
Abstract
Purpose
Human gene icb-1 recently has been reported to be part of a gene expression score predicting response to antiestrogen fulvestrant in breast cancer patients. In the present study, we examined to what extent icb-1 expression would affect the response of breast cancer cells to this antiestrogen in vitro and investigated underlying molecular mechanisms. Using open access mRNA data, we elucidated the significance of icb-1 expression for survival of breast cancer patients.
Methods
Icb-1 gene expression was knocked down by RNAi. Breast cancer cell growth after treatment with fulvestrant was assessed using the Cell Titer Blue assay. Gene expression was analyzed by Western blot analysis or RT-qPCR. Survival analyses were performed using bioinformatical online tools and data.
Results
Knockdown of icb-1 in T-47D breast cancer cells significantly increased growth of this cell line and also elevated the growth-stimulatory effect of E2 (p < 0.001). After treatment with different concentrations of fulvestrant, icb-1 knockdown cells exhibited a significantly enhanced response to this drug (p < 0.01). On the molecular level, icb-1 knockdown led to elevated expression of ESR1 and its target gene TFF1 (pS2) and enhanced E2-triggered up-regulation of proliferation genes. Finally, bioinformatical meta-analysis of gene expression data of 3951 breast cancer patients revealed that high icb-1 expression increases their relapse-free survival (HR = 0.87, p < 0.05).
Conclusion
The presented data further support a tumor-suppressive role of icb-1 in breast cancer and suggest an inhibitory effect of this gene on fulvestrant action, which both are suggested to be mediated by suppression of cellular E2 response.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Human gene icb-1 (THEMIS2) has been originally cloned and described by our group to be involved in differentiation processes of gynecological cancer cells and leukocytes [1,2,3]. Later, it has been identified as member of a new metazoan gene family called THEMIS coding for cytosolic proteins binding to the Grb2 adaptor protein involved in receptor tyrosine kinase signaling [4]. Whereas leukocytes exhibit the highest expression levels of icb-1, this gene is also expressed in all human tissues and in a variety of cancer types and cell lines including breast cancer cells. A recent proteomic study detected ICB-1 protein in the membrane of breast cancer tissue [5]. We were able to show that icb-1 is a component of signaling pathway-mediating differentiating effects of vitamin D3 and ATRA on breast cancer cells [2]. Other studies demonstrating that icb-1 is an interferon-γ responsive gene which in turn inhibits the effects of this cytokine on tumor cells showed that the view on icb-1 function had to be broadened [6]. This was confirmed by identification of an estrogen response element (ERE) in the promoter region of this gene regulating it in an ERα-dependent manner [7]. In turn, icb-1 gene was reported to inhibit estrogen responsiveness of ovarian and breast cancer cells [8]. The proposed role of icb-1 in breast cancer was further corroborated by results, suggesting that single-nucleotide polymorphisms in this gene affect breast cancer susceptibility [9]. RNAi studies revealed a growth-inhibitory effect of icb-1 on ovarian cancer cells [10] and MCF-7 breast cancer cells [8]. A recent study also demonstrated icb-1 to mediate apoptotic effects on MCF-7 cells, and transcriptome analyses revealed that siRNA-triggered icb-1 knockdown led to down-regulation of a network of apoptosis genes, but to up-regulation of an oncogenic network of genes associated with poor prognosis, invasion, and metastasis [11]. From these in vitro data, icb-1 was suggested to exert tumor-suppressing functions in breast cancer cells.
Icb-1 has recently been reported to be part of a gene expression score predicting response to the selective estrogen receptor degrader (SERD) fulvestrant (ICI 182,780, Faslodex®) in breast cancer patients [12]. In the present study, we examined the role of icb-1 expression in the response of breast cancer cells to this antiestrogen in vitro and investigated underlying molecular mechanisms. Furthermore, we intended to elucidate the significance of icb-1 expression for survival of breast cancer patients by means of bioinformatical analyses.
Methods
Materials
Phenol red-free DMEM culture medium was obtained from Invitrogen (Karlsruhe, Germany), and FCS was purchased from PAA (Pasching, Austria). T-47D breast cancer cells were obtained from American Type Culture Collection (ATCC®, HTB-133™, Manassas, USA). RNeasy Mini Kit, RNase Free DNase Set and Quantitect SYBR Green PCR Kit were obtained from Qiagen (Hilden, Germany). PCR primers were synthesized at Metabion (Planegg-Martinsried, Germany). Transfectin reagent was obtained from Bio-Rad (Hercules, USA). Platinum Pfx Polymerase and OptiMEM medium were purchased at Invitrogen (Karlsruhe, Germany). Cell Titer Blue (CTB) Assay was from Promega (Madison, USA).
Cell culture, transfection, and proliferation assays
T-47D breast cancer cells were maintained in DMEM/F12 medium supplemented with 10% FCS and 0.2 units/ml insulin. Cells were cultured with 5% CO2 at 37 °C in a humidified incubator. Cells were transfected in 6-well plates containing 2 × 105 freshly seeded cells using 20 nM of icb-1 siRNA (Silencer Select siRNA s18156, ThermoFisher) or the same amount negative control siRNA (Silencer™ Select Negative Control No. 1, Invitrogen™) and 7 µl Transfectin in OptiMEM medium. 72 h after transfection, total mRNA was isolated using RNeasy Mini Kit Qiagen (Hilden, Germany). For cell proliferation assays, the transfected cells were serum-starved by changing medium to 1% FCS and 1 × SR2 (serum replacement 2, Sigma) the day after transfection and after 16 h were seeded in phenol red-free DMEM/1xSR2 into 96-well plates in triplicates (2000 cell /well). For the fulvestrant experiments, culture medium contained 1 nM E2 + vehicle as control or E2 + 3, 10, 30, 100, 300, or 1000 nM fulvestrant. On days 0, 4, 5, and 6, relative numbers of viable cells were measured using the fluorimetric, resazurin-based Cell Titer Blue assay (Promega) according to the manufacturer’s instructions at 560Ex/590Em nm in a Victor3 multilabel counter (PerkinElmer, Germany). Cell growth was expressed as percentage of day 0 or in percentage of vehicle (± E2), as indicated.
Western blot analysis
T-47D (icb-1 KD) and T-47D (control) cells were lysed in RIPA buffer (1% (v/v) Igepal CA-630, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulfate (SDS) in phosphate-buffered solution (PBS) containing aprotinin and sodium orthovanadate 72 h after transfection. Aliquots containing 10 µg of protein were resolved by 10% (w/v) SDS–polyacrylamide gel electrophoresis, followed by electrotransfer to a PVDF hybond (Amersham, UK) membrane. Immunodetection was carried out using anti-ICB-1 antibody (C-term) (AP9910b, Abgent, USA) in a dilution of 1:100 and β-actin antibody (8226, Abcam, Germany) diluted 1:5000 in PBS containing 5% skim milk (w/v) followed by horseradish peroxidase-conjugated secondary antibody (1:20,000) which was detected using chemiluminescence (ECL) system (Amersham, Buckinghamshire, UK). Western blot bands were quantified using ImageJ software [13] and band densities were calculated in relation to β-actin.
Reverse transcription and qPCR
Total RNA from the transfected tumor cells was isolated 72 h after transfection, using RNeasy Mini Kit Qiagen (Hilden, Germany), according to the manufacturer´s instructions. From 0.3 µg total RNA, cDNA was synthesized using 100 U M-MLV-P reverse transcriptase (Promega), 2.5 mM dNTP mixture, and 50 pM random primers (Invitrogen). For real-time PCR detection of gene expression in an intron-spanning manner (primer sequences in Table 1), 2 µl cDNA were amplified using Light Cycler®FastStart DNA master mix SYBR Green I and the LightCyler 2.0 PCR device (Roche Diagnostics, Mannheim, Germany). The PCR program was 95 °C for 15 min, followed by 40 PCR cycles (95 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s) and a final extension for 5 min at 72 °C, followed by a standard melting curve analysis. In all RT-PCR experiments, a 190 bp β-actin fragment was amplified as reference gene using intron-spanning primers actin-2573 and actin-2876. Data were analyzed using the comparative ΔΔCT method [14, 15] calculating the difference between the threshold cycle (CT) values of the target and reference gene of each sample and then comparing the resulting ΔCT values between different samples. In these experiments, mRNA not subjected to reverse transcription was used as a negative control to distinguish cDNA and genomic DNA amplification.
Bioinformatical analysis
For correlation of icb-1 expression with ESR1, TFF1, CCND1, CCNA2, and MKI67 in 1085 breast cancer patients, we used the correlation function of the GEPIA online tool analyzing publicly available gene expression data http://gepia.cancer-pku.cn/index.html [16]. For survival analyses with regard to icb-1 expression, we used the Kaplan–Meier Plotter online tool http://kmplot.com/analysis/ including gene expression data of 3951 breast cancer patients [17].
Statistical analysis
Statistical analysis of gene expression between cells transfected with icb-1 siRNA and control siRNA was performed by means of Student’s t test. Multiple comparisons were analyzed using ANOVA F-test to test the null hypothesis, followed by Tukey’s test. Correlation analyses were performed by means of Spearman’s rank correlation. For statistics, we used Graph Pad Prism Version 7.04 Software (Graph Pad, San Diego, USA). Statistical significance was stated in case of p values being lower than 0.05.
Results
Icb-1 knockdown increases basal and E2-triggered growth of T-47D breast cancer cells
In a previous study, we observed knockdown of icb-1 to strongly increase basal and E2-triggered proliferation of SK-OV-3 ovarian cancer cells and to a weaker extent of MCF-7 breast cancer cells [8]. To corroborate these data using an additional cell line and to prepare the fulvestrant experiments performed in this study, we knocked down icb-1 expression in a second ERα-positive breast cancer cell line, T-47D, by means of siRNA transfection. 96 h after transfection, icb-1 protein levels by means of Western blot analysis were found to be reduced down to 24.8%, and transcript levels as assessed by RT-qPCR were decreased down to 18.6% when compared to cells transfected with negative control (NC) (both p < 0.01) (Fig. 1). This knockdown of icb-1 raised the number of viable cells 2.16-fold 5 days after transfection (Fig. 2a) and increased the growth-stimulatory effect of E2 (3 nM) by up to 69.4% on day 6 (p < 0.001), when cells were cultured in DMEM with serum- and hormone-free serum replacement (SR2, Sigma) (Fig. 2b).

Knockdown of icb-1 expression. a Western blot analysis of ICB-1 protein expression 96 h after transfection with 20 nM icb-1-specific siRNA (Silencer Select siRNA s18156, ThermoFisher) or negative control siRNA (NC) using anti-ICB-1 antibody (C-term) (Biomol, WA-AP9910b) (1:100). Left panel: representative Western blot result. Right panel: illustration of mean band densities (measured with ImageJ software) of three independent experiments normalized to respective ACTB levels and expressed in percentage of icb-1 expression in NC-siRNA transfected cells. b RT-qPCR analysis of icb-1 mRNA levels using the ΔΔ(CT) method [14, 15]. Data are expressed in percentage of icb-1 levels in cells transfected with negative control (NC) siRNA. (n = 3)

Icb-1 knockdown effect on proliferation of T-47D cells. a Time-dependent increase of viable cell numbers after transfection with icb-1 or NC (negative control) siRNA. Values are expressed in arbitrary units. b Effect of an icb-1 knockdown on E2 response, compared to cells with normal icb-1 levels (transfected with NC-siRNA). Experiments were performed in serum-and hormone-free defined SR2 medium (Sigma). Relative numbers of viable cells were measured using the Cell Titer Blue assay (Promega) and values are expressed in percent of E2 vehicle. (n = 4), *p < 0.05, **p < 0.01 vs. NC-siRNA. NC-siRNA negative control siRNA
Knockdown of icb-1 increases the growth-inhibitory effect of fulvestrant
In dose–response experiments, T-47D cells transfected with icb-1 or NC-siRNA were now treated with different concentrations of fulvestrant (1, 3, 10, 30, 100, 300, and 1000 nM) in serum-free SR2 medium in the presence of 3 nM E2. Icb-1 knockdown cells exhibited a significantly increased response to this SERD when used in concentrations between 10 and 1000 nM compared to cells transfected with NC-siRNA (Fig. 3b). After 5 days of treatment, the strongest increase of fulvestrant response was observed at concentrations of 30, 100, and 300 nM (all p < 0.01) and 1000 nM (p < 0.05), ranging from a decrease of viable cell numbers by 21.7% (NC-siRNA) to 36.7% (icb-1 siRNA) (p < 0.01) at 30 nM fulvestrant to a reduction from 44.8% (NC-siRNA) to 63.5% (icb-1 siRNA) at 1000 nM (p < 0.05). To avoid off-target effects of the highest fulvestrant concentrations tested, we now performed a time-course analysis with the lowest concentration of this drug strongly affected by icb-1 knockdown. Treatment of T-47D cells with 30 nM fulvestrant in serum-free medium containing 3 nM E2 resulted in a time-dependent decrease of viable cell numbers from day 4 to 6, which was significantly more pronounced in cells transfected with icb-1 siRNA (Fig. 3a). The maximum effect was observed on day 6, when the growth-inhibitory effect of 30 nM fulvestrant on control cells (26.4%) was increased to 43.7% after icb-1 knockdown (p < 0.01).

Effect of an icb-1 knockdown on fulvestrant response of T-47D breast cancer cells. a Time-course analysis of the effect of an icb-1 knockdown on fulvestrant (30 nM) action in the presence of 3 nM E2. b Dose–response analysis of the influence of an icb-1 knockdown on the effect of the indicated concentrations of fulvestrant in the presence of 3 nM E2 5 days after treatment. Relative numbers of viable cells were measured using the Cell Titer Blue assay (Promega) and are expressed in percentage of cells cultured in E2 plus vehicle. (n = 5), *p < 0.05, **p < 0.01 vs. NC-siRNA. NC-siRNA negative control siRNA
Icb-1 knockdown affects expression of estrogen pathway and proliferation genes
To examine molecular mechanisms which might underlie the observed effect of icb-1 gene expression on basal and E2-triggered growth as well as the fulvestrant sensitivity of T-47D cells, we analyzed the expression of six candidate genes, three of them part of the ERα pathway [ESR1, PR, and TFF1 (pS2)] and three proliferation genes (CCND1, CCNA2, and KI67). Knockdown of icb-1 in T-47D cells increased mRNA expression of ESR1 (1.6-fold, p < 0.01) and TFF1 (2.37-fold, p < 0.01) (Fig. 4a), but did not affect PR expression (data not shown). In serum-free medium, addition of 3 nM E2 resulted in a 1.63-fold increase of TFF1 (pS2) mRNA expression in icb-1 knockdown cells when compared to control cells, but did not affect ESR1 or PR (Fig. 4b). In cells transfected with icb-1 siRNA, E2 strongly increased Ki67 expression (6.07-fold, p < 0.001) and to a lower extent mRNA levels of cell cycle regulators CCND1 (1.59-fold) and CCNA2 (1.50-fold) (both p < 0.05) when compared to control cells.

Gene expression after icb-1 knockdown, as assessed by RT-qPCR analysis. a Effect of icb-1 knockdown. b Effect of E2 stimulation (3 nM) on gene expression. Expression values were processed in relation to housekeeping gene ACTB and the ΔΔC(t) method and are expressed in percent of the values of cells treated with negative control (NC) siRNA
Icb-1 negatively correlates with expression of ESR1, TFF1, and CCND1 in breast cancer tissue
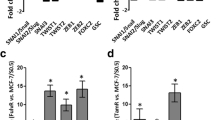
To examine to what extent our in vitro gene expression results from T-47D knockdown experiments could be verified in breast cancer tissue, we next analyzed correlation between expression of icb-1 and the genes affected by icb-1 knockdown in 1085 breast cancer tissues by means of the GEPIA online tool for meta-analysis of publicly available DNA microarray data [16]. Spearman rank correlation analyses between expression of icb-1 and these genes revealed a negative correlation of icb-1 with mRNA expression of ESR1 (Spearman’s rho = − 0.38, p = 7.8e-38) in breast cancer tissues and a weaker, but statistically highly significant negative correlation with ERα target TFF1 (pS2) (Spearman´s rho = − 0.26, p = 1.2e-18). With regard to the proliferation-associated genes tested, these analyses revealed no significant correlation of icb-1 with CCNA2 or KI67, but a moderate negative association with CCND1 (Spearman´s rho = − 0.25, p = 1.7e-16).
Icb-1 expression affects survival of breast cancer patients
To examine the role of icb-1 gene in breast cancer survival, we used an online tool for meta-analysis of publicly available DNA microarray data from 3951 breast cancer patients [17]. Kaplan–Meier plotter software revealed a longer relapse-free survival (RFS) of patients with high icb-1 expression (HR = 0.87, p = 0.015) (Fig. 5), whereas their overall survival (OS) was not affected. With regard to the four “classical” molecular intrinsic subtypes of breast cancer [18, 19], high levels of icb-1 expression increased both RFS and OS of patients with basal-like breast cancer (HR = 0.7, p = 0.0059 and HR = 0.52, p = 0.0093, respectively) (Fig. 6). In patients with luminal B-type cancer, high icb-1 expression considerably increased RFS (HR = 0.62, p = 1.3e-06), but not their OS. In the HER2-enriched intrinsic subgroup, high icb-1 expression notably elevated RFS of the patients (HR = 0.53, p = 0.0014). Whereas a similar trend also was observed with regard of OS, it did not reach statistical significance due to low case numbers. In contrast, in patients with Luminal A breast cancer, no effect of icb-1 expression on RFS or OS was observed.

Kaplan–Meier analysis of icb-1 expression in 3951 breast cancer patients and their relapse-free survival using the online tool http://kmplot.com/analysis/

Kaplan–Meier analysis of icb-1 expression in tumors of four different intrinsic subtypes and patients´ relapse-free survival (RFS) and overall survival (OS) using the online tool http://kmplot.com/analysis/
Discussion
For the first time, we demonstrate that knockdown of icb-1 is able to increase the response of ERα-positive breast cancer cells to antiestrogen fulvestrant in vitro, which supports the results of a previous study suggesting icb-1 to be part of a gene expression score predicting response of breast cancer patients to this SERD [12]. Our observation that knockdown of icb-1 strongly increases both basal growth and the effect of E2 on T-47D breast cancer cell growth is in line with a previous study reporting the same effects on MCF-7 cells [8]. Our analysis of genes of the ERα pathway as well as of proliferation-associated genes in icb-1 knockdown cells provided possible molecular mechanisms underlying the observed increased fulvestrant response of these cells. The observed effects of an icb-1 knockdown on T-47D gene expression, like E2-independent up-regulation of ESR1 mRNA and of ERα target gene TFF1 (pS2), are also in line with our previous study using MCF-7 breast cancer cells [8, 20]. When we examined the effects of icb-1 siRNA on E2-triggered expression of proliferation genes, in this study, we broadened gene selection. The finding that icb-1 knockdown enhanced E2-triggered up-regulation of its target gene CCND1 gene in T-47D cells is in line with similar results of experiments using MCF-7 cells [8, 20]. In the present study, we additionally examined the E2 effect on proliferation-associated genes CCNA2 and MKI67 subject to icb-1 expression, which demonstrated up-regulation of both genes, which was most pronounced with regard to MKI67, suggesting that icb-1 suppresses ERα activity, which not only affects its target gene TFF1 (pS2), but also E2-triggered induction of CCND1, CCNA2, and MKI67 expression [20]. Thus, the increased fulvestrant response in icb-1 knockdown cells is suggested to be the consequence of increased ERα levels, resulting in an elevated E2 effect on expression of proliferation genes leading to a considerable increase of T-47D cell proliferation. Thus, icb-1 is suggested to suppress E2-triggered proliferation via restriction of ERα expression, leading to a limitation of the growth-inhibitory response to fulvestrant.
To verify our in vitro results of an icb-1 knockdown on gene expression of T-47D cells, we examined the correlation of icb-1 expression with ESR1, TFF1 (pS2), CCND1, CCNA2, and MKI67 in 1085 breast cancer tissues by means of the GEPIA online tool for meta-analysis of publicly available DNA microarray data [16]. The fact that we found a considerable negative correlation of icb-1 with ESR1 and TFF1 (pS2) in this high number of breast cancer tissues clearly suggests that the restrictive action of icb-1 on ESR1 levels and activity we found by in vitro knockdown experiments with T-47D cells is also present in the in vivo situation. The observed negative correlation of icb-1 with CCND1 in breast cancer tissues might support a previous hypothesis that icb-1 exerts tumor-suppressing properties in this cancer entity [11]. The fact that we did not find significant correlations of icb-1 with CCNA2 and MKI67 is suggested to result from the fact that these proliferation genes are not regulated by ERα only, but by a multitude of other growth-regulatory pathways [21, 22].
Finally, the in silico analysis of gene expression data of 3951 breast cancer patients using an online survival tool (Kaplan–Meier Plotter) [17], revealing a longer RFS of patients with high icb-1 expression for the first time, demonstrated a significant beneficial role of icb-1 gene expression in breast cancer survival. This finding strongly supports the proposed tumor-suppressing role of this gene in breast cancer [11]. With regard to the “classical” intrinsic molecular subtypes of breast cancer [18, 19], which could be analyzed by means of the Kaplan–Meier Plotter online tool [17], in the luminal B subgroup, high icb-1 expression significantly increased patients’ RFS, which might be explained by its restricting effect on ESR1 and MKI67 highly expressed in this subtype. In contrast, in luminal A cancer, which also expresses ESR1 but low levels of MKI67, icb-1 expression did not affect patients´ survival, which might be due to the fact that in this low-grade and slowly growing cancer subgroup, the inhibiting effect of this gene on E2-triggered proliferation might be too low to be visible. The fact that high icb-1 expression is associated with a longer RFS in the HER2-enriched, ESR1-negative subtype and with a longer RFS and OS in basal-like cancer, which also lacks ESR1 expression, might be explained by the fact that icb-1 has been reported to also exert ESR1-independent anti-tumoral effects in breast cancer, like up-regulation of apoptosis genes [11].
Conclusion
The results of this study suggest that icb-1 not only decreases the sensitivity of T-47D breast cancer cells to antiestrogen fulvestrant, but also exerts considerable beneficial effects on the survival of breast cancer patients. Considering the gene expression data from our knockdown experiments and the observed correlations in breast cancer tissue, our data suggest both findings to be mediated by icb-1 triggered restriction of ERα expression and activity.
Data availability
Data and materials are available from the corresponding author on reasonable request.
References
Treeck O, Strunck E, Vollmer G (1998) A novel basement membrane-induced gene identified in the human endometrial adenocarcinoma cell line HEC1B. FEBS Lett 425(3):426–430. https://doi.org/10.1016/s0014-5793(98)00278-6
Treeck O (2011) Silencing of the icb-1 gene inhibits the induction of differentiation-associated genes by vitamin D3 and all-trans retinoic acid in gynecological cancer cells. Int J Mol Med. https://doi.org/10.3892/ijmm.2011.663
Treeck O, Odani T, Itoh N et al (2002) Detection of increased icb-1 transcript levels in maturing HL-60 cells: a novel marker for granulocytic and monocytic in vitro differentiation. Leuk Res 26(8):765–769. https://doi.org/10.1016/s0145-2126(02)00005-x
Peirce MJ, Brook M, Morrice N et al (2010) Themis2/ICB1 is a signalling scaffold that selectively regulates macrophage toll-like receptor signalling and cytokine production. PLoS ONE 5(7):e11465. https://doi.org/10.1371/journal.pone.0011465
Muraoka S, Kume H, Adachi J et al (2013) In-depth membrane proteomic study of breast cancer tissues for the generation of a chromosome-based protein list. J Proteome Res 12(1):208–213. https://doi.org/10.1021/pr300824m
Treeck O, Kindzorra I, Pauser K et al (2005) Expression of icb-1 gene is interferon-gamma inducible in breast and ovarian cancer cell lines and affects the IFN gamma-response of SK-OV-3 ovarian cancer cells. Cytokine 32(3–4):137–142. https://doi.org/10.1016/j.cyto.2005.08.008
Bollmann J, Ortmann O, Treeck O (2008) Expression of differentiation-associated gene icb-1 is estrogen-responsive in ovarian and breast cancer cell lines. J Steroid Biochem Mol Biol 109(1–2):16–21. https://doi.org/10.1016/j.jsbmb.2007.12.007
Konwisorz A, Springwald A, Haselberger M et al (2010) Knockdown of ICB-1 gene enhanced estrogen responsiveness of ovarian and breast cancer cells. Endocr Relat Cancer 17(1):147–157. https://doi.org/10.1677/ERC-09-0095
Springwald A, Lattrich C, Seitz S et al (2009) Single nucleotide polymorphisms in human gene icb-1 and breast cancer susceptibility. Cancer Invest 27(6):669–672. https://doi.org/10.1080/07357900802620877
Treeck O, Schüler S, Häring J et al (2013) icb-1 Gene counteracts growth of ovarian cancer cell lines. Cancer Lett 335(2):441–446. https://doi.org/10.1016/j.canlet.2013.02.049
Treeck O, Belgutay D, Häring J et al (2012) Network analysis of icb-1 gene function in human breast cancer cells. J Cell Biochem 113(9):2979–2988. https://doi.org/10.1002/jcb.24175
Knudsen S, Jensen T, Hansen A et al (2014) Development and validation of a gene expression score that predicts response to fulvestrant in breast cancer patients. PLoS ONE 9(2):e87415. https://doi.org/10.1371/journal.pone.0087415
Girish V, Vijayalakshmi A (2004) Affordable image analysis using NIH Image/ImageJ. Indian J Cancer 41(1):47
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3(6):1101–1108. https://doi.org/10.1038/nprot.2008.73
Tang Z, Li C, Kang B et al (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45(W1):W98–W102. https://doi.org/10.1093/nar/gkx247
Györffy B, Lanczky A, Eklund AC et al (2010) An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 123(3):725–731. https://doi.org/10.1007/s10549-009-0674-9
Perou CM, Sørlie T, Eisen MB et al (2000) Molecular portraits of human breast tumors. Nature 406(6797):747–752. https://doi.org/10.1038/35021093
Sorlie T, Perou CM, Tibshirani R et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci 98(19):10869–10874. https://doi.org/10.1073/pnas.191367098
Ikeda K, Horie-Inoue K, Inoue S (2015) Identification of estrogen-responsive genes based on the DNA binding properties of estrogen receptors using high-throughput sequencing technology. Acta Pharmacol Sin 36(1):24–31. https://doi.org/10.1038/aps.2014.123
Blanchard JM (2000) Cyclin A2 transcriptional regulation: modulation of cell cycle control at the G1/S transition by peripheral cues. Biochem Pharmacol 60(8):1179–1184. https://doi.org/10.1016/s0006-2952(00)00384-1
Sales Gil R, Vagnarelli P (2018) Ki-67: more hidden behind a “classic proliferation marker.” Trends Biochem Sci 43(10):747–748. https://doi.org/10.1016/j.tibs.2018.08.004
Acknowledgements
We would like to thank Mrs. Bettina Federhofer for expert technical assistance.
Funding
None.
Author information
Authors and Affiliations
Contributions
MR: data collection and management, and data analysis. MS, SS: data analysis and manuscript editing. OO: providing essential project support and manuscript editing. OT: protocol/project development, data collection and management, data analysis, and manuscript writing/editing.
Corresponding author
Ethics declarations
Conflict of interest
All authors state that they do not have any conflicts of interest with regard to this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Riedmeier, M., Skrzypczak, M., Schüler-Toprak, S. et al. Icb-1 expression inhibits growth and fulvestrant response of breast cancer cells and affects survival of breast cancer patients. Arch Gynecol Obstet 304, 203–213 (2021). https://doi.org/10.1007/s00404-020-05902-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00404-020-05902-x