
Abstract
Germ cell tumors constitute a heterogeneous group that displays a broad spectrum of morphology. They often arise in testes; however, extragonadal occurrence, in particular brain, is not uncommon, and whether they share a common pathogenesis is unknown. We performed whole exome sequencing in 41 pairs of central nervous system germ cell tumors (CNS GCTs) of various histology and their matched normal tissues. We then performed targeted sequencing of 41 selected genes in a total of 124 CNS GCTs, 65 testicular germ cell tumors (tGCTs) and 8 metastatic GCTs to the CNS. The results showed that mutually exclusive mutations of genes involved in the MAPK pathway were most common (48.4 %), typically in KIT (27.4 %), followed by those in the PI3K pathway (12.9 %), particularly in MTOR (6.5 %), among the 124 CNS GCTs. Pure germinomas and non-germinomatous germ cell tumors (NGGCTs), as well as CNS and testicular GCTs, showed similar mutational profiles, suggesting that GCTs share a common molecular pathogenesis. Mutated MTOR identified in CNS GCTs upregulated phosphorylation of the AKT pathway proteins including AKT and 4EBP1 in nutrient-deprived conditions and enhanced soft-agar colony formation; both events were suppressed in a dose-dependent manner by addition of the MTOR inhibitor pp242. Our findings indicate that the dominant genetic drivers of GCTs regardless of the site of origin are activation of the MAPK and/or PI3K pathways by somatic point mutations. Mutated MTOR represents a potential target for novel targeted therapies for refractory GCTs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Central nervous system germ cell tumors (CNS GCTs) predominantly arise in children and adolescents with a strong male preponderance [11]. CNS GCTs are rare in Western countries, accounting for only 3.7 % of all primary brain and CNS tumors under the age of 14 in the United States [14]. However, they are the second most common pediatric brain tumors in Japan, making up nearly 16.9 % of all primary brain tumors under the age of 19 [6]. It has been reported that some rare germline variants in JMJD1C, a histone H3K9 demethylase, are enriched among Japanese CNS GCT patients as compared to a non-Japanese cohort, suggesting the existence of a genetic predisposition [24]. However, the result needs to be validated in a much larger cohort.
CNS GCTs, like other GCTs, constitute a very heterogeneous group of tumors that displays a broad spectrum of morphology. The WHO classification recognizes several subtypes of CNS GCTs, e.g., germinoma, teratoma, yolk sac tumor, embryonal carcinoma and choriocarcinoma. Germinomas commonly appear as a single histology, referred to as pure germinomas, while non-germinomatous germ cell tumors (NGGCTs) often manifest as a mixture of two or more subtypes which may include germinoma [10]. It is well documented that CNS GCTs may change histological subtype at recurrence. The mechanism underlying such a dynamic histological diversity, both spatially and temporally, within the same tumor tissue is unknown. Primordial germ cells (PGC) have been suggested as a likely candidate for the cellular origin of GCTs in general; however, the exact evolutional mechanism of how each subtype of CNS GCTs develops is still a matter of debate [13, 20, 22].
CNS GCTs generally respond well to combined chemotherapy and radiotherapy. The prognosis of pure germinoma patients is generally good, their 10-year overall survival rate being over 90 % [1, 11]. However, non-germinomatous GCTs (NGGCTs) and approximately 10 % of pure germinomas are resistant to therapy [11]. Patients with GCTs consisting mainly of malignant components, i.e., yolk sac tumor, embryonal carcinoma or choriocarcinoma, have a particularly poor prognosis, the 5-year survival rate being below 10 % [11]. To identify a novel therapeutic target for therapy-refractory CNS GCTs is critical to further improve patients’ outcome.
We have previously reported in our preliminary study that mutually exclusive mutations of KIT/RAS are found in 60 % of pure germinomas; they are, however, rare among NGGCTs, suggesting that there may be some genetic factors that determine the phenotypical diversity of GCTs [3]. A recent investigation using whole exome sequencing confirmed that mutations of KIT or RAS are the most frequent event in CNS GCTs, as well as identified several other genes including CBL, BCORL1 and MTOR as mutated in a subset of these tumors [24]. However, the number of cases examined was rather small, and the incidence and impact of these mutations remained to be explored. The limitation inherent to the genome analysis of CNS GCTs is largely attributable to the scarcity of specimen, due in part to their low incidence except in East Asia and the preference for a non-surgical approach to the treatment of CNS GCTs applied in many countries. In addition, the surgical specimens, if at all, are most often small biopsy, which may not be sufficient for elaborate biological analyses.
In this study, we collected a large number of fresh-frozen surgical specimens for CNS GCTs of diverse histological subtypes and analyzed them by combined whole exome and targeted sequencing. Testicular GCTs (tGCTs) were also analyzed to investigate whether GCTs arising at different locations share the same pathogenesis. Our results showed that genes involved in the MAPK or the PI3K pathways are mutated in more than half of all GCTs regardless of location or histology, indicating that these genetic abnormalities are the main genetic drivers of GCTs.
Materials and methods
Tumor samples and clinical information
All tumor samples and patients’ clinical information were collected through The Intracranial Germ Cell Tumor Genome Analysis Consortium in Japan (The iGCT Consortium, Supplementary Information). Fresh surgical tumor specimens were stored at −80 °C until DNA/RNA extraction. A total of 124 CNS GCT from 123 patients were collected. GCT112 initially arose as a teratoma and recurred as a germinoma after radio-chemotherapy (GCT103). All 124 CNS GCT underwent a central pathology review by a single pathologist (YN) according to the WHO Classification 2007 [16]. As a result, a total of 197 GCTs from 196 patients including 124 CNS GCTs (70 pure germinomas and 54 NGGCTs), 65 tGCTs (39 seminomas and 26 non-germinomatous GCTs) and 8 metastatic GCTs in the CNS from 13 centers participating in The iGCT Consortium were entered in the study (Supplementary Tables S1–S4). Matched blood from 68 primary CNS GCTs as well as 3 matched and 2 unrelated non-cancerous testicular tissues were also included in the analysis.
DNA extraction
Genomic DNA from tumor tissue was extracted using a DNeasy Blood & Tissue kit (Qiagen, Tokyo, Japan) from all 197 tumor specimen (194 frozen tissues and 3 formalin-fixed paraffin-embedded (FFPE) tissues). Genomic DNA was extracted from 68 blood samples from primary CNS GCT patients using FlexiGene DNA Kit (Qiagen, Tokyo, Japan). Extracted DNA was quality controlled using a spectrophotometer (NanoDrop 1000; Thermo Scientific, Delaware, DE) and quantified using a fluorometer (Qubit® 2.0; Life Technologies, Tokyo, Japan).
Whole exome sequencing
We selected 41 CNS GCTs for which sufficient materials for tumor and paired blood DNA were available for whole exome sequencing (WES). WES was performed essentially as described elsewhere [23]. The details of library preparation, exon capture, Illumina GAIIx or HiSeq sequencing, SNV/indel calling, estimation of significantly mutated genes (SMG) and gene set enrichment analysis (GSEA) are described in Supplementary Information.
Ion Torrent sequencing
We selected 41 candidate genes generally based on the following criteria: presence of either recurrent (more than 2 tumors) non-synonymous somatic single nucleotide mutations among the 41 exome-sequenced tumors, low-frequency (1–2) mutation with q-value below 0.05 by SMG (Supplementary Table S8) or mutation in a single case whether the gene is involved either in the MAPK or PI3K pathway, the most frequently targeted pathways identified by GSEA (Supplementary Table S7). For these genes, exon-by-exon targeted sequencing was performed using an Ion Torrent system (Life Technologies Japan Ltd., Tokyo, Japan). Design of the Ion AmpliSeq primers, Ion Torrent sequencing and the mutation call are described in the Supplementary Information. Selected mutations detected by either WES or Ion Torrent sequencing were confirmed by Sanger sequencing performed as previously described [3].
Antibodies and reagents
Antibodies against Akt, phospho-Akt (Ser472), p44/42 MAPK (ERK1/2), phospho-ERK1/2 (Thr202/Tyr204), GSK-3β, phospho-GSK-3β (Ser9), phospho-4E-BP1 (Thr37/46) and phosho-p70 S6 Kinase (p70S6K, Thr389) were purchased from Cell Signaling Technology. Antibodies against GAPDH, p70 S6 Kinase, and 4E-BP1 were purchased from Santa Cruz Biotechnology. Other antibodies used were mTOR (N′) (Abcam, Cambridge, UK) and Halo-tag (Promega, Tokyo, Japan). The chemical inhibitor pp242 was purchased from Calbiochem (Merck Millipore, Darmstadt, Germany).
Generation of MTOR constructs
A Halotag ORF Clone pFN21AB1123 (Cat.# FHC01207, Promega, Tokyo, Japan) was used as wild-type full-length MTOR cDNA. Each of three selected MTOR mutations (L1453P, M2327I and L2334V, see “Results and discussion”) was introduced using a modified site-directed mutagenesis technique [25]. The inserts were sequence-verified and transferred to a pFN28K HaloTag CMV-neo vector to generate wild-type or mutant MTOR constructs for transfection.
Cell culture and transfection
HEK293T human embryonic kidney cells or YT-nu neuroblastoma cells were maintained in RPMI 1640 medium (ThermoFisher Scientific, Tokyo, Japan) supplemented with 10 % fetal bovine serum (FBS, ThermoFisher Scientific, Tokyo, Japan), 100 units/mL penicillin and 100 μg/mL streptomycin (PS, Nacalai Tesque, Kyoto, Japan) at 37 °C in a humidified atmosphere containing 5 % CO2. For serum deprivation, the cells were washed twice with PBS and cultured with RPMI and PS without FBS for 24 h. For nutrient deprivation, the cells were washed twice with D-PBS(−) (Nacalai Tesque, Kyoto, Japan) and the medium was replaced with D-PBS(−) for 5 min.
The cDNAs encoding MTOR mutant genes were transfected into HEK293T or YT-nu cells by electroporation using the NEON system (Thermo Fisher Scientific, Tokyo, Japan). The conditions used for the NEON system are as follows: voltage, 1100 V; pulse width, 30 mA; pulse number, 1.
Immunoblotting
Cells were washed twice with ice-cold PBS and lysed in lysis buffer (0.5 M Tris–HCl [pH 6.8], 10 % glycerol, 2 % sodium dodecyl sulfate [SDS]) and sonicated. The protein concentration of the cell lysates was determined using the BCA protein assay reagents (Thermo Fisher Scientific, Tokyo, Japan). The samples were resolved by SDS-PAGE and transferred to PVDF membranes (Merck Millipore, Darmstadt, Germany). The membranes were blocked with Blocking One reagent (Nakalai Tesque, Kyoto, Japan) for 30 min at room temperature (RT) and incubated with primary antibodies for 2 h at RT followed by incubation with HRP-conjugated secondary antibodies (GE healthcare Japan, Tokyo, Japan) for 30 min at RT. The immunoreactive bands on the membrane were visualized using ECL Plus Western Blotting Substrate (GE Healthcare Japan, Tokyo, Japan) on X-ray films. The bands were quantified using Image J software, and relative expression levels of the phosphoproteins for each MTOR mutant were calculated as a ratio of phosphoprotein level/total protein level.
Cell migration assay
HEK293T or YT-nu cells transiently transfected with empty vector (Vec), wild-type mTOR (WT), mutant mTOR L1453P (M1), M2327I (M2) or L2334V (M3) (1 × 104 cells) were seeded onto the upper part of the transwell insert (BD Japan, Tokyo, Japan), the lower surface of which was coated with fibronectin (Sigma-Aldrich, St. Louis, MO, USA). After 7 h of culture with either pp242 (200 nM, 1 or 10 μM) or DMSO (vehicle), the migrated cells on the lower surface of the filter were fixed and stained with Giemsa’s stain solution (Merck Millipore, Darmstadt, Germany). The filter was mounted on a glass slide, and the total number of migrated cells was counted under bright field using a BX51 microscope (Olympus, Tokyo, Japan).
Data for all the quantitative experiments are described as mean and standard deviation from three independent experiments. Graphing and testing of difference of means by Student’s t test were performed using Microsoft Excel 2007 software. p values of <0.01 were considered as statistically significant.
Soft-agar colony assay
Six-well tissue culture plates were coated with a layer of RPMI 1640-PS containing 0.5 % ultrapure agarose (Thermo Fisher Scientific, Tokyo, Japan). HEK293T cells were transiently transfected with empty vector (Vec), wild-type mTOR (WT), mutant mTOR L1453P (M1), M2327I (M2) or L2334V (M3) for 24 h followed by additional treatment with either pp242 (10 μM) or DMSO (vehicle) for 3 h. The cells were trypsinized and resuspended in RPMI 1640-PS at 5 × 103 cells/ml. A 500-μl cell suspension was then added to 1 ml of RPMI 1640-PS containing 0.5 % ultrapure agarose (final concentration, 0.33 %). The cells were plated on the coated culture plates, allowed to solidify and then placed in a 37 °C incubator with continuous treatment by pp242 (10 μM) or DMSO. After 14 days, phase-contrast images of colonies were visualized under an IX81 microscope (Olympus, Tokyo, Japan). The numbers of total colonies or colonies with diameter more than 100 um were counted in 20 random fields.
Survival analysis
Clinical information including the site of occurrence, tumor markers, treatment, clinical history and the patients’ outcome were collected for all but 2 primary CNS GCT patients (Supplementary Table S1). The median age of onset was 16.0 and male to female ratio 8.5:1 (male 110, female 13). Patient characteristics are summarized in Supplementary Table S4. Only minimal clinical information was available for tGCTs as described in Supplementary Table S2. The study was approved by the Institutional Review Board (IRB) of the National Cancer Center (Protocol No. 2012-043) and the respective local IRBs. All survival analyses were carried out using the JMP 10.0.0 software (SAS Institute Inc., Cary, NC).
Results and discussion
Mutation profiling of GCTs
All 197 GCTs from 196 patients including 124 primary CNS GCTs, 65 primary tGCTs and 8 metastatic germ cell tumors collected through The iGCT Consortium in Japan were subjected to mutation profiling (Supplementary Information, Supplementary Tables S1–S4). Among them, WES was performed on 41 selected CNS GCTs (mean age of onset = 15.8, male/female = 36/5, Supplementary Table S5). The mean coverage of tumor DNA was 123 achieving >20-fold coverage of 87 % of the coding region (89× coverage and 76 % for the matched normal DNA) (Supplementary Table S5). A total of 662 non-synonymous somatic mutations were identified (range 0–286, mean = 16.1/median = 8.0 per tumor) (Supplementary Table S6). One notable case was GCT27, which had an exceptionally high number of mutations (286) compared with all the others (range 0–41, mean = 14.9/median = 7.5 in 40 CNS GCTs excluding GCT27). GCT27 was a treatment-naïve newly diagnosed germinoma at the time of operation and has not recurred after 40 months of clinical follow-up. The mean somatic non-synonymous mutation rate in the coding region was 0.48 per megabase, which was comparable to that of medulloblastoma and treatment-naïve pediatric gliomas [7, 15, 21]. A C>T/G>A substitution was the most common somatic mutation (including synonymous/non-synonymous in the coding/non-coding region) in both pure germinomas and NGGCT (Supplementary Fig. S3a). There was no significant difference in the patterns of nucleotide substitutions between these two groups of GCTs when compared in the 96-pattern sequence context (Supplementary Fig. S3b, c). Tumors in female patients (2 pure germinomas, 1 mature teratoma, 1 mixed GCT, 1 unspecified GCT) had more somatic mutations than that in male patients (p = 0.00071 for all somatic mutations and p = 0.036 for somatic non-synonymous mutations, Wilcoxon test, Supplementary Table S5). The finding needs to be validated in a larger cohort.
A gene set enrichment analysis (GSEA) of mutated genes indicated that the MAPK and PI3K pathways are frequently targeted (Supplementary Table S7). All the remaining GCTs were then studied by targeted sequencing for mutations in the 41 selected genes (Supplementary Information, Supplementary Tables S8–S13). Among the 41 genes examined, NOTCH1 and PLEC were excluded from further analysis because the reading depth was less than 100 for 50 % or more of their amplicons (as compared to the overall mean read depth of 622 for all genes), most likely due to the high GC-content (mean = 68.1 ± 3.9 %) of almost all amplicons in these two genes. SEC16A, FLG and DNAH17 were also excluded from further analysis because a high frequency of non-synonymous sequence variation was observed in normal DNA, which made the significance of mutations in these genes uncertain (Supplementary Table S13). MLL3 was excluded because a potential pseudogene effect that may lead to erroneous identification of mutation has recently been suggested (https://ash.confex.com/ash/2014/webprogram/Paper76200.html). As a result, a total of 150 non-synonymous mutations were identified by Ion Torrent sequencing (Supplementary Table S12). Among the 37 tumors where matched normal DNA was available, the somatic origin of mutations was confirmed. For all other tumors, the somatic origin of the mutations was presumed based on their absence from the common SNPs and from the 37 normal DNAs tested. The targeted sequencing results were combined with the WES data to generate a full mutational profile (Supplementary Tables S14, S15).
MAPK pathway alterations
When all results from WES and Ion Torrent sequencing were compiled, KIT was by far the most frequently mutated gene across all subtypes of CNS GCTs (34/124, 27.4 %, Fig. 1; Supplementary Tables S14, S15). Mutations of KIT are clustered in the juxtamembrane region (11/34 = 32.3 %) and the second tyrosine kinase domain (18/34 = 52.9 %), where a number of mutation hotspots were observed (e.g., W557, D816 and N822, Fig. 2; Supplementary Table S14). These findings are consistent with previous reports [18, 24]. A predominant theory, though yet to be proven, is that GCTs, at least in part, may originate from primordial germ cells (PGC) [20]. Expression of KIT and its ligand KITLG is essential for the maintenance and survival of PGCs during their migration to the gonads [2]. Down-regulation of KITLG induces apoptotic death in PGCs that failed to migrate and are left in the midline [17]. Oncogenic mutations of KIT that may lead to ligand-independent activation would overcome this sequence of events and may allow the survival of mis-migrated PGCs and their aberrant development into a germinoma.

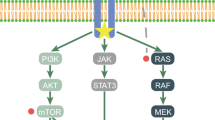
Landscape of somatic mutations in CNS GCTs. a Somatic mutations of 32 genes in 124 CNS GCTs are shown from the combined results of whole exome and targeted sequencing. A complete mutational profile for all 197 CNS and testicular GCTs is shown in Supplementary Fig. S1. Each row represents either a category or a gene, and each column corresponds to an individual tumor. The histological diagnosis has been centrally reviewed and determined according to the WHO classification [10] for all CNS GCTs. GCTs are also classified as germinoma or non-germinomatous germ cell tumors (NGGCT). The availability of matched normal DNA is indicated in the “Matched N” row so that the “true” and “presumed” somatic mutations where the absence of mutation was confirmed in matched normal DNA in the former while not confirmed in the latter are readily discernable. Mutations in genes involved in the MAPK pathway are indicated by filled brown boxes and those in the PI3K pathway by filled dark green boxes. All the remaining individual gene mutations are indicated by filled blue boxes. Indels/truncating mutations or mutations that affect splice consensus sites are indicated as such, while missense mutations are represented by a closed box without text (for detail see Supplementary Table S14). The MAPK or PI3K row indicates cases that have alterations in at least one gene of either pathway. The frequency of mutations in 124 CNS GCTs is indicated on the right-hand side of the panel except for USP28, which was not examined in all tumors (NA, not applicable). KIT protein expression determined by immunohistochemistry (IHC) is indicated where available (KIT IHC). b Schematic diagram of the genes involved in the MAPK (red box) and PI3K (green box) pathways and the frequency of mutations in pure germinomas (Germ, middle line left), NGGCT (middle line right) and all 197 GCTs (bottom line)

Somatic mutations in the MAPK/PI3K genes. The positions of somatic mutations in selected genes are shown by arrow heads. The class of mutation (missense or truncating/indel) as well as the site of the tumors that harbor the mutations (CNS GCT, tGCT or both) are color-coded as indicated. Amino acid changes are shown on top or beside the arrow heads. The number of recurrent mutations is indicated in brackets. Protein domains are indicated as a colored box with an abbreviated domain name below (for more details on each domain see InterPro, http://www.ebi.ac.uk/interpro). Mutations of KIT are clustered in two regions, the juxtamembrane region (exon 11) and the second tyrosine kinase region (exon 17), where a number of mutation hotspots are observed. The majority of CBL mutations (7/10 = 70 %) are located within the RING-type Zinc-finger domain. Mutational hotspots are less evident for MTOR; nonetheless, 50 % (5/10) of mutations are located within the PI3/4 kinase domain
The RAS genes (KRAS, NRAS, HRAS, RRAS2) were collectively second most commonly mutated in this cohort (24/197, 12.2 %). Mutations of KIT and the four RAS genes were mutually exclusive to each other except in two tumors which had both KIT and NRAS mutations (Fig. 1). Mutations of other genes involved in the MAPK pathway including NF1, CBL, FGD6, FGFR2, TRAF6 and F2R were also found (Fig. 1). Among them, CBL is notable as it is one of the few tumor suppressors in this pathway. CBL is a RING finger E3 ubiquitin ligase that negatively regulates activated receptor tyrosine kinases including KIT through ubiquitination and degradation [19]. CBL mutations, which mostly occur within the RING finger domain (Fig. 2), would lead to prolonged RTKs signaling as has been reported in myelodysplastic/myeloproliferative neoplasms [19], thus presumably upregulating the MAPK pathway.
Altogether, alterations in the MAPK pathway were observed in 60 CNS GCTs (48.4 %), mostly in a mutually exclusive manner, being particularly common among germinomas (64.3 %) (Fig. 3a, b; Supplementary Tables S14, S15). These facts consolidate the central role of altered MAPK signaling in the development of CNS GCTs.

Comparisons of mutation profiles in GCTs. a Frequencies of the MAPK and PI3K pathway gene mutations in each histological subtype among CNS GCTs are compared (shown when the number of tumor is 2 or more). Ge germinoma, MT mature teratoma, iMT immature teratoma, Mix(ge) mixed GCT with germinoma, YST yolk sac tumor, GCTnos GCT not otherwise specified. The incidence of mutations in selected genes is compared between either CNS germinoma and NGGCT (b), CNS and testicular GCT (c), CNS germinoma and testicular seminoma (d), testicular seminoma and non-seminoma (e) or CNS GCTs with and without recurrence (f)
PI3K pathway alterations
The PI3K pathway genes including MTOR, PTEN, PIK3C2B and PIK3R2 were the second most common target of alteration in CNS GCTs. Mutations were mutually exclusive except in 2 tumors, resulting in 12.1 % (15/124) of CNS GCTs having one or more alterations of the PI3K pathway. The most frequently mutated gene in this pathway was MTOR (8/124, 6.5 %, Figs. 1, 3b, c; Supplementary Tables S14, S15). MTOR is an atypical serine/threonine kinase that interacts with two distinct protein complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2) [8], each consisting of several proteins including raptor (mTORC1) and rictor (mTORC2). mTORC1 phosphorylates proteins such as 4EBP or S6K and facilitates cell cycle progression while mTORC2 phosphorylates AKT (not mutated in our series) and promotes cell survival, collectively controlling cell proliferation.
Mutated MTOR has been found in a variety of cancers and considered as an oncogene [5, 12]. However, unlike other archetypal oncogenes such as KRAS or IDH1 in which clear mutation hotspots exist, MTOR mutations were rather scattered across the C-terminal half of the protein (Fig. 2). We, therefore, investigated the functional consequences and the possibility of a therapeutic exploitation of three selected MTOR mutations (L1453P, M2327I and L2334V) which have not previously been functionally investigated [4, 12]. When the mutated MTOR constructs were transiently transfected into HEK293T cells and cultured under nutrient-supplemented conditions, phosphorylation of p70S6K, one of many downstream effectors of mTORC1, did not increase compared with the wild-type (wt) MTOR construct. However, when cultured under nutrient-deprived conditions, all three mutant constructs upregulated the phosphorylation of not only p70S6K but also 4EBP1, another known substrate of mTORC1 (Fig. 4a). Moreover, phosphorylation of Akt (Ser473), the main effector of mTORC2, and that of GSK3B, a major substrate of Akt, was also significantly upregulated in these cells at the same time (Fig. 4a). This effect was completely abrogated by addition of pp242, a dual inhibitor of mTORC1 and mTORC2, in a dose-dependent manner (Fig. 4b, Supplementary Fig. S5). Cell migration was enhanced by introduction of mutant MTOR under nutrient-deprived conditions and was also suppressed by pp242, in a dose-dependent manner (Fig. 4c, Supplementary Fig. S5). There was no difference in cell death rate between the cells treated with pp242 (10 μM) and those treated with DMSO (vehicle) for 7 h, indicating that pp242 does not have cellular toxicity at this concentration (Supplementary Fig. S5c). A similar result was obtained using another cell line, the neuroblastoma cell line YT-nu (Supplementary Fig. S6). To further assess the transforming effect of the mutant MTOR, anchorage-independent growth of the mutant MTOR transfectants was investigated using a soft-agar colony formation assay. A significant increase of the number of large colonies (diameter >100 μm) was observed in the mutant MTOR transfectants compared to those expressing the wt-MTOR construct (Fig. 5, Supplementary Fig. S7). The anchorage-independent growth was completely suppressed by addition of pp242 (Fig. 5). These findings indicate that these mutations in MTOR, together with other types of mutation which have previously been investigated [4, 12], are oncogenic and would contribute to GCT development.

Upregulation of mTOR signaling and increased cell migration by mutated MTOR. a HEK293T cells were transiently transfected with empty vector (Vec), wild-type MTOR (WT), mutant MTOR L1453P (M1), M2327I (M2) or L2334V (M3) for 24 h. The cells were further cultured with or without nutrients. The cell lysates were analyzed by immunoblotting using the indicated antibodies (left panel). Plots of relative phosphoprotein levels are shown in the right panel as a mean of three independent experiments. *p < 0.05. b HEK293T cells were transiently transfected with empty vector (Vec), wild-type MTOR (WT), mutant MTOR L1453P (M1), M2327I (M2), or L2334V (M3) for 24 h. The cells were further cultured under serum deprivation for 24 h followed by treatment with mTORC1/mTORC2-dual inhibitor pp242 (10 μM) or DMSO (vehicle) for 3 h. The cell lysates were analyzed by immunoblotting using the indicated antibodies. c HEK293T cells were transiently transfected with empty vector (Vec), wild-type MTOR (WT), mutant MTOR L1453P (M1), M2327I (M2), or L2334V (M3) for 24 h. The cells were further cultured under serum-deprived conditions for 24 h with additional treatment by pp242 (10 μM) or DMSO for 3 h. Migration of the cells was analyzed and quantified as described in Supplementary Information. *p < 0.01

Promotion of anchorage-independent cell growth by mutated MTOR. a HEK293T cells were transiently transfected with empty vector (Vec), wild-type mTOR (WT), mutant mTOR L1453P (M1), M2327I (M2) or L2334V (M3) for 24 h. The cells were further cultured under serum-deprived conditions for 24 h with additional treatment by pp242 (10 μM) or DMSO for 3 h. Anchorage-independent cell growth was analyzed as described in “Materials and methods”. Phase-contrast microscopy images of colonies are shown. Bar 100 μm. *p < 0.01. b Mutant transfectants formed larger colonies compared with the wild-type transfectants (colony size not measured). The colony counts were significantly increased in the mutant MTOR transfectants. These effects were completely abrogated by addition of pp242
Altogether, 68 tumors (54.8 %) had alterations of either the MAPK and/or PI3K pathways, while seven had alterations of both pathways. In these seven tumors, the frequency of mutated alleles was consistently and significantly higher for MAPK mutations than for PI3K mutations (p = 0.001, t test, Supplementary Fig. S4a). In addition, the allele frequency of MAPK mutations, particularly KIT, was significantly higher than any other mutations as observed by pair-wise comparison of each exome-sequenced tumor (p = 0.0001, Supplementary Fig. S4b). Our findings that MAPK alterations are the most clonally dominant mutational event in CNS GCTs strongly suggest that they are the earliest, and therefore, most fundamental and probably causative genetic event in their development.
Other mutations
Among the 124 primary CNS GCTs, 56 tumors (45.2 %) had no mutations in either MAPK or PI3K pathway. Of these, 22 tumors (17.7 %) had mutations in genes belonging to other pathways. These mutations are mostly non-recurrent, except for USP28, which was found mutated in 5 tumors by WES, three of which had neither MAPK nor PI3K mutations (Fig. 1). A total of 34 tumors (27.4 %), including 2 tumors (GCT43, GCT66) investigated by WES, showed no mutations among the genes studied. The molecular mechanisms that operate in these tumors are currently unclear. One possibility is that either MAPK or PI3K pathways may be activated by a mechanism other than point mutations. We have previously reported that all germinomas and more than half of NGGCTs showed KIT protein expression detectable by immunohistochemistry (IHC) regardless of the mutation status of KIT [3]. When the KIT IHC results (available in 73 cases) were compared with the mutation profile, positive KIT expression was observed in the majority of CNS GCTs (59/73, 80.8 %, Fig. 1; Supplementary Table S14), in particular among pure germinomas (35/35, 100 %) and mixed GCTs with germinoma components (15/17, 88.2 %) (Supplementary Fig. S8a). Even among the GCTs with no mutations in the MAPK/PI3K genes, 66.7 % of them also showed KIT expression (Supplementary Fig. S8b). These results strongly suggest that upregulation of the MAPK pathway plays a central role in the majority of CNS GCTs, especially among germinomatous GCTs.
Mutation profiles among subtypes of CNS GCTs
There was surprisingly little difference in the somatic mutational profile between germinomas and NGGCTs (Figs. 1, 3a, b, Supplementary Fig. S1). Excluding GCT27 (see above), there was no significant difference in the number of mutations detected by WES between pure germinomas (mean = 8.7/median = 7.0) and NGGCTs (mean = 9.9/median = 7.5) (p = 0.64, t test, Supplementary Fig. 2a, b). The only notable difference was that KIT mutation was more common in germinoma than in NGGCT (p < 0.001, Fig. 3b), although 6 out of 54 NGGCTs (11.1 %) did harbor a KIT mutation. This is in contrast with the report by Wang et al., in which no KIT mutations were found in NGGCTs [24] (Supplementary Table S15). In our previous study, protein expression of KIT was positive in all pure germinomas as well as in about 50 % of NGGCTs of various histology [3], suggesting that KIT does play a role in all subtypes of GCTs. The frequency of MAPK pathway mutations is thus higher in germinoma than in NGGCT (p < 0.001, Fig. 3b). The incidence of PI3K pathway mutations was similar between the two groups (p = 0.395). Mutations of other genes such as NF1 or MTOR were generally observed in almost all histological subtypes of CNS GCT (Figs. 1, 3a; Supplementary Tables S14, S15). These facts may suggest that germinomas and NGGCTs share at least in part the same pathogenesis, possibly developing from the same cell of origin by acquiring the same types of mutations, thereafter diverging through a mechanism other than genetic alterations such as epigenomic modifications. Further investigation in a larger number of samples is warranted to support this hypothesis.
Mutational profiles of CNS and testicular GCTs
CNS GCTs and tGCTs shared a very similar mutation profile, although the mutation frequencies of KIT (18.5 %), the MAPK pathway (43.1 %) and the PI3K pathway (10.8 %) are slightly lower than that of CNS GCTs (Fig. 3c, Supplementary Fig. S1; Supplementary Table S15). This tendency was replicated when germinoma and seminoma, considered as the histological counterparts of CNS GCT and tGCT, were compared separately (Fig. 3d; Supplementary Table S15). Mutational profiles are similar between seminomas and non-seminomas amongst tGCTs, except for the absence of KIT mutations among non-seminomas (p = 0.002) (Fig. 3e; Supplementary Table S15). This supports the hypothesis that GCTs develop through common genetic mechanisms regardless of the site of origin. Although recurrent somatic mutations in chromatin remodeling genes were not evident in our series, somatic mutations in KDM2A or germline variants of JMJD1C have been reported in CNS GCTs in a similar study [24]. Interestingly, among the 8 CNS metastases of tGCT, none showed mutations of KIT or the PI3K pathway, although the difference with the primary tGCT was not statistically significant (Supplementary Fig. 1; Supplementary Tables S14, S15).
Recently, a whole exome sequencing study on 42 tGCTs was published [9]. When the exome mutation profiles are compared with our current study, few genes are shared between the two cohorts except KIT and KRAS, which were found mutated at a comparable frequency. The inconsistency may be due to the low frequency of the mutations and the relatively small sample size of each study.
Clinical association
The impact of genetic abnormalities on the clinical course of the patients with CNS GCTs was then investigated. Survival data were available for 120 patients out of 123 primary CNS GCTs. A total of 13 patients died, 96 are alive and the fate of 11 patients is unknown. Recurrence was recorded in 15 pure germinomas and 16 NGGCTs (including GCT103 and GCT112, see below). The median follow-up was 73.6 months (0.1–283.0). The median survival of pure germinomas and NGGCTs were 283.0 and 163.2 months, respectively. Pure germinomas had significantly longer overall survival (OS) than NGGCT (p = 0.027, log-rank test, Supplementary Fig. S9a). There was no difference in progression-free survival (PFS) between the two groups (Supplementary Fig. S9b). When the patients’ survival was compared with the mutation status of either each gene or the MAPK or PI3K pathways, no statistically significant difference of survival was found. There was a tendency for longer OS in pure germinomas with MAPK pathway mutations (p = 0.171); however, the tendency was opposite among NGGCTs (Supplementary Fig. S9c, d). There was a tendency for shorter OS and PFS in the tumors with PI3K/MTOR pathway alterations, either among pure germinomas or all tumors, but not in NGGCTs (Supplementary Fig. S9e, f). The impact of MAPK/PI3K pathway mutations on the clinical course of CNS GCT patients remains thus uncertain. This may well be due to non-uniform treatments each patient received in different centers over the years, a limitation inherent to a retrospective study such as this one. An independent investigation in a prospective cohort is required to clarify the role of the genetic aberrations identified in this study as a prognostic marker.
Among the 41 exome-sequenced CNS GCTs, 7 cases experienced recurrence. When GCT27 which had 286 mutations was excluded, the number of somatic mutations detected by WES was marginally higher among the cases that recurred (mean = 17.3) than those which did not (mean = 7.7, p = 0.09, t test). When the mutation profile was compared in all 121 CNS GCTs, of which 31 cases recurred, there was no significant difference in the mutation frequency between the tumors from recurrent cases and the tumors without recurrence (Fig. 3f). Paired tumors from two different time of recurrence were analyzed for one patient. GCT112 was a neurohypophyseal teratoma taken at recurrence of a tumor initially diagnosed as germinoma. This patient eventually developed a metastatic tumor in the spine, which was diagnosed as a germinoma (GCT103), and later succumbed. These two tumors had exactly the same mutation profile among the genes examined, sharing two identical mutations, one in FGD6 (c.3695C>T, p.1232Thr>Ile) and the other in TTLL4 (c.868G>A, p.290Ala>Thr) (Supplementary Tables S1, S14). TTLL4 (Tubulin Tyrosine Ligase-Like Family Member 4) is a polyglutamylase which modifies beta-tubulin and nucleosome assembly proteins NAP1 and NAP2 (Q14679, http://www.uniprot.org). TTLL4 mutations have been found at a low frequency in diverse types of cancers (cancer.sanger.ac.uk/cosmic); however, their significance is currently unclear. Whether genetic mutations play a role in the recurrence of CNS GCTs will remain unanswered until a significant number of paired initial and recurrent tumors are compared. This will, however, prove difficult as CNS GCTs are seldom operated more than once.
Among the CNS GCT that relapsed, two tumors had MTOR mutations. One of them (GCT01), a patient with a yolk sac tumor who died, had a L1453P mutation, which we have shown to respond to the MTOR inhibitor pp242 (Figs. 4, 5, see above). A further eight tumors harbored various KIT mutations. Many of them would potentially be sensitive to one or another kinase inhibitors as suggested [24], in particular a patient with a germinoma that harbored a microdeletion in exon 11 (GCT79, Supplementary Table S14). The targeted therapy of MTOR or KIT could thus stand as a viable option for refractory GCTs.
Here we present the largest molecular study of CNS GCTs to date. Our results as well as other studies [3, 9, 24] indicate that activation of the MAPK and/or PI3K pathways by somatic point mutations is one of the main driving forces of GCT development regardless of histological subtype or site of origin. We showed that the function of activating MTOR mutations may be effectively suppressed by a specific inhibitor suggesting that these mutations can be exploited to treat at least a subset of CNS GCTs that shows resistance to conventional chemotherapy. Our data at the same time demonstrated that a considerable proportion of GCTs have no mutations in the MAPK/PI3K pathway or any of the other genes studied, suggesting that mutations alone may not be sufficient to explain the remarkable diversity and temporal dynamism of CNS GCTs. Our study thus stands as a mile stone in the further elucidation of the molecular pathogenesis of CNS GCTs and the future development of novel targeted therapies.
References
Acharya S, DeWees T, Shinohara ET, Perkins SM (2015) Long-term outcomes and late effects for childhood and young adulthood intracranial germinomas. Neuro Oncol 17:741–746. doi:10.1093/neuonc/nou311
Boldajipour B, Raz E (2007) What is left behind—quality control in germ cell migration. Sci STKE Signal Transduct Knowl Environ 2007:pe16. doi:10.1126/stke.3832007pe16
Fukushima S, Otsuka A, Suzuki T, Yanagisawa T, Mishima K, Mukasa A, Saito N, Kumabe T, Kanamori M, Tominaga T, Narita Y, Shibui S, Kato M, Shibata T, Matsutani M, Nishikawa R, Ichimura K (2014) Mutually exclusive mutations of KIT and RAS are associated with KIT mRNA expression and chromosomal instability in primary intracranial pure germinomas. Acta Neuropathol 127:911–925. doi:10.1007/s00401-014-1247-5
Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, Jordan A, Beck AH, Sabatini DM (2014) A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 4:554–563. doi:10.1158/2159-8290.CD-13-0929
Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer Cell 12:9–22. doi:10.1016/j.ccr.2007.05.008
Japan TCoBTRo (2014) Report of brain tumor registry of Japan (2001–2004). Neurol Med Chir (Tokyo) 54(Suppl):1–102
Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ et al (2012) ICGC PedBrain: dissecting the genomic complexity underlying medulloblastoma. Nature 488:100–105
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. doi:10.1016/j.cell.2012.03.017
Litchfield K, Summersgill B, Yost S, Sultana R, Labreche K, Dudakia D, Renwick A, Seal S, Al-Saadi R, Broderick P, Turner NC, Houlston RS, Huddart R, Shipley J, Turnbull C (2015) Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun 6:5973. doi:10.1038/ncomms6973
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO classification of tumours of the central nervous system, 4th edn. International Agency for Research on Cancer, Lyon
Matsutani M (2004) Clinical management of primary central nervous system germ cell tumors. Semin Oncol 31:676–683
Murugan AK, Alzahrani A, Xing M (2013) Mutations in critical domains confer the human mTOR gene strong tumorigenicity. J Biol Chem 288:6511–6521. doi:10.1074/jbc.M112.399485
Oosterhuis JW, Looijenga LH (2005) Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer 5:210–222. doi:10.1038/nrc1568
Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J (2014) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 16(Suppl 4):iv1–iv63. doi:10.1093/neuonc/nou223
Pugh TJ, Weeraratne SD, Archer TC, Krummel DAP, Auclair D, Bochicchio J, Carneiro MO et al (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488:106–110
Rosenblum MK, Nakazato Y, Matsutani M (2007) Chapter 12. Germ cell tumours. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumours of the central nervous system, 4th edn. International Agency for Research on Cancer, Lyon
Runyan C, Schaible K, Molyneaux K, Wang Z, Levin L, Wylie C (2006) Steel factor controls midline cell death of primordial germ cells and is essential for their normal proliferation and migration. Development 133:4861–4869. doi:10.1242/dev.02688
Sakuma Y, Sakurai S, Oguni S, Satoh M, Hironaka M, Saito K (2004) c-kit gene mutations in intracranial germinomas. Cancer Sci 95:716–720
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N, Onodera M, Nobuyoshi M, Hayashi Y, Harada H, Kurokawa M, Chiba S, Mori H, Ozawa K, Omine M, Hirai H, Nakauchi H, Koeffler HP, Ogawa S (2009) Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460:904–908. doi:10.1038/nature08240
Sano K (1999) Pathogenesis of intracranial germ cell tumors reconsidered. J Neurosurg 90:258–264. doi:10.3171/jns.1999.90.2.0258
Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, de Torres C, Cruz O, Mora J, Entz-Werle N, Ingram WJ, Monje M, Hargrave D, Bullock AN, Puget S, Yip S, Jones C, Grill J (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461. doi:10.1038/ng.2925
Teilum G (1965) Classification of endodermal sinus tumour (mesoblatoma vitellinum) and so-called “embryonal carcinoma” of the ovary. Acta pathologica et microbiologica Scandinavica 64:407–429
Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, Yamamoto S, Shinbrot E, Hama N, Lehmkuhl M, Hosoda F, Arai Y, Walker K, Dahdouli M, Gotoh K, Nagae G, Gingras MC, Muzny DM, Ojima H, Shimada K, Midorikawa Y, Goss JA, Cotton R, Hayashi A, Shibahara J, Ishikawa S, Guiteau J, Tanaka M, Urushidate T, Ohashi S, Okada N, Doddapaneni H, Wang M, Zhu Y, Dinh H, Okusaka T, Kokudo N, Kosuge T, Takayama T, Fukayama M, Gibbs RA, Wheeler DA, Aburatani H, Shibata T (2014) Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 46:1267–1273. doi:10.1038/ng.3126
Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, Nakamura H, He Z, Doddapaneni H, Lewis L, Wang M, Suzuki T, Nishikawa R, Natsume A, Terasaka S, Dauser R, Whitehead W, Adekunle A, Sun J, Qiao Y, Marth G, Muzny DM, Gibbs RA, Leal SM, Wheeler DA, Lau CC (2014) Novel somatic and germline mutations in intracranial germ cell tumours. Nature 511:241–245. doi:10.1038/nature13296
Wang W, Malcolm BA (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 26:680–682
Acknowledgments
The authors thank all clinicians who took care of patients and made a great contribution to this study by providing specimen and clinical information. We thank all participating institutions of the iGCT Consortium for their invaluable support. We thank Tomoko Urushidate, Shoko Ohashi, and Wakako Mukai for excellent technical assistance with next-generation sequencing and data analysis. We thank Dr. Sylvia Kocialkowski for critical reading of the manuscript. This study was carried out as a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct) Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (No. 15cm0106066h0005). SF is an awardee of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research (Japan) for the 3rd Term Comprehensive 10-year Strategy for Cancer Control.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest related to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ichimura, K., Fukushima, S., Totoki, Y. et al. Recurrent neomorphic mutations of MTOR in central nervous system and testicular germ cell tumors may be targeted for therapy. Acta Neuropathol 131, 889–901 (2016). https://doi.org/10.1007/s00401-016-1557-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-016-1557-x