
Abstract
Disturbances of glutamatergic neurotransmission and mononuclear phagocyte system activation have been described uni- and bipolar depression (UD/BD). Linking the glutamate and immune hypotheses of depression, quinolinic acid (QUIN) is synthesized by activated microglia and acts as an endogenous N-methyl-D-aspartate glutamate receptor (NMDA-R) agonist with neurotoxic properties. Recently, we observed an increased microglial QUIN expression in the subgenual and supracallosal, but not in the pregenual part of the anterior cingulate cortex in postmortem brains of suicide cases with severe depression. Since several hints point to a role of the hippocampus in depression, we extended our study and addressed the question whether microglial QUIN is also changed in subregions of the hippocampus (CA1 and CA2/3 areas) in these patients. Postmortem brains of 12 acutely depressed patients (UD, n = 6; BD, n = 6) and 10 neuropsychiatric healthy age- and gender-matched control subjects were analyzed using QUIN-immunohistochemistry. Hippocampal volumes were determined in order to assess possible neurotoxic or neurodegenerative aspects. Microglial QUIN expression in the whole group of depressed patients was either comparable (left CA1, right CA2/3) or decreased (right CA1: p = 0.004, left CA2/3: p = 0.044) relative to controls. Post hoc tests showed that QUIN was reduced both in UD and BD in the right CA1 field (UD, p = 0.048; BD, p = 0.031). No loss of hippocampal volume was detected. Our data indicate that UD and BD are associated with a local reduction in QUIN-immunoreactive microglia in the hippocampus and underline the importance of the NMDA-R signaling in depressive disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
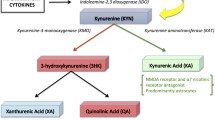
Uni- (UD) and bipolar (BD) depression are common psychiatric diseases. Neither their neurobiology nor their etiologies are understood in detail. Treatment failures frequently occur with existing therapies, indicating the need for a better understanding of the mechanisms underlying depressive disorders. Disturbances of glutamatergic neurotransmission [1–3] and mononuclear phagocyte system activation [1, 4–8] have been associated with the pathogenesis of UD and BD. In this context, kynurenine metabolites generated via the pathway initiated by the enzyme indoleamine-2,3-dioxygenase (IDO) are of particular interest, because they are linking the glutamate and immune hypotheses and neurodegeneration hypothesis of depression [9, 10]. IDO is expressed by activated microglia and induces the production of quinolinic acid (QUIN), an endogenous N-methyl-D-aspartate receptor (NMDA-R) agonist with neurotoxic properties [11, 12]. It is still uncertain how QUIN induces neuronal damage. Since the neurotoxicity of QUIN is considerably greater than can be accounted by the activation of NMDA-Rs, it could act by mechanisms additional to the activation of NMDA-Rs, possibly involving lipid peroxidation [13].
We have previously shown an association between severe depression and increased expression of QUIN in two subregions of the anterior cingulate cortex (ACC) [13]. Depressed patients had an increased density of QUIN-immunopositive microglia in the subgenual and supracallosal ACC compared to the pregenual ACC. These findings were seemingly in parallel to proton magnetic resonance spectroscopy (1H-MRS) studies which showed a relative reduction in pregenual ACC glutamate content that was related to the severity of depression [15–17].
The glutamatergic system has been implicated in depressive disorders since reports from patients with UD described elevated levels of glutamate in serum and plasma [18, 19]. This metabolite was also increased or normal in the cerebrospinal fluid (CSF) [20]. The first evidence that the kynurenine pathway (KP) of tryptophan (TRP) metabolism is involved in the pathophysiology of depression was already published in 1970 [21]: Urinary kynurenine (KYN) was increased in patients suffering from major depression. However, these studies do not allow drawing conclusions on brain-regional differences regarding glutamatergic neurotransmission and kynurenine metabolite (e.g., QUIN) expression in UD and BD.
1H-MRS is a noninvasive neuroimaging technique that facilitates the quantification of metabolites in the central nervous system in vivo, including glutamate-related ones such as glutamate, glutamine, GABA and other metabolites (termed Glx). Studies using 1H-MRS in depressive disorders identified abnormalities in Glx also in the hippocampus, where mainly a decrease in Glx level was described [2, 3, 22]. The hippocampus belongs to the most susceptible brain areas to QUIN [23] and is considered to play an important role in the pathogenesis of depression. It is important for learning and episodic memory for contextual fear conditioning and neuro-endocrine regulation [24]. Reduced hippocampal NMDA receptor-mediated neurotransmission may trigger the occurrence of cognitive deficits in depression [25].
Therefore, we have extended our previous study [14] on the microglial QUIN expression in subregions of the hippocampus. We addressed the question whether microglial QUIN is also changed in the hippocampal CA1 and CA2/3 fields of patients with UD and BD, similar to the observed changes in glutamatergic neurotransmission which have been described by the above-mentioned 1H-MRS studies. Moreover, hippocampal volumes were determined in order to assess possible neurotoxic or neurodegenerative aspects.
Methods
Human brain tissue
Postmortem brains were obtained from the Magdeburg brain bank. Brain collection and scientific histopathological analyses have been approved by the local ethics committee in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written consent was obtained from the next of kin. The donors were acutely depressed patients (n = 12) who had committed suicide (mean age 49 years; 7 males, 5 females) and controls (n = 10) with no neuropsychiatric illness (mean age 56 years; 5 males, 5 females). The cases showed no significant differences regarding age, gender, duration of disease and autolysis time (Table 1). Patients had been diagnosed with either unipolar depression (UD, n = 6) or bipolar depression (BD, n = 6).
The information used for clinical diagnoses was obtained by carefully studying the patients’ clinical records and by structured interviews with physicians involved in patients’ treatment and with persons who either lived with or had frequent contact with the subjects before death. The DSM-IV axis I diagnosis was established using all available information from interviews and clinical records. Brains with lifetime reports of substance abuse, dementia, neurological illness, severe trauma or chronic terminal diseases known to affect the brain were excluded. Additionally, neuropathological changes due to tumors, inflammatory, vascular, traumatic processes or neurodegenerative disorders were excluded by an experienced neuropathologist (CM), using Nissl (cresyl violet)-, myelin (Heidenhain-Wölcke)- and hematoxylin eosin-stained sections as well as beta amyloid- and tau-immunostainings (Braak staging). The determination of suicide was made by a forensic pathologist (T.G.) and was verified based on the individual records. As summarized in Table 2, the mean daily doses of psychotropic medication taken by patients during the last 90 lifetime days were established according to the clinical files [26–28].
Tissue preparation was performed as described previously [29]. Briefly, brains were fixed in 8 % phosphate-buffered formaldehyde (pH 7.0) for 3 months. Subsequently, after separation of the brainstem and the cerebellum, the hemispheres were divided by coronal cuts into three bi-hemispherical coronal blocks comprising the frontal lobe anterior to the genu of the corpus callosum (“anterior” block), the fronto-temporo-parietal lobe extending the entire length of the corpus callosum (“middle” block) and the occipital lobe (“posterior” block). After embedding the brains in paraffin, serial coronal whole brain sections were cut (section thickness: 20 µm) and mounted.
Region selection
Within the hippocampal formation, we analyzed the posterior part, spanning from the lateral geniculate nucleus to the level of the splenium of the corpus callosum. Subregions to be investigated separately were CA (cornu ammonis) 1 and 2–3. According to the literature [30], CA2 and CA3 were lumped together because these small regions are difficult to separate on the microscopic level by histological criteria (Fig. 1a).

Nissl (cresyl violet)- and myelin (Heidenhain–Wölcke)-stained section for anatomical orientation; illustration of the analyzed hippocampal subregions (a). Ramified QUIN-immunopositive microglial cell (b). Negative control staining after preabsorption of the primary antibody with QUIN (c)
The exact thickness of each section was determined by focusing on the upper and lower surfaces of the section and subtracting the z-axis coordinate of the lower surface from that of the upper surface. The movements in the z-axis were measured with a microcator, part of the Leica DM RB microscope (Leica, Gießen, Germany).
Immunohistochemistry
As previously described [14], formalin-fixed tissue sections were deparaffinized, and antigen demasking was performed by boiling the sections for 4 min in 10 mM citrate buffer (pH 6.0). Preincubation with 1.5 % H2O2 for 10 min to block endogenous peroxidase activity was followed by blocking non-specific binding sites with 10 % normal goat serum for 60 min and repeated washings with PBS. Next, a polyclonal rabbit QUIN antibody was used (ab37106, Abcam, Cambridge, UK) at a dilution of 1:150 for 72 h at 4 °C. Sections were then incubated with a biotinylated goat anti-rabbit secondary antibody (Amersham, Little Chalford, UK) for the streptavidin–biotin technique. Chromogen 3,3′-diaminobenzidine (DAB) and ammonium nickel sulfate were used to visualize the reaction product [31]. The specificity of the polyclonal rabbit primary antibody was confirmed by a loss of signal after preabsorption of 2 ml of the primary antibody solution (dilution 1:150) with 1 mg QUIN (Sigma-Aldrich, Munich, Germany) for 24 h and by the supplier’s ELISA competition experiments with QUIN, kynurenic acid and phenylalanine.
Quantification
Immunopositive cells were counted in the delineated brain regions listed above at ×200 magnification (Olympus BH2, Olympus, Hamburg, Germany) by experimenters blind to the donors’ diagnoses (TG and GML). The counting area was measured with the graphical analysis software Digitrace v. 2.10a (Imatec, Miesbach, Germany) using a SZX12 stereomicroscope (Olympus, Hamburg, Germany). The cytological classification of immunopositive cells as microglia, astrocytes, oligodendrocytes or neurons was performed according to established cytomorphological criteria [32]. Cells visibly located inside vessels were classified as monocytes; only cells that were clearly outside the vessels and situated in tissue were evaluated. Cell densities were calculated by dividing the cell number by the counting area multiplied by the section thickness [cells/mm3].
Assessment of hippocampal volumes
The measurement of the hippocampal formation included the subiculum, the fascia dentata and the cornu ammonis. The hippocampal gyrus was separated from the hippocampal formation at the most medial extent of the hippocampus–parahippocampus junction.
The measurements of the cross-sectional areas of the structures were performed using a planimetry from fourfold magnifications of the sections. The volumes were calculated by multiplying the cross-sectional areas by the distance between the sections and adding up volumes obtained by the procedure along the entire rostro-caudal axis. The volumes of the structures in the fixed brain were determined by multiplying the measured volumes of the respective structures in the paraffin block by the individual shrinkage factors for each brain.
Statistical analysis
Statistical analyses were performed with the SPSS 15.0 program (Statistical Product and Service Solutions, Chicago, IL, USA). Demographic data were compared by the Chi-square test, t test and analysis of variance (ANOVA). QUIN data were not normally distributed, as indicated by the Shapiro–Wilk test. Therefore, Spearman’s rank correlation coefficient, the Kruskal–Wallis H test and the Mann–Whitney U test were employed. These nonparametric tests were further used to explore potential confounds due to age, autolysis time, sex, duration of disease and medication dosage.
Results
Qualitative evaluation
Strong QUIN-immunoreactivity was observed in monocytes, pericytes and microglial cells (Fig. 1b). In contrast, faint staining was only occasionally observed in fibers and other cell types, such as pyramidal neurons and astroglia.
Quantitative evaluation of QUIN-positive cells
Comparing QUIN-immunopositive microglia between depressed patients and healthy controls revealed a region-specific pattern with group effects in the right CA1 and left CA2/3 subregion of the hippocampus. Depressed patients had significantly decreased QUIN-positive cells in the right CA1 (p = 0.004) and left CA2/3 (p = 0.044). In contrast, QUIN-positive cell counts in the left CA1 (p = 0.539) and right CA2/3 (p = 0.792) did not differ between groups (Fig. 2a).

a Depressed patients had significantly decreased QUIN-positive cells in the right CA1 and left CA2/3. b Reduced QUIN-positive cells in the right CA1 subregion in BD compared to controls or in UD compared to controls. Annotation: The box plots show the median, interquartile range, sample minimum and sample maximum, *p < 0.05, **p < 0.01
Diagnostic subgroups were analyzed by Kruskal–Wallis H tests. As illustrated in Fig. 2b, significant differences of QUIN-positive cells densities between BD and UD patients and controls were only found in the right CA1 region (H Test: p = 0.017). Post hoc tests of this effect showed reduced QUIN-positive cells in the right CA1 subregion in BD compared to controls (p = 0.031, corrected by Bonferroni–Holm) or in UD compared to controls (p = 0.048, corrected by Bonferroni–Holm). The reported significant diagnosis-related effects were checked for the potential confounding factors of age, autolysis time, sex, duration of disease and medication dosage (Supplementary file).
Quantitative evaluation of the hippocampal volume
In contrast to the differences detected in the QUIN immunostaining, we did not find any changes (left: p = 0.962; right: p = 1.000) regarding the volume of this brain area between controls (mean hippocampal volume left: 3,248 mm3; right: 3,550 mm3), UD patients (mean hippocampal volume left: 3,346 mm3; right: 3,555 mm3) and BD patients (mean hippocampal volume left: 3,223 mm3; right: 3,565 mm3).
Discussion
In a recently published report, increased CSF levels of QUIN, but not KYNA, have been associated with glutamate agonism in suicide attempters [33]. However, such glutamatergic abnormalities may be brain region-specific. The aim of the present study was to further contribute to clarify the role of microglial QUIN expression in the hippocampal CA1 and CA2/3 fields of the postmortem brains of suicidal patients with UD and BD. A decrease in QUIN-immunopositive microglia was detected in the CA1 right and the CA2/3 left subregions of the hippocampus with high NMDA-R densities. This decrease in QUIN-positive microglial cell densities was found in unipolar as well as in bipolar patients. However, we did not observe any significant difference between UD and BD (Fig. 2b).
Neurotoxic effects of QUIN are caused by several mechanisms: It agonizes glutamate receptors sensitive to NMDA [34], has pro-oxidant capacities and exacerbates the neurotoxic effects by corticosterone and cytokines [35, 36]. Additionally, QUIN acts pro-inflammatory by enhancing the IFNγ/IL-10 ratio and therefore further triggering an initiated inflammatory response [37]. Increased levels of QUIN, which is produced by microglia, macrophages and monocytes, have been found in the blood and CSF of patients with cytokine-induced depression or UD [38–40]. Therefore, an increase in microglial QUIN expression in suicidal UD patients could be linked with a systemic MPS activation during acute disease phases of depression [4, 7, 41–43]. We had observed an increased microglial QUIN expression in two subregions of the ACC, namely the subgenual and supracallosal ACC, in postmortem brains of suicide UD and BD patients [14]. Unlike in these brain regions, we found a reduced QUIN-immunoreactivity in the hippocampus of these individuals. Notably, the present study showed an overlap of 21 out of 22 cases with the previous study (measures of QUIN-immunoreactivity in subregions of the ACC) [14].
While some imaging studies showed a selective hippocampal volume reduction in depressive disorders [44–46], others were unable to find significant group differences between UD and control subjects [47–53]. The results in BD were also inconsistent, with reports of increased [54], decreased [55] and unchanged hippocampal volumes [56]. Most studies that reported hippocampal structural abnormalities have been restricted to certain subgroups of depressed subjects, such as patients with treatment resistant depression [57–59], preferentially women [52], elderly depressed patients [45, 60] or patients with multiple depressive episodes [61].
QUIN-induced neurodegenerative changes do not seem to play a major role in suicide patients with UD or MD in the present study, since no diagnosis-dependent differences regarding hippocampal volume were observed. Apart from depression, based on the glutamate hypothesis, schizophrenia may be caused by altered glutamate signalling. Notably, reduced microglial QUIN expression was also observed in the hippocampus of schizophrenia patients by our work group [29]. Similar to the present study, this finding was neither associated with diagnosis-related changes in hippocampal volume nor differences in numerical density of human leukocyte antigen-DR/HLA-DR-expressing microglial cells.
Kynurenine is mainly metabolized into QUIN when microglia is activated, otherwise the local induction of IDO expression could induce a negative feedback loop, potentially underlying the self-limitation of autoimmune inflammation in neurological disorders [62]. NMDA-Rs are expressed on neurons throughout the brain, their highest densities are found, e.g., in the hippocampus. Since QUIN acts directly as an agonist, an accumulation of QUIN is one of the events associated with depression or Alzheimer’s disease.
Since we observed reduced microglial QUIN-immunoreactivity in the hippocampus of depressed suicide cases without changes in hippocampal volume, we assume that local anti-inflammatory and neuroprotective compensatory responses may play a role in these particular patients. It is difficult to connect these findings with previous MRI spectroscopy studies, suggesting a decreased Glx (glutamate, glutamine and GABA mix) in the ACC and hippocampus [22, 63, 64]. First, the human brain is not reacting uniformly and brain-regional differences may occur. Accordingly, our previous paper on QUIN-immunoreactive microglial cells did not observe an upregulated QUIN-immunoreactivity in the whole anterior cingulate cortex (ACC), but only in the subgenual ACC (sACC) or anterior midcingulate cortex (aMCC), and not in the pregenual anterior cingulated cortex (pACC) subregion of the ACC. Second, the used MRI techniques cannot be easily compared to our measures of microglial QUIN-immunoreactivity.
The present study has certain limitations that need to be considered: (1) Our findings are based on a relatively small number of UD and BD cases and must be confirmed in a larger sample size. (2) We used immunohistochemistry to determine QUIN-positive microglia. Protein expression or activity of the kynurenine pathway enzyme QUIN phosphoribosyltransferase which is involved in the degradation of QUIN may add important facts on QUIN-immunopositive cell densities. Future studies may focus on the expression of this enzyme. (3) It was not possible to track data on drug exposure or the history of inflammation and infection across the patients’ entire life spans, as we could only collect data on psychotropic medication in the 3 months prior to death; thus, we cannot completely exclude that patients’ medication has influenced our findings. (4) We did not find a significant influence of age as interfering variable (Supplementary file). However, due to the observation of an age-dependent increase of QUIN (and decrease in QUIN phosphoribosyltransferase) in the brains of Wistar rats [65] and due to the trend toward an older age of the control cohort compared to the depression cohort in our presented study, we cannot completely exclude an influence of age on our results.
Summary
Here, we present data providing evidence that suicidal patients with UD or BD show decreased QUIN-immunoreactive cells in CA1 and CA2/3 along with unchanged hippocampal volume size. These results add a novel link regarding region specificity to the immune hypotheses of depression and underline the importance of the NMDA-R signaling in depressive disorders. Further work in this area could result in a better understanding of the pathophysiology of depressive disorders.
References
Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG, Myint AM (2012) Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: potential role of glial NMDA receptor modulators and impaired blood–brain barrier integrity. World J Biol Psychiatry 13(7):482–492
Milne A, MacQueen GM, Yucel K, Soreni N, Hall GB (2009) Hippocampal metabolic abnormalities at first onset and with recurrent episodes of a major depressive disorder: a proton magnetic resonance spectroscopy study. Neuroimage 47(1):36–41
Block W, Traber F, von Widdern O, Metten M, Schild H, Maier W, Zobel A, Jessen F (2009) Proton MR spectroscopy of the hippocampus at 3 T in patients with unipolar major depressive disorder: correlates and predictors of treatment response. Int J Neuropsychopharmacol 12(3):222–415
Drexhage RC, Knijff EM, Padmos RC, Heul-Nieuwenhuijzen L, Beumer W, Versnel MA, Drexhage HA (2010) The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Rev Neurother 10(1):59–76
Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, McCorkle R, Seligman DA, Schmidt K (2001) The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun 15(3):199–226
Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, de Ridder D, Kupka RW, Nolen WA, Drexhage HA (2008) A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry 65(4):395–407
Seidel A, Arolt V, Hunstiger M, Rink L, Behnisch A, Kirchner H (1996) Major depressive disorder is associated with elevated monocyte counts. Acta Psychiatr Scand 94(3):198–204
Maes M, Van der Planken M, Stevens WJ, Peeters D, DeClerck LS, Bridts CH, Schotte C, Cosyns P (1992) Leukocytosis, monocytosis and neutrophilia: hallmarks of severe depression. J Psychiatr Res 26(2):125–134
Myint AM, Kim YK (2003) Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses 61(5–6):519–525
Myint AM, Kim YK (2014) Network beyond IDO in psychiatric disorders: revisiting neurodegeneration hypothesis. Prog Neuropsychopharmacol Biol Psychiatry 48:304–313
Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew BJ (2003) Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol 527:105–112
Guillemin GJ, Smythe G, Takikawa O, Brew BJ (2005) Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 49(1):15–23
Stone TW, Behan WM, MacDonald M, Darlington LG (2000) Possible mediation of quinolinic acid-induced hippocampal damage by reactive oxygen species. Amino Acids 19(1):275–281
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C, Brisch R, Bielau H, Meyer zu Schwabedissen L, Bogerts B, Myint AM (2011) Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation 8(94):1–9
Horn DI, Yu C, Steiner J, Buchmann J, Kaufmann J, Osoba A, Eckert U, Zierhut KC, Schiltz K, He H, Biswal B, Bogerts B, Walter M (2010) Glutamatergic and resting-state functional connectivity correlates of severity in major depression—the role of pregenual anterior cingulate cortex and anterior insula. Front Syst Neurosci 4(33):10
Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F (2000) Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry 47(4):305–313
Rosenberg DR, Macmaster FP, Mirza Y, Smith JM, Easter PC, Banerjee SP, Bhandari R, Boyd C, Lynch M, Rose M, Ivey J, Villafuerte RA, Moore GJ, Renshaw P (2005) Reduced anterior cingulate glutamate in pediatric major depression: a magnetic resonance spectroscopy study. Biol Psychiatry 58(9):700–704
Maes M, Verkerk R, Vandoolaeghe E, Lin A, Scharpe S (1998) Serum levels of excitatory amino acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant depression: modulation by treatment with antidepressants and prediction of clinical responsivity. Acta Psychiatr Scand 97(4):302–308
Kim JS, Schmid-Burgk W, Claus D, Kornhuber HH (1982) Increased serum glutamate in depressed patients. Archiv fur Psychiatrie und Nervenkrankheiten 232(4):299–304
Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW (2000) Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry 47(7):586–593
Curzon G, Bridges PK (1970) Tryptophan metabolism in depression. J Neurol Neurosurg Psychiatry 33(5):698–704
Yüksel C, Ongur D (2010) Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry 68(9):785–794
Schwarcz R, Kohler C (1983) Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci Lett 38(1):85–90
MacQueen G, Frodl T (2011) The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry 16(3):252–264
Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, Tidball P, Fang G, Irvine MW, Costa BM, Monaghan DT, Bortolotto ZA, Molnar E, Lodge D, Jane DE (2013) The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 64:13–26
Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C (1999) Effectiveness of antidepressants. Meta-analysis of dose-effect relationships in randomised clinical trials. Br J Psychiatry 174:297–303
Rey MJ, Schulz P, Costa C, Dick P, Tissot R (1989) Guidelines for the dosage of neuroleptics. I: chlorpromazine equivalents of orally administered neuroleptics. Int Clin Psychopharmacol 4(2):95–104
Perry PJ, Alexander B (1986) Sedative/hypnotic dependence: patient stabilization, tolerance testing, and withdrawal. Drug Intell Clin Pharm 20(7–8):532–537
Gos T, Myint AM, Schiltz K, Meyer-Lotz G, Dobrowolny H, Busse S, Müller UJ, Mawrin C, Bernstein HG, Bogerts B, Steiner J (2014) Reduced microglial immunoreactivity for endogenous NMDA receptor agonist quinolinic acid in the hippocampus of schizophrenia patients. Brain Behav Immun (in press)
Ellison-Wright I, Glahn DC, Laird AR, Thelen SM, Bullmore E (2008) The anatomy of first-episode and chronic schizophrenia: an anatomical likelihood estimation meta-analysis. Am J Psychiatry 165(8):1015–1023
Hsu SM, Soban E (1982) Color modification of diaminobenzidine (DAB) precipitation by metallic ions and its application for double immunohistochemistry. J Histochem Cytochem 30(10):1079–1082
Polak M, Haymaker W, Johnson JE, D´Amelio F (1982) Neuroglia and their reactions. In: Haymaker W, Adams RD (eds) Histology and histopathology of the nervous system. Charles C Thomas Publishing, Springfield, pp 363–480
Erhardt S, Lim CK, Linderholm KR, Janelidze S, Lindqvist D, Samuelsson M, Lundberg K, Postolache TT, Traskman-Bendz L, Guillemin GJ, Brundin L (2013) Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 38(5):743–752
Stone TW, Behan WM (2007) Interleukin-1beta but not tumor necrosis factor-alpha potentiates neuronal damage by quinolinic acid: protection by an adenosine A2A receptor antagonist. J Neurosci Res 85(5):1077–1085
Platenik J, Stopka P, Vejrazka M, Stipek S (2001) Quinolinic acid-iron (ii) complexes: slow autoxidation, but enhanced hydroxyl radical production in the fenton reaction. Free Radical Res 34(5):445–459
Ngai LY, Herbert J (2005) Glucocorticoid enhances the neurotoxic actions of quinolinic acid in the striatum in a cell-specific manner. J Neuroendocrinol 17(7):424–434
Maes M, Mihaylova I, Ruyter MD, Kubera M, Bosmans E (2007) The immune effects of trycats (tryptophan catabolites along the IDO pathway): relevance for depression—and other conditions characterized by tryptophan depletion induced by inflammation. Neuro Endocrinol Lett 28(6):826–831
Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B (2007) Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord 98(1–2):143–151
Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, Spivey JR, Saito K, Miller AH (2010) CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry 15(4):393–403
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56
Kaestner F, Hettich M, Peters M, Sibrowski W, Hetzel G, Ponath G, Arolt V, Cassens U, Rothermundt M (2005) Different activation patterns of proinflammatory cytokines in melancholic and non-melancholic major depression are associated with HPA axis activity. J Affect Disord 87(2–3):305–311
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65(9):732–741
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bernstein HG, Bogerts B (2008) Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42(2):151–157
Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS (2000) Hippocampal volume reduction in major depression. Am J Psychiatry 157(1):115–118
Sheline YI (1996) Hippocampal atrophy in major depression: a result of depression-induced neurotoxicity? Mol Psychiatry 1(4):298–299
Frodl T, Meisenzahl EM, Zetzsche T, Born C, Groll C, Jager M, Leinsinger G, Bottlender R, Hahn K, Moller HJ (2002) Hippocampal changes in patients with a first episode of major depression. Am J Psychiatry 159(7):1112–1118
Ashtari M, Greenwald BS, Kramer-Ginsberg E, Hu J, Wu H, Patel M, Aupperle P, Pollack S (1999) Hippocampal/amygdala volumes in geriatric depression. Psychol Med 29(3):629–638
Axelson DA, Doraiswamy PM, McDonald WM, Boyko OB, Tupler LA, Patterson LJ, Nemeroff CB, Ellinwood EH Jr, Krishnan KR (1993) Hypercortisolemia and hippocampal changes in depression. Psychiatry Res 47(2):163–173
Coffey CE, Wilkinson WE, Weiner RD, Parashos IA, Djang WT, Webb MC, Figiel GS, Spritzer CE (1993) Quantitative cerebral anatomy in depression. A controlled magnetic resonance imaging study. Arch Gen Psychiatry 50(1):7–16
Pantel J, Schroder J, Essig M, Popp D, Dech H, Knopp MV, Schad LR, Eysenbach K, Backenstrass M, Friedlinger M (1997) Quantitative magnetic resonance imaging in geriatric depression and primary degenerative dementia. J Affect Disord 42(1):69–83
Rusch BD, Abercrombie HC, Oakes TR, Schaefer SM, Davidson RJ (2001) Hippocampal morphometry in depressed patients and control subjects: relations to anxiety symptoms. Biol Psychiatry 50(12):960–964
Vakili K, Pillay SS, Lafer B, Fava M, Renshaw PF, Bonello-Cintron CM, Yurgelun-Todd DA (2000) Hippocampal volume in primary unipolar major depression: a magnetic resonance imaging study. Biol Psychiatry 47(12):1087–1090
Monkul ES, Hatch JP, Nicoletti MA, Spence S, Brambilla P, Lacerda AL, Sassi RB, Mallinger AG, Keshavan MS, Soares JC (2007) Fronto-limbic brain structures in suicidal and non-suicidal female patients with major depressive disorder. Mol Psychiatry 12(4):360–366
Kemmerer M, Nasrallah HA, Sharma S, Olson SC, Martin R, Lynn MB (1994) Increased hippocampal volume in bipolar disorder. Biol Psychiatry 35(9):626
Swayze VW II, Andreasen NC, Alliger RJ, Yuh WT, Ehrhardt JC (1992) Subcortical and temporal structures in affective disorder and schizophrenia: a magnetic resonance imaging study. Biol Psychiatry 31(3):221–240
Hauser P, Altshuler LL, Berrettini W, Dauphinais ID, Gelernter J, Post RM (1989) Temporal lobe measurement in primary affective disorder by magnetic resonance imaging. J Neuropsychiatry Clin Neurosci 1(2):128–134
Hsieh MH, McQuoid DR, Levy RM, Payne ME, MacFall JR, Steffens DC (2002) Hippocampal volume and antidepressant response in geriatric depression. Int J Geriatr Psychiatry 17(6):519–525
Mervaala E, Fohr J, Kononen M, Valkonen-Korhonen M, Vainio P, Partanen K, Partanen J, Tiihonen J, Viinamaki H, Karjalainen AK, Lehtonen J (2000) Quantitative MRI of the hippocampus and amygdala in severe depression. Psychol Med 30(1):117–125
Shah PJ, Ebmeier KP, Glabus MF, Goodwin GM (1998) Cortical grey matter reductions associated with treatment-resistant chronic unipolar depression. Controlled magnetic resonance imaging study. Br J Psychiatry 172:527–532
Steffens DC, Byrum CE, McQuoid DR, Greenberg DL, Payne ME, Blitchington TF, MacFall JR, Krishnan KR (2000) Hippocampal volume in geriatric depression. Biol Psychiatry 48(4):301–309
MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmias C, Young LT (2003) Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA 100(3):1387–1392
Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I (2005) Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. Faseb J 19(10):1347–1349
Sanacora G, Treccani G, Popoli M (2012) Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62(1):63–77
Block W, Traber F, von Widdern O, Metten M, Schild H, Maier W, Zobel A, Jessen F (2009) Proton MR spectroscopy of the hippocampus at 3 T in patients with unipolar major depressive disorder: correlates and predictors of treatment response. Int J Neuropsychopharmacol 12(3):415–422
Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Grant R (2011) Changes in kynurenine pathway metabolism in the brain, liver and kidney of aged female wistar rats. FEBS J 278(22):4425–4434
Acknowledgments
Gabriela Meyer-Lotz and Kathrin Paelchen provided excellent technical assistance. Prof. Christian Mawrin (Department of Neuropathology, University of Magdeburg, Germany) performed the routine neuropathological examinations of all tested brains.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Mandy Busse and Stefan Busse have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Busse, M., Busse, S., Myint, A.M. et al. Decreased quinolinic acid in the hippocampus of depressive patients: evidence for local anti-inflammatory and neuroprotective responses?. Eur Arch Psychiatry Clin Neurosci 265, 321–329 (2015). https://doi.org/10.1007/s00406-014-0562-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-014-0562-0