Natl Cancer Inst 57:289–293
Yuan J, Ghosal G, Chen J (2009) The annealing helicase HARP protects stalled replication forks. Genes Dev 23:2394–2399
Zegerman P, Diffley JF (2010) Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467:474–478
Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208:563–579
Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16:2–9
Zlotorynski E, Rahat A, Skaug J, Ben-Porat N, Ozeri E, Hershberg R, Levi A, Scherer SW, Margalit H, Kerem B (2003) Molecular basis for expression of common and rare fragile sites. Mol Cell Biol 23:7143–7151
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548
Acknowledgments
We thank all members of the laboratory for useful discussions and apologize to the authors whose works are not cited due to space restrictions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This review article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no competing interests.
Funding
Research in JM lab is supported by grants BFU2013-49153P and Consolider CSD2007-00015 from the Spanish Ministry of Economy and Competitiveness.
Rights and permissions
About this article

Cite this article
Muñoz, S., Méndez, J. DNA replication stress: from molecular mechanisms to human disease. Chromosoma 126, 1–15 (2017). https://doi.org/10.1007/s00412-016-0573-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-016-0573-x