
Abstract
Rare and common GBA variants are risk factors for both Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). However, the degree to which GBA variants are associated with neuropathological features in Lewy body disease (LBD) is unknown. Herein, we assessed 943 LBD cases and examined associations of 15 different neuropathological outcomes with common and rare GBA variants. Neuropathological outcomes included LBD subtype, presence of a high likelihood of clinical DLB (per consensus guidelines), LB counts in five cortical regions, tyrosine hydroxylase immunoreactivity in the dorsolateral and ventromedial putamen, ventrolateral substantia nigra neuronal loss, Braak neurofibrillary tangle (NFT) stage, Thal amyloid phase, phospho-ubiquitin (pS65-Ub) level, TDP-43 pathology, and vascular disease. Sequencing of GBA exons revealed a total of 42 different variants (4 common [MAF > 0.5%], 38 rare [MAF < 0.5%]) in our series, and 165 cases (17.5%) had a copy of the minor allele for ≥ 1 variant. In analysis of common variants, p.L483P was associated with a lower Braak NFT stage (OR = 0.10, P < 0.001). In gene-burden analysis, presence of the minor allele for any GBA variant was associated with increased odds of a high likelihood of DLB (OR = 2.00, P < 0.001), a lower Braak NFT stage (OR = 0.48, P < 0.001), a lower Thal amyloid phase (OR = 0.55, P < 0.001), and a lower pS65-Ub level (β: −0.37, P < 0.001). Subgroup analysis revealed that GBA variants were most common in LBD cases with a combination of transitional/diffuse LBD and Braak NFT stage 0-II or Thal amyloid phase 0–1, and correspondingly that the aforementioned associations of GBA gene-burden with a decreased Braak NFT stage and Thal amyloid phase were observed only in transitional or diffuse LBD cases. Our results indicate that in LBD, GBA variants occur most frequently in cases with greater LB pathology and low AD pathology, further informing disease–risk associations of GBA in PD, PD dementia, and DLB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lewy body disease (LBD) is a neuropathologically defined disorder that is characterized by occurrence of Lewy bodies (LBs) and Lewy neurites in the brainstem (brainstem-predominant LBD) that can extend to the limbic (transitional LBD) and neocortical (diffuse LBD) regions [21]. Common clinical presentations of LBD include Parkinson’s disease (PD), PD with dementia (PDD), and dementia with Lewy bodies (DLB); the presence of Lewy body (LB) pathology is considered a pathological hallmark of these α-synucleinopathies [21]. Additionally, Alzheimer’s disease (AD) neuropathological changes are commonly observed in LBD [21], and conversely up to 50% of AD patients display LB pathology [8, 9]. Unlike PD, PDD, and DLB, LB pathology is not thought to play a major role in the pathogenesis of AD [9].
The GBA gene encodes glucocerebrosidase, a lysosomal enzyme involved in the metabolism of glucosylceramide [35]. Importantly, impaired activity of glucocerebrosidase is associated with an accumulation of α-synuclein [35], which is the main component of LBs [21]. Although initially discovered to cause Gaucher’s disease in a recessive manner [35], GBA variants also play an important role in LBD on several levels. First, presence of rare exonic GBA variants is associated with a markedly increased risk of both PD (odds ratio [OR] ~ 5) [37] and DLB (OR ~ 8) [7, 31], where disease risk is predominantly driven by the p.L483P and p.N409S variants. Second, common exonic GBA variants, including p.E365K and p.T408M, are risk factors for PD (both variants) and DLB (p.E365K) [31, 41]. Third, GBA is a susceptibility locus for both PD and DLB in unbiased genome-wide association studies (GWAS) [13, 30], where evidence suggests that the GWAS signal is driven by the p.E365K variant [4, 13]. On the other hand, GBA variation has not been linked with susceptibility to AD [40, 42].
Presence of rare GBA variants has also been associated with specific clinical features in individuals with LB disorders, including (but not limited to) a younger age at onset, shorter survival, and more frequent cognitive impairment/dementia [12, 36]. However, studies assessing whether GBA variants may also influence neuropathology in individuals who have developed LBD have been limited by small sample sizes [1, 32, 34]. With this in mind, in the current study we evaluated associations of GBA variants with a variety of neuropathological features in a large series of LBD cases.
Materials and methods
Study patients
A total of 943 neuropathologically confirmed LBD cases from the Mayo Clinic Florida brain bank for neurodegenerative disorders were included in this study. All LBD cases were evaluated by a single neuropathologist (D.W.D). LBD cases were excluded if they had significant coexisting non-AD neurodegenerative conditions (e.g., progressive supranuclear palsy, corticobasal degeneration, multiple system atrophy, amyotrophic lateral sclerosis, etc.), had amygdala-predominant LBD in the setting of advanced AD, or did not have information available for any of the neuropathological outcome measures examined in this study. Demographic and clinical information was collected regarding age at death, sex, and presence of dementia, with age at disease onset and disease duration collected in a subset of 384 LBD cases. Fifteen different neuropathological outcomes were assessed and included LBD subtype (N = 943), presence of high likelihood of clinical DLB [27] (N = 943), LB counts in five different regions (N = 753 to 762), dorsolateral and ventromedial putaminal tyrosine hydroxylase immunoreactivity (TH-ir) (N = 443), neuronal loss in the ventrolateral part of the substantia nigra (SN) (N = 486), Braak neurofibrillary tangle (NFT) stage (N = 943), Thal amyloid phase (N = 785), phospho-ubiquitin (pS65-Ub) level (N = 723), TDP-43 pathology (N = 751), and vascular disease (VaD) (N = 942). Characteristics of LBD cases are shown in Table 1.
Neuropathological assessment
Formalin-fixed brains underwent systematic and standardized sampling with neuropathologic evaluation. Regions sampled for histopathologic assessment included six regions of the neocortex, two levels of the hippocampus, a basal forebrain section that includes the amygdala, lentiform nucleus and hypothalamus, anterior corpus striatum, thalamus at the level of the subthalamic nucleus, midbrain, pons, medulla, and two sections of the cerebellum, one including the deep nuclei. Paraffin-embedded 5-μm thick sections mounted on glass slides were stained with hematoxylin and eosin (H&E) and thioflavin S (Sigma-Aldrich, St. Louis, MO). Sections of the cortex, hippocampus, and basal forebrain, and brainstem were immunostained with anti-α-synuclein antibody (NACP; rabbit polyclonal; 1:3000; a gift from Dr. Petrucelli, Mayo Clinic, Jacksonville, Florida; formic acid pretreatment) [10] to establish a neuropathological diagnosis of LBD [27].
LBD subtype was classified as brainstem, transitional, or diffuse as described by McKeith et al. [23, 28] Presence of a high likelihood of clinical DLB (Braak NFT stage 0–II and transitional/diffuse LBD, or Braak NFT stage III–IV and diffuse LBD) was assessed according to the criteria of the fourth report of the DLB consortium [27]. LBs were manually counted in the middle frontal (MF), superior temporal (ST), inferior parietal (IP), cingulate (CG), and para-hippocampal (PH) gyrus in the field of highest density at x200 magnification. The TH-ir in the dorsolateral and ventromedial putamen was assessed using the antibody against TH (rabbit polyclonal, 1:600; Affinity Bioreagents, Golden, Colorado), as previously described [18]; a lower TH-ir value corresponds to a greater degree of putaminal dopaminergic degeneration The neuronal loss in the ventrolateral part of the substantia nigra (SN) was semi-quantitatively scored on H&E-stained sections as follows: 0 = none, 0.5 = minimal, 1 = mild, 1.5 = mild-to-moderate, 2 = moderate, 2.5 = moderate-to-severe, 3 = severe.
Braak NFT stage and Thal amyloid phase were assessed with thioflavin S fluorescence microscopy, a method validated in our prior work and recognized by the National Institute on Aging-Alzheimer’s Association guidelines for its consistency with tau immunohistochemistry results, particularly in large-scale studies [5, 22, 25, 29, 38]. The mitophagy alteration was assessed in the hippocampus on immunostained slides of the mitophagy marker phosphorylated ubiquitin on serine 65 (pS65-Ub, in-house rabbit polyclonal antibody, 1:650), and pS65-Ub positive cell density was quantified using established algorithm via digital pathology (Aperio ImageScope v12) [11, 15,16,17]. TDP-43 pathology, such as neuronal or glial cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear inclusion, spheroid, or perivascular inclusion, was screened in the amygdala based on immunostained slides of phospho-TDP-43 (pS409/410, mouse monoclonal, 1:5000, Cosmo Bio, Tokyo, Japan) [19]. VaD included lacunar infarcts, microscopic infarcts, hemorrhages, cerebral amyloid angiopathy, and leukoencephalopathy, which were assessed on H&E-stained sections or thioflavin S fluorescent microscopy [20]. All immunohistochemistry was done using IHC Autostainer 480S (Thermo Fisher Scientific Inc., Waltham, MA) and DAKO EnVision™ + reagents (Dako, Carpinteria, CA).
Genetic analysis
All cases underwent automated DNA extraction from frozen cerebellar tissue using the Autogen 245 T and Autogen FlexStar + (Holliston, MA). Sanger sequencing of GBA (NM_ 001005742) was performed. GBA was amplified in 3 amplicons (exons 1–5, exons 5–7, exons 8–11; primer sequences available on request) to avoid amplification of the pseudogene (GBAP). All PCR products were cleaned using Agencourt AMPure XP magnetic beads and the Biomek FXP (Beckman Coulter). The individual exons were then bidirectionally cycle-sequenced using internal primers and ABI BigDye Terminator chemistry. The cycle sequence products were cleaned with Agencout CleanSEQ magnetic beads and analyzed on an ABI 3730xl DNA Sequencer. Sequences were analyzed using Seqscape v3.0 Software. All coding and flanking regions within 25 bp of GBA were analyzed, and call rates for each variant were > 98%. LBD cases with call rates < 95% were excluded.
Additionally, GWAS data were available from other studies performed involving these LBD cases (see the Supplemental Information for methodological details). We utilized this GWAS data in the current study in order to exclude subjects with non-European ancestry, and also to extract principal components (PCs) in order to adjust for population stratification in regression analyses.
Statistical analysis
For common GBA variants with a minor allele frequency (MAF) of > 0.5%, associations of presence of the minor allele with neuropathological outcomes were evaluated using regression models appropriate for the nature of the given outcome (continuous, binary, ordinal, or count) that were adjusted for age at death, sex, and the top five PCs of available GWAS data. Specifically, proportional odds logistic regression models were used for ordinal outcomes (LBD subtype, ventrolateral SN neuronal loss score, Braak NFT stage, and Thal amyloid phase), linear regression models were used for continuous outcomes (dorsolateral putaminal TH-ir, ventromedial putaminal TH-ir, and pS65-Ub), binary logistic regression models were used for binary outcomes (high likelihood of clinical DLB, TDP-43 pathology, VaD), and negative binomial regression models were used for count outcomes (MF, ST, IP, CG, and PH LB counts). Associations of presence of the minor allele of common GBA variants with dementia and male sex were assessed using the aforementioned binary logistic regression models; the associations with male sex were assessed due to recent reports of differences in the frequency of GBA mutations according to sex [33, 39]. Owing to their skewed distributions, in linear regression analysis, dorsolateral putaminal TH-ir and pS65-Ub were examined on the natural logarithm scale, while ventromedial putaminal TH-ir was examined on the square root scale.
For LBD subtype, ventrolateral SN neuronal loss score, Braak NFT stage, and Thal amyloid phase, ORs and 95% confidence intervals (CIs) are interpreted as the multiplicative increase in the odds of a more severe category for cases with presence of the minor allele for the given GBA variant. For dorsolateral putaminal TH-ir, ventromedial putaminal TH-ir, and pS65-Ub, regression coefficients (denoted as β), and 95% CIs are interpreted as the additive increase in the mean value of the given outcome measure (on the natural logarithm scale or square root scale) for cases with presence of the minor allele for the given GBA variant. For high likelihood of DLB, TDP-43 pathology, VaD, dementia, and male sex, ORs, and 95% CIs are interpreted as the multiplicative increase in the odds of occurrence of the given outcome for cases with presence of the minor allele for the given GBA variant. For MF, ST, IP, CG, and PH LB counts, multiplicative effects and 95% CIs are interpreted as the multiplicative increase in the mean outcome measure for cases with presence of the minor allele for the given GBA variant.
For rare variants with a MAF < 0.5%, single-variant analysis was not performed due to the low power such analysis would have to detect associations with neuropathological outcomes. Instead, gene-burden tests were performed by collapsing across all rare variants and evaluating the associations of presence of the minor allele for any rare variant with neuropathological outcomes using the previously described regression models that were adjusted for age at death, sex, and the top five PCs [24]. A similar gene-burden analysis was also employed when considering all GBA variants, regardless of MAF. P-values < 0.0029 are considered as statistically significant after applying a Bonferroni correction for the 17 different outcome measures that were examined. All statistical tests were two-sided. Statistical analyses were performed using SAS (version 9.4; SAS Institute, Inc., Cary, North Carolina).
Results
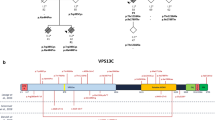
A summary of GBA variants that were observed in our series of 943 LBD cases is provided in Table 2. Of the 42 different variants that were observed, four (rs140335079 x7−18 bp [MAF 1.49%]; rs2230288 p.E365K [MAF 3.19%]; rs75548401 p.T408M [MAF 1.38%]; rs76763715 p.N409S [MAF 0.69%]) were common variants with a MAF > 0.5%, and the remaining 38 were rare. Also of note, the MAF of rs421016 p.L483P was 0.48%. Of the 943 LBD cases, 165 (17.5%) had a copy of the minor allele for at least one GBA variant, while 79 (8.4%) had a copy of the minor allele for at least one GBA variant with a MAF < 0.5%.
Associations of the four common (MAF > 0.5%) GBA variants with neuropathological outcome measures (as well as dementia and male sex) are displayed in Table 3 for outcomes where a significant (P < 0.0029 after multiple testing adjustment) or nominally significant (P < 0.05) association was observed, with results for the remaining neuropathological outcomes shown in Supplemental Table 1. Additionally, we also assessed associations with outcomes for the rs421016 p.L483P PD risk variant as its MAF was only slightly below the > 0.5% threshold used to identify common variants. After adjusting for multiple testing, a significant association was observed between rs421016 p.L483P and lower Braak NFT stage (≥ 4: 11.1% vs. 52.5%, OR = 0.10, P < 0.001), with nominally significant associations observed toward a less severe LBD subtype (Diffuse: 33.3% vs. 51.4%, OR = 0.23, P = 0.021), a lower Thal amyloid phase (≥ 3: 22.2% vs. 72.8%, OR = 0.19, P = 0.008), and a lower pS65-Ub level (Median: 0.83 vs. 3.23, β: −0.98, P = 0.009). Additional nominally significant associations were observed of rs2230288 p.E365K with an increased odds of a high likelihood of clinical DLB (57.9% vs. 41.8%, OR = 1.88, P = 0.025) and greater likelihood of male sex (70.2% vs. 57.0%, OR = 1.85, P = 0.042), and of rs7673715 p.N409S with a decreased Braak NFT stage (≥ 4: 15.4% vs. 52.6%, OR = 0.23, P = 0.005).
Table 4 shows gene-burden analysis assessing associations with neuropathological outcomes for presence of the minor allele for any GBA variant, and presence of the minor allele for any GBA variant with a MAF < 0.5%; only outcomes where at least a nominally significant association are displayed, with results for other outcomes provided in Supplemental Table 2. After correcting for multiple testing, presence of any GBA variant was associated with a greater odds of a high likelihood of clinical DLB (58.8% vs. 39.2%, OR = 2.00, P < 0.001), a lower Braak NFT stage (≥ 4: 35.2% vs. 55.7%, OR = 0.48, P < 0.001), a lower Thal amyloid phase (≥ 3: 58.5% vs. 75.1%, OR = 0.55, P < 0.001), and a lower pS65-Ub level (Median: 1.88 vs. 3.56, β: −0.37, P < 0.001), with similar findings observed when considering presence of any GBA variant with a MAF < 0.5% (all P ≤ 0.001, Table 4). These results are further illustrated in Fig. 1 (presence of any GBA variant) and Supplemental Fig. 1 (presence of any GBA variant with a MAF < 0.5%). Of note, results of rare-variant (MAF < 0.5%) gene-burden analyses were relatively similar when excluding rs421016 p.L483P (Table 4), indicating that the aforementioned findings were not driven by this rare variant. Additionally, although only nominally significant, presence of any GBA variant was associated with a lower dorsolateral putaminal TH-ir (Median: 2.83 vs. 4.39, β: −0.28, P = 0.014). A similar association with dorsolateral putaminal TH-ir was noted when examining presence of any GBA variant with a MAF < 0.5% (Median: 2.63 vs. 4.37, β: −0.41, P = 0.007).

Proportion of LBD cases with presence of any GBA variant according to high likelihood of clinical DLB, Braak NFT stage, Thal amyloid phase, and pS65-Ub level. pS65-Ub level was dichotomized based on the sample median
Given the significant association between GBA gene-burden measures and an increased odds of a high likelihood of clinical DLB, in Table 5, we further explored this finding by assessing the frequencies of gene-burden measures in nine different subgroups defined by a combination of LBD subtype (brainstem, transitional, or diffuse) and Braak NFT stage (0–II, III–IV, or V–VI), as these same subgroups are used to define likelihood of DLB [27]. Subgroups defined by a combination of LBD subtype and Thal amyloid phase (0–1, 2–3, or 4–5) were also examined (Table 5). When considering presence of any GBA variant, observed frequencies were the highest in LBD subgroups with Braak NFT stage 0–II and either transitional LBD (37.8%) or diffuse LBD (28.4%); in comparison to a reference group of brainstem LBD and Braak NFT stage 0–II patients (in analysis adjusted for age at death, sex, and the top 5 PCs), the likelihood of presence of any GBA variant was higher for transitional LBD and Braak NFT stage 0–II (OR = 3.24, P = 0.007), with a slightly weaker finding for diffuse LBD and Braak NFT stage 0–II (OR = 2.20, P = 0.077). Findings were similar when assessing LBD subtype and Thal amyloid phase, with the highest frequencies of GBA gene-burden noted for the transitional LBD and Thal amyloid phase 0–1 (OR [vs. brainstem LBD/Thal 0–1] = 4.52, P = 0.001) and diffuse LBD and Thal amyloid phase 0–1 (OR = 5.14, P = 0.005) subgroups. Results were consistent when examining the presence of any GBA variant with a MAF < 0.5% (Supplemental Table 3).
The results shown in Table 5 suggest the presence of interactions between GBA variants and LBD subtype with regard to association with AD-related neuropathology. Therefore, we assessed this possibility in Table 6, where associations of GBA gene burden measures with Braak NFT stage, Thal amyloid phase, and pS65-Ub were assessed separately for each LBD subtype. There were significant interactions between presence of any GBA variant and LBD subtype regarding the association with Braak NFT stage (interaction P < 0.001) and Thal amyloid phase (interaction P < 0.001), where presence of GBA variants was only associated with a lower Braak NFT stage and Thal amyloid phase for cases with transitional or diffuse LBD. No significant interaction between presence of any GBA variant and LBD subtype in relation to pS65-Ub level (interaction P = 0.21) was noted. These results were relatively consistent when assessing presence of any GBA variant with a MAF < 0.5% (Table 6). Of note, the results of all of the aforementioned regression analyses were similar when adjusting regression models for age at disease onset instead of age at death in the 384 LBD cases who had information available for both measures (data not shown), which is to be expected given the very strong degree of correlation between these two age-related variables (Spearman’s r = 0.85, P < 0.001).
Discussion
GBA variants are well-known risk factors for both PD and DLB [7, 13, 30, 31, 37, 41]. However, little is known regarding whether GBA variation may modify neuropathology in LBD cases. Herein, we assessed associations between GBA variants and 15 different neuropathological features in a series of 943 LBD cases. One significant association was observed when examining common variants after correcting for multiple testing, which was between p.L483P and a lower Braak NFT stage. In gene-burden analysis, we observed significant associations of presence of the minor allele for any GBA variant (rare variants only, and also all variants regardless of MAF) with a greater odds of a high likelihood of clinical DLB, a less severe Braak NFT stage and Thal amyloid phase, and a lower pS65-Ub level in the overall series of LBD cases. In subgroup analysis, GBA gene-burden measures were most common in cases with either transitional or diffuse LBD and low levels of AD pathology (Braak NFT stage and Thal amyloid phase), and correspondingly the aforementioned associations of presence of any GBA variant with lower Braak NFT stage and Thal amyloid phase were observed predominantly for transitional and diffuse LBD cases. Importantly, all findings were independent of age at death, sex, and top PCs of genome-wide genetic data.
Some of the strongest findings of our study were the aforementioned protective associations that we observed in gene-burden analysis of the overall LBD series for the AD-related measures of Braak NFT stage, Thal amyloid phase, and pS65-Ub level. Previous studies of PD patients have failed to identify associations between presence of GBA mutations and AD-related neuropathological outcomes [1, 32, 34]. However, sample sizes have been very small. It is also worth noting that GBA variants are not associated with a decreased risk of AD from large population case–control GWAS and sequencing studies [3, 14]. Thus, our findings likely reflect a higher frequency of GBA mutation carriers in PD and DLB (where Braak, Thal, and pS65-Ub level are lower) compared to clinical AD (where Braak, Thal, and pS65-Ub level are higher). To further investigate this, we also assessed gene-burden associations vs. the outcome of clinically assessed dementia. The fact that no notable associations with dementia were observed suggests that our significant findings regarding Braak NFT stage, Thal amyloid phase, and S65-Ub level are indeed indicative of the disease–risk associations for PD and DLB but not AD, rather than a general independent association between GBA variants and dementia-related measures in LBD.
Interestingly, we also identified a strong association between presence of GBA mutations and increased risk of a high likelihood of DLB. This initially seems to reflect the aforementioned associations between presence of GBA variants and a lower Braak NFT stage, lower Thal amyloid phase, and lower pS65-Ub level, as a high likelihood of DLB is defined in part by presence of low AD pathology (Braak NFT stage 0–II). However, subsequent examination of the frequency of GBA variants according to specific subgroups defined by combinations of LBD subtype and either Braak NFT stage or Thal amyloid phase reveals important insights into all of these findings, in that LBD subtype appears to interact with severity of AD pathology in regard to association with GBA gene-burden measures. Specifically, GBA gene-burden measures were only associated with Braak NFT stage and Thal amyloid phase in LBD cases with transitional or diffuse LBD, with no notable association observed in the brainstem LBD subgroup. The aforementioned associations of presence of GBA variants with a less severe Braak NFT stage and Thal amyloid phase in the overall series appear to have been driven mostly by the very high frequency of such variants in LBD cases with transitional or diffuse LBD and low levels of AD pathology, who of course comprise part of the high likelihood of DLB subgroup. These findings are in agreement with those of Tsuang et al., who noted a higher frequency of GBA mutations in 80 cases with “pure DLB” (dementia, transitional or diffuse LBD, no/low levels of AD pathology) compared to 231 cases with “LBD-AD” (dementia, transitional or diffuse LBD, high levels of AD pathology) [40].
In our LBD series, GBA variants were not strongly associated with severity of LB pathology (as measured by LB counts and LBD subtype), with striatal dopaminergic degeneration (as measured by putaminal TH-ir), or with substantia nigra neuronal loss. Though again limited by small sample size, previous studies have mostly observed a similar lack of association [1, 34], with the exception of one report of a more severe LBD subtype for 17 PD GBA mutation carriers compared to 16 PD non-carriers [32]. The negative findings of our large study of LBD cases could indicate that while GBA variants are strong risk factors for developing the LB disorders of PD and DLB, they do not act as disease modifiers for PD/DLB-related neuropathological outcomes in individuals who have already developed LBD. Alternatively, these results could suggest that GBA variation may influence specific aspects of disease that may occur earlier in the disease course, rather than severity of disease at the later stages of disease as measured by our neuropathological outcomes. For example, it may be that GBA mutations play a role in the initial accumulation, seeding and aggregation of α-synuclein, or the spread of pathology to the cortical regions; further mechanistic studies are needed to tease out the interplay of GBA and α-synuclein [6].
Our detailed neuropathologic characterization of the frequency of GBA mutations across the spectrum of both LB and AD pathology in LBD also further informs disease–risk associations involving PD, PDD, and DLB. For example, OR estimates regarding GBA mutations have typically been higher for DLB than for PD [31, 37] (with DLB more commonly having severe LB pathology), and also GBA mutations are associated with an increased likelihood of dementia in PD [12, 26] (with PDD also more frequently presenting with greater LB pathology compared to PD without dementia). Of note, recent reports have suggested a possible sex-specific difference in the frequency of GBA mutations in PD [33, 39]; with the exception of the p.E365K mutation that was nominally more common in males, we were unable to demonstrate such differences in our LBD series.
Strengths of our study include the large number of neuropathological features that were assessed, the relatively large number of LBD cases that were included, and the fact that we were able to adjust for population stratification using top PCs of genome-wide data. However, although the sample size is relatively large for an LBD series, it is still somewhat limited for a genetic association study. Therefore, the possibility of a type II error (i.e., a false-negative finding) is important to consider, particularly for the dichotomous outcomes of TDP-43 pathology and VaD, for outcomes with a greater amount of missing data, and when examining individual common GBA variants, as power would have been lowest in these scenarios. Additionally, our study examined only individuals of Caucasian ancestry; future studies evaluating associations between GBA variants and neuropathology of LBD in other racial/ethnic groups will be important.
Several other limitations of our study are important to draw attention to. The possibility of ascertainment bias in our autopsy-based cohort must be acknowledged; our LBD series is likely over-represented with AD patients, as evidenced by the high proportion of cases with a Thal amyloid phase equal to 5 (42.0%). However, as we directly analyzed AD-related neuropathological outcomes in our study, we do not feel that this would have had any noticeable impact on our results. Another limitation to highlight is the presence of missing data for a number of our neuropathological outcomes, as well as for disease duration and age at disease onset. Regarding age at onset, it is noteworthy to mention that although we could not directly account for the potential confounding influence of this variable in our regression analyses due to the extent of missing data, we did adjust for age at death in all of our models. Though these two age-related variables capture different components of disease, due to their very high degree of correlation, the choice of which age-related variable is adjusted for in multivariable analysis has a minimal impact on the results involving individual GBA variants and GBA gene-burden measures. Furthermore, TDP-43 pathology was assessed only in the amygdala; evaluation of other regions, such as hippocampus, midbrain, and frontal lobe, could have impacted our findings regarding this neuropathological outcome. Finally, though the guidelines utilized in our study for evaluation of Lewy pathology are well-established and widely used, other guidelines that have recently been proposed would also have been a reasonable alternative [2], noting that this would have only changed the analyses regarding LBD subtype.
In conclusion, the results of our study shed light on the role of GBA variation in determining severity of neuropathology in LBD and also aid in our understanding of the role of GBA mutations in susceptibility to specific LB disorders, such as PD, PDD, and DLB. Specifically, the presence of GBA mutations is associated with a lower severity of AD-related neuropathological features in LBD, with associations involving Braak NFT stage and Thal amyloid phase observed solely in cases with transitional or diffuse LBD. The latter observation is reflective of the high frequencies of GBA gene-burden measures in LBD cases with either transitional or diffuse LBD and also low levels of AD pathology (Braak NFT stage 0–II, or Thal amyloid phase 0–1). Interestingly, GBA variation was not strongly associated with neuropathological features that are more specific to PD and DLB. Unbiased GWAS or whole-genome sequencing studies will be important in order to more fully characterize genetic drivers of neuropathological variation in LBD cases.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- LBD:
-
Lewy body disease
- LB:
-
Lewy body
- PD:
-
Parkinson’s disease
- DLB:
-
Dementia with Lewy bodies
- AD:
-
Alzheimer’s disease
- GWAS:
-
Genome-wide association study
- TH-ir:
-
Tyrosine hydroxylase immunoreactivity
- SN:
-
Substantia nigra
- NFT:
-
Neurofibrillary tangle
- pS65-Ub:
-
Phospho-ubiquitin
- TDP-43:
-
TAR DNA-binding protein-43
- VaD:
-
Vascular disease
- H & E:
-
Hematoxylin and eosin
- MF:
-
Middle frontal
- ST:
-
Superior temporal
- IP:
-
Inferior parietal
- CG:
-
Cingulate
- PH:
-
Parahippocampal
- SN:
-
Substantia nigra
- PC:
-
Principal component
- MAF:
-
Minor allele frequency
- OR:
-
Odds ratio
- CI:
-
Confidence interval
References
Adler CH, Beach TG, Shill HA, Caviness JN, Driver-Dunckley E, Sabbagh MN et al (2017) GBA mutations in Parkinson disease: earlier death but similar neuropathological features. Eur J Neurol 24:1363–1368. https://doi.org/10.1111/ene.13395
Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G et al (2021) Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol 141:159–172. https://doi.org/10.1007/s00401-020-02255-2
Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N et al (2022) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54:412–436. https://doi.org/10.1038/s41588-022-01024-z
Blauwendraat C, Bras JM, Nalls MA, Lewis PA, Hernandez DG, Singleton AB (2018) Coding variation in GBA explains the majority of the SYT11-GBA Parkinson’s disease GWAS locus. Mov Disord 33:1821–1823. https://doi.org/10.1002/mds.103
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Chatterjee D, Krainc D (2023) Mechanisms of Glucocerebrosidase Dysfunction in Parkinson’s Disease. J Mol Biol 435:168023. https://doi.org/10.1016/j.jmb.2023.168023
Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH, Gustavsson E et al (2021) Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 53:294–303. https://doi.org/10.1038/s41588-021-00785-3
Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC (2015) Clinical Features of Alzheimer Disease With and Without Lewy Bodies. JAMA Neurol 72:789–796. https://doi.org/10.1001/jamaneurol.2015.0606
DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14:32. https://doi.org/10.1186/s13024-019-0333-5
Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R et al (1999) Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol 155:1241–1251. https://doi.org/10.1016/s0002-9440(10)65226-1
Fiesel FC, Ando M, Hudec R, Hill AR, Castanedes-Casey M, Caulfield TR et al (2015) (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep 16:1114–1130. https://doi.org/10.15252/embr.201540514
Gan-Or Z, Liong C, Alcalay RN (2018) GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr Neurol Neurosci Rep 18:44. https://doi.org/10.1007/s11910-018-0860-4
Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD et al (2018) Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. The Lancet Neurology 17:64–74. https://doi.org/10.1016/s1474-4422(17)30400-3
Holstege H, Hulsman M, Charbonnier C, Grenier-Boley B, Quenez O, Grozeva D et al (2022) Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat Genet 54:1786–1794. https://doi.org/10.1038/s41588-022-01208-7
Hou X, Chen TH, Koga S, Bredenberg JM, Faroqi AH, Delenclos M et al (2023) Alpha-synuclein-associated changes in PINK1-PRKN-mediated mitophagy are disease context dependent. Brain Pathol. https://doi.org/10.1111/bpa.13175
Hou X, Fiesel FC, Truban D, Castanedes Casey M, Lin WL, Soto AI et al (2018) Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 14:1404–1418. https://doi.org/10.1080/15548627.2018.1461294
Hou X, Watzlawik JO, Cook C, Liu CC, Kang SS, Lin WL et al (2020) Mitophagy alterations in Alzheimer’s disease are associated with granulovacuolar degeneration and early tau pathology. Alzheimer’s Dementia 17:417–430. https://doi.org/10.1002/alz.12198
Kasanuki K, Heckman MG, Diehl NN, Murray ME, Koga S, Soto A et al (2017) Regional analysis and genetic association of nigrostriatal degeneration in Lewy body disease. Mov Disord 32:1584–1593. https://doi.org/10.1002/mds.27184
Koga S, Lin WL, Walton RL, Ross OA, Dickson DW (2018) TDP-43 pathology in multiple system atrophy: colocalization of TDP-43 and α-synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol 44:707–721. https://doi.org/10.1111/nan.12485
Koga S, Roemer SF, Tipton PW, Low PA, Josephs KA, Dickson DW (2020) Cerebrovascular pathology and misdiagnosis of multiple system atrophy: An autopsy study. Parkinsonism Relat Disord 75:34–40. https://doi.org/10.1016/j.parkreldis.2020.05.018
Koga S, Sekiya H, Kondru N, Ross OA, Dickson DW (2021) Neuropathology and molecular diagnosis of Synucleinopathies. Mol Neurodegener 16:83. https://doi.org/10.1186/s13024-021-00501-z
Koga S, Zhou X, Dickson DW (2021) Machine learning-based decision tree classifier for the diagnosis of progressive supranuclear palsy and corticobasal degeneration. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12710
Kosaka K, Yoshimura M, Ikeda K, Budka H (1984) Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol 3:185–192
Li B, Leal SM (2008) Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 83:311–321. https://doi.org/10.1016/j.ajhg.2008.06.024
Liesinger AM, Graff-Radford NR, Duara R, Carter RE, Hanna Al-Shaikh FS, Koga S et al (2018) Sex and age interact to determine clinicopathologic differences in Alzheimer’s disease. Acta Neuropathol 136:873–885. https://doi.org/10.1007/s00401-018-1908-x
Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM et al (2016) GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord 31:95–102. https://doi.org/10.1002/mds.26359
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D et al (2017) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89:88–100. https://doi.org/10.1212/wnl.0000000000004058
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. https://doi.org/10.1212/01.wnl.0000187889.17253.b1
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123:1–11. https://doi.org/10.1007/s00401-011-0910-3
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D et al (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18:1091–1102. https://doi.org/10.1016/s1474-4422(19)30320-5
Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF et al (2013) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70:727–735. https://doi.org/10.1001/jamaneurol.2013.1925
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH et al (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132:1783–1794. https://doi.org/10.1093/brain/awp044
Ortega RA, Bressman SB, Raymond D, Ozelius LJ, Wang C, Bennett SAL et al (2023) Differences in Sex-specific frequency of glucocerebrosidase variant carriers and familial parkinsonism. Mov Disord 38:714–715. https://doi.org/10.1002/mds.29353
Parkkinen L, Neumann J, O’Sullivan SS, Holton JL, Revesz T, Hardy J et al (2011) Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson’s disease. Mol Genet Metab 103:410–412. https://doi.org/10.1016/j.ymgme.2011.04.015
Riboldi GM, Di Fonzo AB (2019) GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells. https://doi.org/10.3390/cells8040364
Shiner T, Mirelman A, Gana Weisz M, Bar-Shira A, Ash E, Cialic R et al (2016) High frequency of gba gene mutations in dementia with lewy bodies among ashkenazi jews. JAMA Neurol 73:1448–1453. https://doi.org/10.1001/jamaneurol.2016.1593
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661. https://doi.org/10.1056/NEJMoa0901281
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Thaler A, Mirelman A, Alcalay RN (2023) Differences in sex-specific frequency of glucocerebrosidase variant carriers and familial parkinsonism. Mov Disord 38:713–714. https://doi.org/10.1002/mds.29348
Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA et al (2012) GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 79:1944–1950. https://doi.org/10.1212/WNL.0b013e3182735e9a
Vieira SRL, Schapira AHV (2022) Glucocerebrosidase mutations and Parkinson disease. J Neural Transm (Vienna) 129:1105–1117. https://doi.org/10.1007/s00702-022-02531-3
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D et al (2021) A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 53:1276–1282. https://doi.org/10.1038/s41588-021-00921-z
Acknowledgements
We would like to thank all those who have contributed to our research, particularly the patients and families who donated brain, blood, and DNA samples for this work. We would like to acknowledge the continuous commitment, technical support and teamwork offered by Linda G. Rousseau, Virginia R. Phillips, and Monica Castanedes-Casey. This work was supported by the Mayo Clinic LBD Center WithOut Walls (CWOW; U54-NS110435), Ted Turner and family, the Little Family Foundation, and the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy body dementia program, and in part by the Mayo Clinic Florida Morris K. Udall Parkinson's Disease Research Center of Excellence (NINDS P50 #NS072187), Alzheimer’s disease Research Center (P50 AG016574), an American Parkinson Disease Association (APDA) Mayo Clinic Information and Referral Center, an APDA Center for Advanced Research and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence.
Funding
OAR and DWD are both supported by NINDS Tau Center without Walls Program (U54-NS100693) and NIH (UG3-NS104095). OAR is supported by NIH (P50-NS072187; R01- NS078086; U54-NS100693; U54- NS110435), DOD (W81XWH-17-1-0249), The American Brain Foundation, The Michael J. Fox Foundation, The Little Family Foundation, Ted Turner and family, the Mayo Clinic Foundation, and the Center for Individualized Medicine. DWD receives research support from the NIH (P50-AG016574; P30-AG062677; U54-NS100693; P01-AG003949), CurePSP, the Tau Consortium, and the Robert E. Jacoby Professorship. SK is supported by the State of Florida Ed and Ethel Moore Alzheimer’s Disease Research Program and Mayo Clinic ADRC Research Grant. ZKW is partially supported by the NIH/NIA and NIH/NINDS (1U19AG063911, FAIN: U19AG063911), Mayo Clinic Center for Regenerative Medicine, the gifts from the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and The Albertson Parkinson's Research Foundation. He serves as PI or Co-PI on Biohaven Pharmaceuticals, Inc. (BHV4157-206) and Vigil Neuroscience, Inc. (VGL101-01.002, VGL101-01.201, PET tracer development protocol, Csf1r biomarker and repository project, and ultra-high field MRI in the diagnosis and management of CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia) projects/grants. He serves as Co-PI of the Mayo Clinic APDA Center for Advanced Research and as an external advisory board member for the Vigil Neuroscience, Inc., and as a consultant on neurodegenerative medical research for Eli Lilli & Company. WS is supported by NIH [U54 NS110435, R01 NS085070, R01 NS110085, and R56 AG062556], the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) [W81XWH-17-1-0248], the Michael J. Fox Foundation for Parkinson’s Research (MJFF), the Ted Nash Long Life Foundation, Mayo Clinic Foundation, the Center for Biomedical Discovery (CBD), and the Robert and Arlene Kogod Center on Aging. XH is supported by a pilot grant and a developmental project award from the Mayo Clinic Alzheimer Disease Research Center (ADRC, P30 AG062677) and fellowships awarded by the APDA and Alzheimer’s Association [AARF-22–973152]. FCF is the recipient of fellowships from the Younkin Scholar Program and the APDA and is supported in part by the Florida Department of Health—Ed and Ethel Moore Alzheimer’s Disease Research Program [22A07], the MJFF, a Gerstner Family Career Development Award from the Center for Individualized Medicine (CIM) and an auxiliary award from the CBD at Mayo Clinic. VJL serves as consultant for Bayer Schering Pharma, Philips Molecular Imaging, Piramal Imaging, AVID Radiopharmaceuticals, Eisai Inc., Eli Lilly, and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (NIA, NCI), and the MN Partnership for Biotechnology and Medical Genomics. TJF is supported by NIH (P30-AG062677, U01-NS100620, U19-AG071754) and the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy body dementia program. The funding organizations and sponsors had no role in any of the following: design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
RLW performed genotyping and quality control assessments on all samples, and assisted in drafting the manuscript. SK, KK, and DWD provided brain tissue samples for all cases and provided manuscript improvements. DWD also performed all neuropathological assessments of LBD cases. AIB, TG, MEM, XH, FCF, WS, RJU, JAF, HB, VKR, KK, VJL, CRJ, NE, RS, JG, RCP, JEP, RRR, NRG, TJF, BFB, and ZKW provided manuscript improvements. LJW performed the statistical analysis. OAR led the study, oversaw all methodological developments, and approved the final manuscript. MGH performed the statistical analysis and drafted the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
All authors declare that they have no competing interests.
Ethical approval
This study was approved by the Mayo Clinic Institutional Review Board. All subjects or legal next of kin provided written informed consent.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Walton, R.L., Koga, S., Beasley, A.I. et al. Role of GBA variants in Lewy body disease neuropathology. Acta Neuropathol 147, 54 (2024). https://doi.org/10.1007/s00401-024-02699-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02699-w