
Abstract
Triggered by the ground-breaking finding that ketamine exerts robust and rapid-acting antidepressant effects in patients with treatment-resistant depression, glutamatergic systems have attracted attention as targets for the development of novel antidepressants. Among glutamatergic systems, group II metabotropic glutamate (mGlu) receptors, consisting of mGlu2 and mGlu3 receptors, are of interest because of their modulatory roles in glutamatergic transmission. Accumulating evidence has indicated that mGlu2/3 receptor antagonists have antidepressant-like effects in rodent models that mirror those of ketamine and that mGlu2/3 receptor antagonists also share underlying mechanisms with ketamine that are responsible for these antidepressant-like actions. Importantly, contrary to their antidepressant-like profile, preclinical studies have revealed that mGlu2/3 receptor antagonists are devoid of ketamine-like adverse effects, such as psychotomimetic-like behavior, abuse potential and neurotoxicity. Despite some discouraging results for an mGlu2/3 receptor antagonist decoglurant (classified as a negative allosteric modulator [NAM]) in patients with major depressive disorder, clinical trials of two mGlu2/3 receptor antagonists, a phase 2 trial of TS-161 (an orthosteric antagonist) and a phase 1 trial of DSP-3456 (a NAM), are presently on-going. mGlu2/3 receptors still hold promise for the development of safer and more efficacious antidepressants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) remains a prevalent world-wide health problem. Although a number of antidepressant medications, most of which stem from monoamine-based drug discovery, are currently available, they are far from ideal in terms of efficacy and the onset of antidepressant action. Therefore, new antidepressants with increased efficacy and a shorter therapeutic lag time are required. Because dysfunctions of the glutamatergic system have been implicated in the pathophysiology of depression, glutamatergic systems have recently gained significant attention as a target for the drug discovery of novel antidepressants [1, 2]. The importance of the glutamatergic systems for developing novel antidepressants has been highlighted by the ground-breaking findings of the antidepressant effects of ketamine, a non-competitive antagonist of N-methyl-D-aspartate (NMDA) receptor. Since the rapid and robust antidepressant effects of ketamine in patients with MDD, including those with treatment-resistant depression (TRD), were first shown in 2000 [3] and 2006 [4], the antidepressant effects of ketamine and their mechanisms have been intensively investigated. Currently, Spravato®, a nasal spray that delivers esketamine which is a stereoisomer of ketamine, is available in the US and European markets as an adjunctive therapy for the treatment of TRD patients and for depressive symptoms in adults with MDD who exhibit acute suicidal ideation or behavior. However, esketamine retains the unwanted side effects of ketamine, including dissociative/psychotomimetic symptoms and abuse potential, which hampers the adoption of esketamine in daily practice. Therefore, novel approaches that can dissociate the ketamine-like antidepressant effects from ketamine-like adverse effects have been actively investigated. These approaches target proposed mechanisms underlying the antidepressant effects of ketamine, most of which are molecules within the glutamatergic system including GluN2B-containing NMDA receptor and α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA) receptor [5]. In addition to these approaches, the possible use of another stereoisomer of ketamine, arketamine, and a metabolite of ketamine, (2R,6R)-hydroxynorketamine, have been rigorously investigated. To date, arketamine looks promising as a novel antidepressant without exerting ketamine-like adverse effects based on data in both rodents and TRD patients [6, 7]. Among the components of the glutamatergic system, we have been focusing on group II metabotropic glutamate (mGlu2/3) receptors, which have a modulatory role in glutamatergic activity mainly the presynaptic regulation of glutamate flow. In this review, we will summarize the role of mGlu2/3 receptor in depression and the possibility of mGlu2/3 receptor antagonists as an alternative of ketamine by introducing both the efficacy in animal models and the underlying mechanisms. Moreover, we will touch on clinical trials of mGlu2/3 receptor antagonists.
Pharmacology of mGlu2/3 receptor and mGlu2/3 receptor antagonists
On a molecular basis, mGlu receptors have eight different subtypes. These receptors are divided into three groups (group I, II and III) based on amino acid similarity and signal transduction mechanisms [8, 9]. Among these subtypes, much attention has been paid to group II mGlu receptors, which consist of mGlu2 and mGlu3 receptors. mGlu2/3 receptors couple with Gi/Go proteins, leading to the inhibition of adenylyl cyclase activity. mGlu2/3 receptors are mainly localized at the perisynaptic area of glutamatergic terminals in the frontal cortex and limbic systems, where they negatively modulate the release of glutamate [10]. Therefore, mGlu2/3 receptors play important roles in ameliorating glutamatergic tones when they are perturbed, as observed in several psychiatric conditions including schizophrenia [11], depression [12, 13] and addiction [14]. Indeed, mGlu2/3 receptor agonists were proven to be effective for a certain group of patients with schizophrenia [15] and generalized anxiety disorder [16]. A role of mGlu2/3 receptors in the pathophysiology of depression has also been reported. For example, the level of mGlu2/3 receptor protein was reportedly elevated in rodent models of depression [17, 18] and in the postmortem brains of patients with MDD [19]; in contrast, other reports have described reductions in mGlu2/3 receptor protein or mRNA in MDD patients [20] and in rodent models [21, 22]. These observations suggest that mGlu2/3 receptor abnormalities are involved in depression. In rodent studies, accumulating evidence suggests that mGlu2/3 receptor blockade may be beneficial for the treatment of depression, although limited data also suggest the opposite direction.
To date, several mGlu2/3 receptor antagonists have been synthesized (Fig. 1), and in vitro activities of representative compounds are summarized in Table 1. These antagonists are divided into two categories according to differences in the binding site of the receptor: orthosteric antagonists [23, 24] and negative allosteric modulators (NAMs) [25]. Orthosteric antagonists bind to glutamate binding sites, while NAMs bind to allosteric sites localized in the transmembrane regions of the receptor. Of note, the maximal binding value of an mGlu2/3 receptor ligand that binds to an allosteric site was nearly half that of the radioligand binding of an orthosteric ligand of the mGlu2/3 receptor [26]. Therefore, the binding modes of both classes mGlu2/3 receptor antagonists are apparently different, which may explain the differences in the in vivo outcomes of both types of antagonists.

Representative mGlu2/3 receptor antagonists. To date, two types of mGlu2/3 receptor antagonists have been synthesized: orthosteric antagonists (TP0178894, MGS0039, LY3020371, LY341495), and negative allosteric modulators (Decoglurant, RO04491533). Orthosteric antagonists contain a glutamate moiety in their molecule and bind to glutamate binding sites, which reside in the N-terminus of the receptor. Negative allosteric modulators bind to allosteric sites of the receptor that are located in the transmembrane regions
Antidepressant profiles of mGlu2/3 receptor antagonists
The antidepressant-like effects of mGlu2/3 receptor antagonists were first reported by our group in 2004 [23], triggering subsequent studies on the antidepressant-like characteristics of mGlu2/3 receptor antagonists [27, 28]. To date, accumulating lines of evidence have indicated similarities between the antidepressant-like effects of mGlu2/3 receptor antagonists and those of ketamine [27], which is summarized in Table 2. First, mGlu2/3 receptor antagonists (MGS0039, TP0178894, LY341495) exert rapid-acting and long-lasting antidepressant effects in rodent models such as the chronic social defeat stress model [17, 29, 30] and the chronic unpredictable stress model [31,32,33]; in both of these models, monoamine-based conventional antidepressants must be administered for more than a week for the exertion of the full-blown antidepressant effects. mGlu2/3 receptor antagonists, on the other hand, reversed the depressive-like behaviors in these models within 24 h after administration, and the effects lasted for more than a week after a single administration, as with ketamine. Also similar to ketamine, mGlu2/3 receptor antagonists exerted the antidepressant-like effects in rodent models that were resistant to conventional antidepressants, including a corticosterone-treated model [34]. Therefore, mGlu2/3 receptor antagonists are effective in animal models that are pharmacologically regarded as TRD models. Furthermore, mGlu2/3 receptor antagonists have been shown to have not only curative effects, but also preventive effects. Highland et al. (2019) reported that mGlu2/3 receptor blockade has prophylactic effects in studies involving pharmacological manipulation and the use of knockout mice lacking the mGlu2 or mGlu3 receptor [35]. Both knockout mice lacking the mGlu2 receptor and mice treated with LY341495 during stress were resilient to the development of depressive-like behaviors induced by several stress manipulations. Moreover, a single treatment with LY341495 7 days prior to the commencement of stress exposure prevented not only the initial development of depressive-like behaviors, but also the reinstatement of maladaptive behaviors caused by re-exposure to stress, indicating that the transient blockade of the mGlu2/3 receptor conferred a sustained enhancement of stress resilience. The authors additionally reported that the mGlu2 receptor, but not the mGlu3 receptor, was responsible for these protective effects because the effects in knockout mice lacking the mGlu3 receptor were indistinguishable from those in wild-type mice.
While mGlu2/3 receptor antagonists have ketamine-like antidepressant profiles, they do not show ketamine-like adverse effects in rodents [28]. For example, the mGlu2/3 receptor antagonist LY3020371 does not cause adverse effects known to be problematic with the use of ketamine, such as psychotomimetic-like behavior, cognitive impairment, motor impairment, and abuse liability, in rodent models [36]. In addition, the daily intravenous infusion of a high dose of LY3020371 for 14 days did not produce critical toxicological findings, including brain histochemistry, in either rats or cynomolgus monkeys [36]; therefore, ketamine-like neurotoxic effects were not noted. All these data clearly indicate that mGlu2/3 receptor antagonists are unlikely to have ketamine-like adverse effect profiles. Rather, mGlu2/3 receptor antagonists have been reported to have pro-cognitive effects in some animal models [37, 38], which would support the use of mGlu2/3 receptor antagonists compared with ketamine use for the treatment of MDD patients.
mGlu2/3 receptor antagonists have been reported to possess an additional characteristic that is expected to allow them to be beneficial when they are dosed with other rapid-acting antidepressants. The combination of sub-effective doses of LY341495 and ketamine exerted significant antidepressant-like effects [32, 39, 40], indicating that mGlu2/3 receptor antagonists augment the antidepressant effects of ketamine. In contrast, the combined doses of these substances did not produce the undesirable behavioral effects that are characteristic of ketamine when dosed alone, such as locomotor hyperactivity, impairment of short-term memory and disturbed motor coordination, in rodent studies involving the induction of antidepressant effects [32]. In addition to ketamine, LY341495 has been proven to enhance the antidepressant-like effect of scopolamine [41]. Like ketamine, scopolamine has been shown to exert rapid-onset antidepressant effects in patients with MDD, including those with TRD [42]; however, use of scopolamine is also limited because of adverse effects. Thus, the adjunctive use of mGlu2/3 receptor antagonists may be useful to mitigate the adverse effects of ketamine or scopolamine by allowing their doses to be lowered. Interestingly, LY341495 has recently been demonstrated to enhance the antidepressant-like effects of each ketamine stereoisomer, esketamine and arketamine, differentially [43]. While a sub-effective dose of LY341495 enhanced the antidepressant-like effects of both stereoisomers in a tail suspension test of naïve mice, it potentiated rapid (< 24 h) and sustained (> 3 days) antidepressant effects of arketamine but not esketamine in a chronic unpredictable stress model, suggesting that arketamine may play a key role in the mechanism of the enhancement of the antidepressant-like effects of ketamine by LY341495. Because arketamine has been proposed to have more potent and sustained antidepressant-like effects but fewer adverse effects than esketamine [7, 44, 45], facilitating the effects of arketamine through the use of mGlu2/3 receptor antagonists may be an interesting strategy.
In contrast to the above findings, mGlu2/3 receptor stimulation, rather than blockade, and increased mGlu2 receptor expression have also been reported to exert antidepressant-like behaviors and to confer stress resilience. For example, L-acetylcarnitine, which epigenetically increases mGlu2 receptor expression, exerts antidepressant effects [21], and knockout mice lacking the mGlu2 receptor show increased stress susceptibility [46]. However, evidence involving specific mGlu2/3 receptor agonists or mGlu2 receptor positive allosteric modulators is limited, and the mechanisms involved have not been fully elucidated [47].
Mechanisms underlying the antidepressant-like effects of mGlu2/3 receptor antagonists
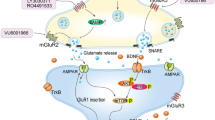
There are striking similarities in the underlying mechanisms between ketamine and mGlu2/3 receptor antagonists, which may explain the ketamine-like antidepressant profiles of mGlu2/3 receptor antagonists [28]. Indeed, they share mechanisms not only at the synaptic level, but also at the network level, and it has been proposed that mGlu2/3 receptor blockade has the same downstream mechanism of ketamine, as explained below. These similarities are depicted in Table 2 and Fig. 2.

Proposed mechanisms for the antidepressant effects of mGlu2/3 receptor antagonists and ketamine. mGlu2/3 receptor antagonists block perisynaptic mGlu2/3 receptors and the postsynaptic mGlu3 receptor, while ketamine blocks the NMDA receptor on GABA interneurons and on extrasynaptic NMDA receptors. Both mechanisms converge to stimulate postsynaptic AMPA receptor signaling, which triggers network and synaptic changes. At the network level, both compounds activate the DRN-mPFC serotonergic system and dopaminergic systems (both mesolimbic and mesocortical dopaminergic systems) through AMPA receptor stimulation. At the synaptic level, both compounds activate BDNF/TrkB/mTORC1 pathways via AMPA receptor stimulation, leading to an increase in spine formation. These mechanisms might contribute to the antidepressant effects of both compounds. NMDA N-methyl-d-aspartate, AMPA α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate, DRN dorsal raphe nucleus, mPFC medial prefrontal cortex, VTA ventral tegmental area, NAC nucleus accumbens, FC frontal cortex, BDNF brain-derived neurotrophic factor, TrkB tropomyosin-related kinase B, mTORC1 mechanistic target of rapamycin complex 1
Similarity at synaptic level
Both mGlu2/3 receptor antagonists and ketamine share underlying synaptic mechanisms that are triggered by an increase in glutamate release in the prefrontal cortex (PFC). mGlu2/3 receptor antagonists are thought to increase glutamate release through the blockade of autoreceptors on glutamate terminals, while ketamine increases glutamate release through the disinhibition of pyramidal neurons [27, 48]. Increased glutamate in the synaptic cleft activates postsynaptic AMPA receptors, which stimulates the secretion of brain-derived neurotrophic factor (BDNF) and, subsequently, the stimulation of tropomyosin-related kinase B (TrkB) receptor signaling. These events lead to an increase in dendritic spine density and synaptic connections, presumably via a mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway [29, 49, 50]. Notably, AMPA receptor stimulation and a subsequent increase in BDNF secretion are considered common pathways for various agents that mimic the antidepressant effects of ketamine [51,52,53], and this mechanism is necessary for mGlu2/3 receptor antagonists to enhance the antidepressant-like effects of arketamine [43]. Indeed, both AMPA receptor positive allosteric modulators and BDNF have been shown to exert antidepressant-like effects [54,55,56,57]. In animal models of depression, the above-mentioned pathways and spine density are reportedly perturbed in the PFC, and both ketamine and mGlu2/3 receptor antagonists ameliorated these abnormalities [29, 58, 59]. Regarding mTORC1 signaling, mTOR knockdown in the infralimbic cortex induced depressive-like behavior in mice [60], and the mTOR pathway was disturbed in the PFC of postmortem MDD patients [61]; therefore, the reversal of dysfunctional mTORC1 signaling may be an important mechanism for the exertion of ketamine-like antidepressant effects. In contrast, an increase in mTOR activity in the PFC of postmortem MDD patients was recently reported [62], and rapamycin, an inhibitor of mTORC1 signaling, prolonged, rather than blocked, the antidepressant effects of ketamine in MDD patients [63]. Therefore, the role of mTORC1 signaling activation in ketamine-like antidepressant actions requires further clarification.
Similarity at network level
Both serotonergic and dopaminergic transmissions are considered to play important roles in exerting antidepressant effects. Dopaminergic and serotonergic pathways are reportedly involved in the antidepressant-like effects of both ketamine and mGlu2/3 receptor antagonists. The systemic administration of ketamine and mGlu2/3 receptor antagonists (LY341495 and LY3020371) increased the number of spontaneously active dopamine neurons in the ventral tegmental area (VTA) and dopamine release in the nucleus accumbens (NAc) and/or frontal cortex [64]. In addition, we reported that the direct injection of mGlu2/3 receptor antagonists (MGS0039 and LY341495) into the NAc increased dopamine release in this region, indicating that mGlu2/3 receptor antagonists may activate the VTA-NAc dopaminergic system [65, 66]. The interaction of mGlu2/3 receptor antagonists with the dopaminergic system is supported by in vivo behavioral results showing that both ketamine and LY341495 potentiated a dopamine D2/3 receptor agonist (quinpirole)-induced increase in locomotor activity [64]. Moreover, the injection of LY341495 into the NAc prevented depressive-like behavior occurring during withdrawal from repeated methamphetamine treatment [67], indicating that mGlu2/3 receptor antagonists may exert antidepressant effects at least partly through the VTA-NAc dopaminergic system. Notably, this enhancement of the effects of mGlu2/3 receptor antagonists on the dopaminergic system has been shown to be mediated through AMPA receptor stimulation [64, 66].
Several lines of evidence imply the involvement of the serotonergic system in the antidepressant-like effects of both ketamine and mGlu2/3 receptor antagonists. Both ketamine and an mGlu2/3 receptor antagonist (MGS0039) increased serotonin release in the medial prefrontal cortex (mPFC) [68,69,70]. The depletion of serotonin cancelled the antidepressant-like effects of ketamine and mGlu2/3 receptor antagonists [70, 71]. Therefore, an increase in serotonergic transmission may be responsible, in part, for the antidepressant-like effects of both ketamine and mGlu2/3 receptor antagonists. We further investigated possible mechanisms through which both ketamine and mGlu2/3 receptor antagonists enhance the activity of the serotonergic system to exert their antidepressant-like effects. We found that the injection of ketamine or LY341495 into the mPFC increased c-Fos positive serotonin neurons in the dorsal raphe nucleus (DRN) [71], indicating a role of the mPFC-DRN pathway in these actions. This presumption was supported by the finding that AMPA receptor stimulation or an increase in glutamate in the mPFC with a glutamate transporter inhibitor (both of these manipulations activate mPFC neurons) increased the number of c-Fos positive serotonin cells in the DRN [72, 73]. Pham et al. reported that the intra-mPFC injection of ketamine increased serotonin release in the mPFC as well as the antidepressant-like effects, both of which were attenuated by AMPA receptor blockade in the DRN [70, 74]. Therefore, the activation of DRN serotonin neurons regulated by the mPFC-DRN pathway is involved in the antidepressant-like effects of both mGlu2/3 receptor antagonists and ketamine. Notably, the optogenetic stimulation of mPFC cells projecting to the DRN has been reported to exert robust antidepressant-like effects [75], and the antidepressant-like effect of ketamine or an mGlu2/3 receptor antagonist was blocked by the silencing of DRN neurons [70, 76], supporting the role of mPFC-DRN projections. Moreover, we further demonstrated that the antidepressant-like effects of an mGlu2/3 receptor antagonist and ketamine were mediated by 5-HT1A receptor stimulation in the mPFC [76, 77]. Interestingly, subsequent activation of the phosphoinositide-3 kinase/Akt/mTORC1 pathway is responsible for the antidepressant-like effects of both compounds [76, 77], and mPFC 5-HT1A receptor activation can mimic the rapid and sustained antidepressant-like effects seen in animal models, including the chronic unpredictable stress model, through mTORC1 signaling [78]. Collectively, these findings imply that stimulation of the mPFC 5-HT1A receptor and the subsequent signaling mechanisms triggered by the release of serotonin through mPFC-DRN-mPFC projections are likely to be involved in the unique antidepressant-like effects of mGlu2/3 receptor antagonists and ketamine.
Role of mGlu2 receptor in the antidepressant actions of ketamine
A role of the mGlu2 receptor in the actions of ketamine has recently been reported. First, the antidepressant-like effects of ketamine were attenuated by an mGlu2/3 receptor agonist [40, 79]. In addition, the antidepressant-like effects of ketamine were no longer observed in mice lacking the mGlu2 receptor [40]. Although the precise molecular mechanism for the role of mGlu2/3 receptor blockade in the action of ketamine needs to be further elucidated, these results indicate the importance of the mGlu2 receptor in the antidepressant-like effects of ketamine and support the concept that mGlu2/3 receptor antagonists may have ketamine-like antidepressant profiles.
The antidepressant-like actions of mGlu2/3 antagonists are often attributed to the inhibition of the mGlu2 receptor, but not the mGlu3 receptor, based on findings that the antidepressant-like effects of mGlu2/3 receptor antagonists are no longer observed in knockout mice lacking the mGlu2 receptor, while they are seen in knockout mice lacking the mGlu3 receptor [40, 79, 80]. Nevertheless, several reports have indicated that mGlu3 receptor blockade may also contribute to the antidepressant-like effects. Conn’s group reported that a selective mGlu3 receptor NAM exerted antidepressant-like effects that were more potent than those of a selective mGlu2 receptor NAM [81]. Moreover, both mGlu2 NAM and mGlu3 NAM reversed depressive-like behavior in a chronic variable stress model [82]. Interestingly, both mGlu2 and mGlu3 NAMs exert antidepressant-like effects via an increase in glutamate transmission in the PFC, but through mechanistically distinct manners: a presynaptic action (for mGlu2 NAM), and postsynaptic action (for mGlu3 NAM). However, no evidence is available regarding whether the blockade of either receptor alone can mimic ketamine-like antidepressant effects and if so, the underlying mechanisms. Therefore, mGlu2/3 receptor antagonists, rather than selective antagonists for the mGlu2 or mGlu3 receptor, might be preferable as an alternative for ketamine.
Clinical trials of mGlu2/3 receptor antagonists
To date, clinical studies of several mGlu2/3 receptor antagonists have been conducted or are being conducted [83], as summarized in Table 3.
Decoglurant (RG1578/RO4995819)
Decoglurant, which was synthesized and developed by F. Hoffmann-La Roche, Ltd., is a NAM of the mGlu2/3 receptor. Decoglurant has been shown to reduce the anhedonic index in a chronic mild stress model of rats and to rescue scopolamine-induced attention deficits in non-human primates [84]; thus, it is expected to be effective for both depression and cognitive dysfunction. A randomized, placebo-controlled, double-blind, multicenter phase 2 trial of decoglurant as an adjunctive treatment to selective serotonin reuptake inhibitors or serotonin- and norepinephrine reuptake inhibitors for 6 weeks in MDD patients with inadequate responses to antidepressant treatment was conducted to evaluate decoglurant’s antidepressant and pro-cognitive effects [84]. In this study, none of the decoglurant doses (5 mg, 15 mg, or 30 mg) demonstrated significant differences from the placebo in terms of the primary endpoint (change from baseline in Montgomery Åsberg Depression Rating Scale total score at the end of 6 weeks of treatment). Furthermore, no pro-cognitive effects, as assessed using the Cambridge Neuropsychological Test Automated Battery, were seen. In addition to a high placebo response and a low prevalence of patients with clinically relevant cognitive impairment at baseline, which were raised by the authors as possible reasons for the lack of efficacy [84], some other reasons can be considered as possible explanations for the failure to show any efficacy in this trial. First, whether optimal dose levels were used is uncertain. Although the authors claimed that the doses were unlikely to be suboptimal given the high brain penetrance observed in preclinical studies, whether the free fraction of decoglurant in the extracellular fluid was sufficient to block the mGlu2/3 receptor was not known. Regrettably, the concentrations of decoglurant in the cerebrospinal fluid (CSF) were not measured, therefore, no data on the target engagement of decoglurant in humans is available. Moreover, the neurophysiological and behavioral properties of decoglurant are quite different from those of ketamine and other mGlu2/3 receptor orthosteric antagonists and NAMs, and decoglurant failed to exert sustained antidepressant effects in preclinical studies [85]. Although detailed pharmacokinetic profiles of decoglurant have not been reported, the long half-life of decoglurant could be responsible for this difference, as the sustained stimulation of AMPA receptors can cause desensitization.
TS-161 (TP0473292)
TS-161 was synthesized and is being developed by Taisho Pharmaceutical Co., Ltd. as an ester prodrug of TP0178894 to improve oral bioavailability. TP0178894 is an mGlu2/3 receptor orthosteric antagonist with potent antagonist activity and high selectivity for mGlu2/3 receptors [86]. TP0178894 and its prodrug have been shown to exert rapid-acting and long-lasting antidepressant-like effects in a rodent model [30] and to exert the antidepressant-like effects in a rodent TRD model [86]. We conducted a randomized, double-blinded, placebo-controlled, single-ascending dose and 10-day multiple ascending dose study in healthy subjects. The clinical study results demonstrated that TS-161 is orally bioavailable and extensively converted into an active metabolite (TP0178894); the Cmax and AUC of the prodrug were approximately 0.1% of those of TP0178894 [86]. In addition, we measured the CSF concentrations of TP0178894 after a single administration of TS-161 and found that TP0178894 penetrated into the CSF, enabling an exposure level that was predicted to be sufficient to block mGlu2/3 receptors and exert the anticipated antidepressant effects [86]. Overall, TS-161 was safe and well tolerated, and only mild adverse events were seen at a dose of 100 mg, which is the dose level expected to exert pharmacological actions. A phase 2 study evaluating the efficacy of TS-161 in patients with TRD is currently on-going with an upper dose level of 100 mg per day (ClinicalTrials.gov Identifier: NCT04821271).
MGS0210 (BCI-838)
MGS0210, synthesized by Taisho Pharmaceutical Co., Ltd., is an ester prodrug of MGS0039. There are several lines of evidence that MGS0039, a selective and potent mGlu2/3 receptor orthosteric antagonist [23], exerts ketamine-like antidepressant effects in rodents and shares the underlying mechanisms of its antidepressant-like actions with ketamine [27]. MGS0210 has been proven to improve the oral bioavailability of MGS0039 in both rats and cynomolgus monkeys [87, 88] and to show antidepressant-like effects in rodent models after oral administration [88]. A phase 1 study was conducted by BrainCells Inc. in healthy subjects. Although the plasma exposure of MGS0039, an active metabolite, increased after single and 7-day multiple doses of MGS0210, the plasma exposure levels of MGS0210 were approximately tenfold higher than that of MGS0039 [89], indicating that MGS0210 is not an ideal prodrug. Nonetheless, it should be noted that dose- and time-dependent effects on some power spectra of quantitative electroencephalograms were observed during a multiple ascending dose study, suggesting that MGS0039 may penetrate the brain to engage the mGlu2/3 receptor. MGS0210 was well tolerated, and adverse events were generally mild and transient in duration [89].
DSP-3456
DSP-3456 is a NAM of the mGlu2/3 receptor that is currently being developed by Sumitomo Pharma Co., Ltd. According to the company’s website [90], DSP-3456 is in the phase 1 stage of development as a potential treatment for TRD in the US. The chemical structure and detailed pharmacological profiles of DSP-3456 have not been disclosed.
Conclusions
Since the first demonstration of the antidepressant-like effects of prototype mGlu2/3 receptor antagonists (MGS0039 and LY341495) in classical animal models [23], accumulating evidence has clearly shown that the blockade of mGlu2/3 receptors is an attractive approach for developing novel antidepressants with ketamine-like antidepressant profiles but with safety profiles that are better than those of ketamine. Among these research activities, several mGlu2/3 receptor antagonists, both orthosteric antagonists and NAMs, have been generated, and some have been tested or are being tested in clinical trials.
In this respect, the results that decoglurant, an mGlu2/3 receptor NAM, failed to show any efficacy in patients with MDD are discouraging. However, many reasons for this failure, including the clinical trial design (dose levels used, patient selection, etc.), and the validity of decoglurant as a tool for a proof-of-concept study for mGlu2/3 receptor antagonists should be considered carefully before drawing any conclusions regarding the efficacy of mGlu2/3 receptor antagonists for the treatment of depression. Currently, at least two mGlu2/3 receptor antagonists are in clinical trials. In particular, the human proof-of-concept study of TS-161 is of importance, because this compound was proven to have ketamine-like antidepressant profiles in rodent studies and the dose levels used in the trial are expected to be sufficient to block mGlu2/3 receptors. Therefore, mGlu2/3 receptor antagonists still hold promise for the development of safer and more efficacious antidepressants.
Data availability
This article does not contain any original data.
References
Gerhard DM, Wohleb ES, Duman RS (2016) Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity. Drug Discov Today 21:454–464. https://doi.org/10.1016/j.drudis.2016.01.016
Skolnick P, Popik P, Trullas R (2009) Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci 30:563–569. https://doi.org/10.1016/j.tips.2009.09.002
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH (2000) Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47:351–354. https://doi.org/10.1016/s0006-3223(99)00230-9
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK (2006) A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63:856–864. https://doi.org/10.1001/archpsyc.63.8.856
Henter ID, Park LT, Zarate CA Jr (2021) Novel glutamatergic modulators for the treatment of mood disorders: current status. CNS Drugs 35:527–543. https://doi.org/10.1007/s40263-021-00816-x
Leal GC, Bandeira ID, Correia-Melo FS, Telles M, Mello RP, Vieira F, Lima CS, Jesus-Nunes AP, Guerreiro-Costa LNF, Marback RF, Caliman-Fontes AT, Marques BLS, Bezerra MLO, Dias-Neto AL, Silva SS, Sampaio AS, Sanacora G, Turecki G, Loo C, Lacerda ALT, Quarantini LC (2021) Intravenous arketamine for treatment-resistant depression: open-label pilot study. Eur Arch Psychiatry Clin Neurosci 271:577–582. https://doi.org/10.1007/s00406-020-01110-5
Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, Dong C, Hashimoto K (2015) R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry 5:e632. https://doi.org/10.1038/tp.2015.136
Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237. https://doi.org/10.1146/annurev.pharmtox.37.1.205
Schoepp DD, Conn PJ (1993) Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci 14:13–20. https://doi.org/10.1016/0165-6147(93)90107-u
Schaffhauser H, Richards JG, Cartmell J, Chaboz S, Kemp JA, Klingelschmidt A, Messer J, Stadler H, Woltering T, Mutel V (1998) In vitro binding characteristics of a new selective group II metabotropic glutamate receptor radioligand, [3H]LY354740, in rat brain. Mol Pharmacol 53:228–233
Moghaddam B, Javitt D (2012) From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37:4–15. https://doi.org/10.1038/npp.2011.181
Sanacora G, Treccani G, Popoli M (2012) Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62:63–77. https://doi.org/10.1016/j.neuropharm.2011.07.036
Palucha A, Pilc A (2007) Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol Ther 115:116–147. https://doi.org/10.1016/j.pharmthera.2007.04.007
Kenny PJ, Markou A (2004) The ups and downs of addiction: role of metabotropic glutamate receptors. Trends Pharmacol Sci 25:265–272. https://doi.org/10.1016/j.tips.2004.03.009
Kinon BJ, Millen BA, Zhang L, McKinzie DL (2015) Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry 78:754–762. https://doi.org/10.1016/j.biopsych.2015.03.016
Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD (2008) Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology 33:1603–1610. https://doi.org/10.1038/sj.npp.1301531
Jing XY, Wang Y, Zou HW, Li ZL, Liu YJ, Li LF (2021) mGlu2/3 receptor in the prelimbic cortex is implicated in stress resilience and vulnerability in mice. Eur J Pharmacol 906:174231. https://doi.org/10.1016/j.ejphar.2021.174231
Kawasaki T, Ago Y, Yano K, Araki R, Washida Y, Onoe H, Chaki S, Nakazato A, Hashimoto H, Baba A, Takuma K, Matsuda T (2011) Increased binding of cortical and hippocampal group II metabotropic glutamate receptors in isolation-reared mice. Neuropharmacology 60:397–404. https://doi.org/10.1016/j.neuropharm.2010.10.009
Feyissa AM, Woolverton WL, Miguel-Hidalgo JJ, Wang Z, Kyle PB, Hasler G, Stockmeier CA, Iyo AH, Karolewicz B (2010) Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry 34:279–283. https://doi.org/10.1016/j.pnpbp.2009.11.018
McOmish CE, Pavey G, Gibbons A, Hopper S, Udawela M, Scarr E, Dean B (2016) Lower [3H]LY341495 binding to mGlu2/3 receptors in the anterior cingulate of subjects with major depressive disorder but not bipolar disorder or schizophrenia. J Affect Disord 190:241–248. https://doi.org/10.1016/j.jad.2015.10.004
Nasca C, Xenos D, Barone Y, Caruso A, Scaccianoce S, Matrisciano F, Battaglia G, Mathé AA, Pittaluga A, Lionetto L, Simmaco M, Nicoletti F (2013) L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA 110:4804–4809. https://doi.org/10.1073/pnas.1216100110
Wierońska JM, Legutko B, Dudys D, Pilc A (2008) Olfactory bulbectomy and amitriptyline treatment influences mGlu receptors expression in the mouse brain hippocampus. Pharmacol Rep 60:844–855
Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, Yoshimizu T, Yasuhara A, Sakagami K, Okuyama S, Nakanishi S, Nakazato A (2014) MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology 46:457–467. https://doi.org/10.1016/j.neuropharm.2003.10.009
Witkin JM, Ornstein PL, Mitch CH, Li R, Smith SC, Heinz BA, Wang XS, Xiang C, Carter JH, Anderson WH, Li X, Broad LM, Pasqui F, Fitzjohn SM, Sanger HE, Smith JL, Catlow J, Swanson S, Monn JA (2017) In vitro pharmacological and rat pharmacokinetic characterization of LY3020371, a potent and selective mGlu2/3 receptor antagonist. Neuropharmacology 115:100–114. https://doi.org/10.1016/j.neuropharm.2015.12.021
Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer-Urios I, Schneider M, Legrand C, Parron D, Girard F, Bessif A, Poli S, Vaugeois JM, Le Poul E, Celanire S (2011) Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J Neurogenet 25:152–166. https://doi.org/10.3109/01677063.2011.627485
Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I, Van der Linden I, Peeters L, Megens A, Wintmolders C, Cid JM, Trabanco AA, Andrés JI, Dautzenberg FM, Lütjens R, Macdonald G, Atack JR (2013) Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H]JNJ-40068782. J Pharmacol Exp Ther 346:514–527. https://doi.org/10.1124/jpet.113.204990
Chaki S (2017) mGlu2/3 receptor antagonists as novel antidepressants. Trends Pharmacol Sci 38:569–580. https://doi.org/10.1016/j.tips.2017.03.008
Witkin JM (2020) mGlu2/3 receptor antagonism: A mechanism to induce rapid antidepressant effects without ketamine-associated side-effects. Pharmacol Biochem Behav 190:172854. https://doi.org/10.1016/j.pbb.2020.172854
Dong C, Zhang JC, Yao W, Ren Q, Ma M, Yang C, Chaki S, Hashimoto K (2017) Rapid and sustained antidepressant action of the mGlu2/3 receptor antagonist MGS0039 in the social defeat stress model: comparison with ketamine. Int J Neuropsychopharmacol 20:228–236. https://doi.org/10.1093/ijnp/pyw089
Dong C, Tian Z, Fujita Y, Fujita A, Hino N, Iijima M, Hashimoto K (2022) Antidepressant-like actions of the mGlu2/3 receptor antagonist TP0178894 in the chronic social defeat stress model: comparison with escitalopram. Pharmacol Biochem Behav 212:173316. https://doi.org/10.1016/j.pbb.2021.173316
Dwyer JM, Lepack AE, Duman RS (2013) mGluR2/3 blockade produces rapid and long-lasting reversal of anhedonia caused by chronic stress exposure. J Mol Psychiatry 1:15. https://doi.org/10.1186/2049-9256-1-15.eCollection2013
Pałucha-Poniewiera A, Podkowa K, Rafało-Ulińska A (2021) The group II mGlu receptor antagonist LY341495 induces a rapid antidepressant-like effect and enhances the effect of ketamine in the chronic unpredictable mild stress model of depression in C57BL/6J mice. Prog Neuropsychopharmacol Biol Psychiatry 109:110239. https://doi.org/10.1016/j.pnpbp.2020.110239
Seo MK, Lee JA, Jeong S, Seog DH, Lee JG, Park SW (2022) Effects of chronic LY341495 on hippocampal mTORC1 signaling in mice with chronic unpredictable stress-induced depression. Int J Mol Sci 23:6416. https://doi.org/10.3390/ijms23126416
Koike H, Iijima M, Chaki S (2013) Effects of ketamine and LY341495 on the depressive-like behavior of repeated corticosterone-injected rats. Pharmacol Biochem Behav 107:20–23. https://doi.org/10.1016/j.pbb.2013.03.017
Highland JN, Zanos P, Georgiou P, Gould TD (2019) Group II metabotropic glutamate receptor blockade promotes stress resilience in mice. Neuropsychopharmacology 44:1788–1796. https://doi.org/10.1038/s41386-019-0380-1
Witkin JM, Monn JA, Li J, Johnson B, McKinzie DL, Wang XS, Heinz BA, Li R, Ornstein PL, Smith SC, Mitch CH, Calligaro DO, Swanson S, Allen D, Phillips K, Gilmour G (2017) Preclinical predictors that the orthosteric mGlu2/3 receptor antagonist LY3020371 will not engender ketamine-associated neurotoxic, motor, cognitive, subjective, or abuse-liability-related effects. Pharmacol Biochem Behav 155:43–55. https://doi.org/10.1016/j.pbb.2017.03.001
Goeldner C, Ballard TM, Knoflach F, Wichmann J, Gatti S, Umbricht D (2013) Cognitive impairment in major depression and the mGlu2 receptor as a therapeutic target. Neuropharmacology 64:337–346. https://doi.org/10.1016/j.neuropharm.2012.08.001
Shimazaki T, Kaku A, Chaki S (2007) Blockade of the metabotropic glutamate 2/3 receptors enhances social memory via the AMPA receptor in rats. Eur J Pharmacol 575:94–97. https://doi.org/10.1016/j.ejphar.2007.08.006
Podkowa K, Pochwat B, Brański P, Pilc A, Pałucha-Poniewiera A (2016) Group II mGlu receptor antagonist LY341495 enhances the antidepressant-like effects of ketamine in the forced swim test in rats. Psychopharmacology 233:2901–2914. https://doi.org/10.1007/s00213-016-4325-7
Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, Morris PJ, Thomas CJ, Moaddel R, Zarate CA Jr, Gould TD (2019) (2R,6R)-hydroxynorketamine exerts mGlu 2 receptor-dependent antidepressant actions. Proc Natl Acad Sci USA 116:6441–6450. https://doi.org/10.1073/pnas.1819540116
Podkowa K, Podkowa A, Sałat K, Lenda T, Pilc A, Pałucha-Poniewiera A (2016) Antidepressant-like effects of scopolamine in mice are enhanced by the group II mGlu receptor antagonist LY341495. Neuropharmacology 111:169–179. https://doi.org/10.1016/j.neuropharm.2016.08.031
Drevets WC, Zarate CA Jr, Furey ML (2013) Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry 73:1156–1163. https://doi.org/10.1016/j.biopsych.2012.09.031
Rafało-Ulińska A, Brański P, Pałucha-Poniewiera A (2022) Combined administration of (R)-ketamine and the mGlu2/3 receptor antagonist LY341495 induces rapid and sustained effects in the CUMS model of depression via a TrkB/BDNF-dependent mechanism. Pharmaceuticals (Basel) 15:125. https://doi.org/10.3390/ph15020125
Fukumoto K, Toki H, Iijima M, Hashihayata T, Yamaguchi JI, Hashimoto K, Chaki S (2017) Antidepressant potential of (R)-ketamine in rodent models: comparison with (S)-ketamine. J Pharmacol Exp Ther 361:9–16. https://doi.org/10.1124/jpet.116.239228
Zhang JC, Li SX, Hashimoto K (2014) R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine. Pharmacol Biochem Behav 116:137–141. https://doi.org/10.1016/j.pbb.2013.11.033
Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS (2015) Mind the gap: glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry 20:755–763. https://doi.org/10.1038/mp.2014.96
Fell MJ, Witkin JM, Falcone JF, Katner JS, Perry KW, Hart J, Rorick-Kehn L, Overshiner CD, Rasmussen K, Chaney SF, Benvenga MJ, Li X, Marlow DL, Thompson LK, Luecke SK, Wafford KA, Seidel WF, Edgar DM, Quets AT, Felder CC, Wang X, Heinz BA, Nikolayev A, Kuo MS, Mayhugh D, Khilevich A, Zhang D, Ebert PJ, Eckstein JA, Ackermann BL, Swanson SP, Catlow JT, Dean RA, Jackson K, Tauscher-Wisniewski S, Marek GJ, Schkeryantz JM, Svensson KA (2011) N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide (THIIC), a novel metabotropic glutamate 2 potentiator with potential anxiolytic/antidepressant properties: in vivo profiling suggests a link between behavioral and central nervous system neurochemical changes. J Pharmacol Exp Ther 336:165–177. https://doi.org/10.1124/jpet.110.172957
Krystal JH, Sanacora G, Duman RS (2013) Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry 73:1133–1141. https://doi.org/10.1016/j.biopsych.2013.03.026
Koike H, Iijima M, Chaki S (2011) Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology 61:1419–1423. https://doi.org/10.1016/j.neuropharm.2011.08.034
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329(5994):959–964. https://doi.org/10.1126/science.1190287
Lepack AE, Bang E, Lee B, Dwyer JM, Duman RS (2016) Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology 111:242–252. https://doi.org/10.1016/j.neuropharm.2016.09.011
Yang C, Yang J, Luo A, Hashimoto K (2019) Molecular and cellular mechanisms underlying the antidepressant effects of ketamine enantiomers and its metabolites. Transl Psychiatry 9:280. https://doi.org/10.1038/s41398-019-0624-1
Zanos P, Gould TD (2018) Mechanisms of ketamine action as an antidepressant. Mol Psychiatry 23:801–811. https://doi.org/10.1038/mp.2017.255
Alt A, Nisenbaum ES, Bleakman D, Witkin JM (2006) A role for AMPA receptors in mood disorders. Biochem Pharmacol 71:1273–1288. https://doi.org/10.1016/j.bcp.2005.12.022
Hara H, Suzuki A, Kunugi A, Tajima Y, Yamada R, Kimura H (2021) TAK-653, an AMPA receptor potentiator with minimal agonistic activity, produces an antidepressant-like effect with a favorable safety profile in rats. Pharmacol Biochem Behav 211:173289. https://doi.org/10.1016/j.pbb.2021.173289
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS (2002) Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22:3251–3261. https://doi.org/10.1523/JNEUROSCI.22-08-03251.2002
Legutko B, Li X, Skolnick P (2001) Regulation of BDNF expression in primary neuron culture by LY392098, a novel AMPA receptor potentiator. Neuropharmacology 40:1019–1027. https://doi.org/10.1016/s0028-3908(01)00006-5
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS (2011) Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry 69:754–761. https://doi.org/10.1016/j.biopsych.2010.12.015
Tang J, Xue W, Xia B, Ren L, Tao W, Chen C, Zhang H, Wu R, Wang Q, Wu H, Duan J, Chen G (2015) Involvement of normalized NMDA receptor and mTOR-related signaling in rapid antidepressant effects of Yueju and ketamine on chronically stressed mice. Sci Rep 5:13573. https://doi.org/10.1038/srep13573
Garro-Martínez E, Fullana MN, Florensa-Zanuy E, Senserrich J, Paz V, Ruiz-Bronchal E, Adell A, Castro E, Díaz Á, Pazos Á, Bortolozzi A, Pilar-Cuéllar F (2021) mTOR knockdown in the infralimbic cortex evokes a depressive-like state in mouse. Int J Mol Sci 22:8671. https://doi.org/10.3390/ijms22168671
Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, Karolewicz B (2011) The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 35:1774–1779. https://doi.org/10.1016/j.pnpbp.2011.05.010
Salort G, Hernández-Hernández E, García-Fuster MJ, García-Sevilla JA (2020) Regulation of cannabinoid CB 1 and CB 2 receptors, neuroprotective mTOR and pro-apoptotic JNK1/2 kinases in postmortem prefrontal cortex of subjects with major depressive disorder. J Affect Disord 276:626–635. https://doi.org/10.1016/j.jad.2020.07.074
Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, Sherif M, Ahn KH, D’Souza DC, Formica R, Southwick SM, Duman RS, Sanacora G, Krystal JH (2020) Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology 45:990–997. https://doi.org/10.1038/s41386-020-0644-9
Witkin JM, Monn JA, Schoepp DD, Li X, Overshiner C, Mitchell SN, Carter G, Johnson B, Rasmussen K, Rorick-Kehn LM (2016) The rapidly acting antidepressant ketamine and the mGlu2/3 receptor antagonist LY341495 rapidly engage dopaminergic mood circuits. J Pharmacol Exp Ther 358:71–82. https://doi.org/10.1124/jpet.116.233627
Karasawa J, Yoshimizu T, Chaki S (2006) A metabotropic glutamate 2/3 receptor antagonist, MGS0039, increases extracellular dopamine levels in the nucleus accumbens shell. Neurosci Lett 393:127–130. https://doi.org/10.1016/j.neulet.2005.09.058
Karasawa J, Kotani M, Kambe D, Chaki S (2010) AMPA receptor mediates mGlu 2/3 receptor antagonist-induced dopamine release in the rat nucleus accumbens shell. Neurochem Int 57:615–619. https://doi.org/10.1016/j.neuint.2010.07.011
Iijima M, Koike H, Chaki S (2013) Effect of an mGlu2/3 receptor antagonist on depressive behavior induced by withdrawal from chronic treatment with methamphetamine. Behav Brain Res 246:24–28. https://doi.org/10.1016/j.bbr.2013.02.039
Karasawa J, Shimazaki T, Kawashima N, Chaki S (2005) AMPA receptor stimulation mediates the antidepressant-like effect of a group II metabotropic glutamate receptor antagonist. Brain Res 1042:92–98. https://doi.org/10.1016/j.brainres.2005.02.032
Nishitani N, Nagayasu K, Asaoka N, Yamashiro M, Shirakawa H, Nakagawa T, Kaneko S (2014) Raphe AMPA receptors and nicotinic acetylcholine receptors mediate ketamine-induced serotonin release in the rat prefrontal cortex. Int J Neuropsychopharmacol 17:1321–1326. https://doi.org/10.1017/S1461145714000649
Pham TH, Mendez-David I, Defaix C, Guiard BP, Tritschler L, David DJ, Gardier AM (2017) Ketamine treatment involves medial prefrontal cortex serotonin to induce a rapid antidepressant-like activity in BALB/cJ mice. Neuropharmacology 112:198–209. https://doi.org/10.1016/j.neuropharm.2016.05.010
Fukumoto K, Iijima M, Chaki S (2016) The Antidepressant Effects of an mGlu2/3 Receptor Antagonist and Ketamine Require AMPA Receptor Stimulation in the mPFC and Subsequent Activation of the 5-HT Neurons in the DRN. Neuropsychopharmacology 41:1046–1056. https://doi.org/10.1038/npp.2015.233
Gasull-Camós J, Tarrés-Gatius M, Artigas F, Castañé A (2017) Glial GLT-1 blockade in infralimbic cortex as a new strategy to evoke rapid antidepressant-like effects in rats. Transl Psychiatry 7(2):e1038. https://doi.org/10.1038/tp.2017.7
Gasull-Camós J, Martínez-Torres S, Tarrés-Gatius M, Ozaita A, Artigas F, Castañé A (2018) Serotonergic mechanisms involved in antidepressant-like responses evoked by GLT-1 blockade in rat infralimbic cortex. Neuropharmacology 139:41–51. https://doi.org/10.1016/j.neuropharm.2018.06.029
Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, Landry DW, Brachman RA, Denny CA, Gardier AM (2018) Common neurotransmission recruited in (R, S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry 84:e3–e6. https://doi.org/10.1016/j.biopsych.2017.10.020
Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, Adhikari A, Tye KM, Frank LM, Deisseroth K (2012) A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492:428–432. https://doi.org/10.1038/nature11617
Fukumoto K, Iijima M, Funakoshi T, Chaki S (2018) 5-HT 1A receptor stimulation in the medial prefrontal cortex mediates the antidepressant effects of mGlu2/3 receptor antagonist in mice. Neuropharmacology 137:96–103. https://doi.org/10.1016/j.neuropharm.2018.05.001
Fukumoto K, Iijima M, Funakoshi T, Chaki S (2018) Role of 5-HT1A receptor stimulation in the medial prefrontal cortex in the sustained antidepressant effects of ketamine. Int J Neuropsychopharmacol 21:371–381. https://doi.org/10.1093/ijnp/pyx116
Fukumoto K, Fogaça MV, Liu RJ, Duman CH, Li XY, Chaki S, Duman RS (2020) Medial PFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant-like effects of 5-HT 1A receptor stimulation. Neuropsychopharmacology 45:1725–1734. https://doi.org/10.1038/s41386-020-0705-0
Witkin JM, Mitchell SN, Wafford KA, Carter G, Gilmour G, Li J, Eastwood BJ, Overshiner C, Li X, Rorick-Kehn L, Rasmussen K, Anderson WH, Nikolayev A, Tolstikov VV, Kuo MS, Catlow JT, Li R, Smith SC, Mitch CH, Ornstein PL, Swanson S, Monn JA (2017) Comparative effects of LY3020371, a potent and selective metabotropic glutamate (mGlu) 2/3 receptor antagonist, and ketamine, a noncompetitive N-methyl-d-aspartate receptor antagonist in rodents: Evidence supporting the use of mGlu2/3 antagonists, for the treatment of depression. J Pharmacol Exp Ther 361:68–86. https://doi.org/10.1124/jpet.116.238121
Gleason SD, Li X, Smith IA, Ephlin JD, Wang XS, Heinz BA, Carter JH, Baez M, Yu J, Bender DM, Witkin JM (2013) mGlu2/3 agonist-induced hyperthermia: an in vivo assay for detection of mGlu2/3 receptor antagonism and its relation to antidepressant-like efficacy in mice. CNS Neurol Disord Drug Targets 12:554–566. https://doi.org/10.2174/18715273113129990079
Engers JL, Bollinger KA, Weiner RL, Rodriguez AL, Long MF, Breiner MM, Chang S, Bollinger SR, Bubser M, Jones CK, Morrison RD, Bridges TM, Blobaum AL, Niswender CM, Conn PJ, Emmitte KA, Lindsley CW (2017) Design and synthesis of N-aryl phenoxyethoxy pyridinones as highly selective and CNS penetrant mGlu 3 NAMs. ACS Med Chem Lett 8:925–930. https://doi.org/10.1021/acsmedchemlett.7b00249.eCollection2017
Joffe ME, Santiago CI, Oliver KH, Maksymetz J, Harris NA, Engers JL, Lindsley CW, Winder DG, Conn PJ (2020) mGlu 2 and mGlu 3 negative allosteric modulators divergently enhance thalamocortical transmission and exert rapid antidepressant-like effects. Neuron 105:46-59.e3. https://doi.org/10.1016/j.neuron.2019.09.044
Witkin JM, Pandey KP, Smith JL (2022) Clinical investigations of compounds targeting metabotropic glutamate receptors. Pharmacol Biochem Behav 219:173446. https://doi.org/10.1016/j.pbb.2022.173446
Umbricht D, Niggli M, Sanwald-Ducray P, Deptula D, Moore R, Grünbauer W, Boak L, Fontoura P (2020) Randomized, double-blind, placebo-controlled trial of the mGlu2/3 negative allosteric modulator decoglurant in partially refractory major depressive disorder. J Clin Psychiatry 81:18m12470. https://doi.org/10.4088/JCP.18m12470
Sheffler DJ, Bicakci MB, Antwan A, Prakash N, Standard EM, Velicelebi G, Gadient RA, Hutchison JH, Panickar DR, Limpert AS, Cosford NDP, Der-Avakian A (2019) In vitro and in vivo characterization of group II mGlu receptor negative allosteric modulators as an alternative to ketamine for depression. Soc Neurosc Abst 684:20
Watanabe M, Marcy B, Hiroki A, Watase H, Kinoshita K, Iijima M, Marumo T, Zarate CA, Chaki S (2022) Evaluation of the safety, tolerability, and pharmacokinetic profiles of TP0473292 (TS-161), a prodrug of a novel orthosteric mGlu2/3 receptor antagonist TP0178894, in healthy subjects and its antidepressant-like effects in rodents. Int J Neuropsychopharmacol 25:106–117. https://doi.org/10.1093/ijnp/pyab062
Nakamura M, Kawakita Y, Yasuhara A, Fukasawa Y, Yoshida K, Sakagami K, Nakazato A (2006) In vitro and in vivo evaluation of the metabolism and bioavailability of ester prodrugs of mgs0039 (3-(3,4-dichlorobenzyloxy)-2-amino-6-fluorobicyclo[3.1.0]hexane-2,6-dicarboxylic Acid), a potent metabotropic glutamate receptor antagonist. Drug Metab Dispos 34:369–374. https://doi.org/10.1124/dmd.105.006213
Yasuhara A, Nakamura M, Sakagami K, Shimazaki T, Yoshikawa R, Chaki S, Ohta H, Nakazato A (2006) Prodrugs of 3-(3,4-dichlorobenzyloxy)-2-amino-6-fluorobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (MGS0039): a potent and orally active group II mGluR antagonist with antidepressant-like potential. Bioorg Med Chem 14:4193–4207. https://doi.org/10.1016/j.bmc.2006.01.060
Gardient RS, Wedel P, Frisbie V, Leuchter AF, Targum SD, Truong C, Hutchinson JH (2012) Safety, pharmacokinetic and pharmacodynamic profile of BCI-632, a selective metabotropic glutamate 2/3 receptor antagonist, in healthy human subjects. Soc Neurosci Abst 841:20
Development Pipeline (As of July 29, 2022) Sumitomo Pharma Co., Ltd. https://www.sumitomo-pharma.com/rd/clinical/pdf/epip20220729.pdf. Accessed Oct 19 2022.
Author information
Authors and Affiliations
Contributions
The first draft of the manuscript was written by Shigeyuki Chaki and Mai Watanabe. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Shigeyuki Chaki is a full-time employee of Taisho Pharmaceutical Co., Ltd., and Mai Watanabe is a full-time employee of Taisho Pharmaceutical R&D Inc.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chaki, S., Watanabe, M. mGlu2/3 receptor antagonists for depression: overview of underlying mechanisms and clinical development. Eur Arch Psychiatry Clin Neurosci 273, 1451–1462 (2023). https://doi.org/10.1007/s00406-023-01561-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-023-01561-6