
Abstract
We investigated the distribution patterns of Lewy body-related pathology (LRP) and the effect of coincident Alzheimer disease (AD) pathology using a data-driven clustering approach that identified groups with different LRP pathology distributions without any diagnostic or researcher’s input in two cohorts including: Parkinson disease patients without (PD, n = 141) and with AD (PD-AD, n = 80), dementia with Lewy bodies subjects without AD (DLB, n = 13) and demented subjects with AD and LRP pathology (Dem-AD-LB, n = 308). The Dem-AD-LB group presented two LRP patterns, olfactory-amygdala and limbic LRP with negligible brainstem pathology, that were absent in the PD groups, which are not currently included in the DLB staging system and lacked extracranial LRP as opposed to the PD group. The Dem-AD-LB individuals showed relative preservation of substantia nigra cells and dopamine active transporter in putamen. PD cases with AD pathology showed increased LRP. The cluster with occipital LRP was associated with non-AD type dementia clinical diagnosis in the Dem-AD-LB group and a faster progression to dementia in the PD groups. We found that (1) LRP pathology in Dem-AD-LB shows a distribution that differs from PD, without significant brainstem or extracranial LRP in initial phases; (2) coincident AD pathology is associated with increased LRP in PD indicating an interaction; (3) LRP and coincident AD pathology independently predict progression to dementia in PD, and (4) evaluation of LRP needs to acknowledge different LRP spreading patterns and evaluate substantia nigra integrity in the neuropathological assessment and consider the implications of neuropathological heterogeneity for clinical and biomarker characterization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly, followed by dementia with Lewy bodies (DLB). If dementia precedes or appears within 1 year after the onset of parkinsonian motor signs, the current convention based on consensus criteria is that subjects are diagnosed clinically as DLB [34], while when the diagnosis of dementia is more than 1 year after the onset of motor signs of Parkinson disease (PD), the clinical diagnosis is PD dementia (PDD) [18, 34]. A clear and objective distinction between DLB and PDD, other than the timing of the appearance of cognitive vs. motor impairments, has not been established [1, 2, 32, 45], while a recent publication has even suggested the two entities be merged [5].
AD is diagnosed neuropathologically by the presence of threshold levels of phosphorylated tau deposits in the form of neurofibrillary tangles (NFT) and dystrophic neurites as well as deposits of amyloid beta (Aβ) aggregates in plaques and other forms of Aβ accumulations [36]. Studies suggest that tau and Aβ pathologies appear to spread in a stereotypical manner, i.e., from the medial temporal lobe to the neocortex for NFTs [7] and from the neocortex to the brainstem and cerebellum for Aβ deposits [41].
Braak and colleagues proposed an α-synuclein staging systems for PD, which hypothesizes that α-synuclein in the form of Lewy body and neurite related pathology (LRP) first appears in the enteric nervous system, dorsal motor nucleus of the vagus nerve and the olfactory bulb [9, 10]. Based on this, LRP would mainly progress rostrally along the brainstem, whereas the α-synuclein in the olfactory bulb would stay within its boundaries. However, this initial staging system has not been able to satisfactorily classify a significant number of cases [17, 31, 38, 47], leading to continuing debates about how LRP truly progresses and evolves [2, 29, 30]. This prompted other α-synuclein staging systems to be proposed for LRP [2, 31, 47].
Similarly, diagnostic criteria for DLB classification based exclusively on the subdivision of LRP into brainstem predominant, limbic and diffuse neocortical types or stages have been proposed [34], but this classification missed an important number of cases with amygdala-predominant LRP without involvement of the brainstem as was later recognized [31]. All these conflicting results indicate that there might be different and independent distribution patterns.
We therefore hypothesize that LRP distribution might be related not only to the primary LRP, but also to the presence or absence of coincident AD pathology. We also expect that these different groups differ in clinical phenotypes, integrity of the nigrostriatal pathway and prognosis. To identify LRP distribution patterns, we applied an unsupervised data-driven approach, hierarchical clustering, in each cohort that included all patients and was not influenced by clinical or neuropathological diagnosis, as diagnostic grouping was not used in the clustering approach. Hierarchical clustering sequentially grouped the subjects with the most similar characteristics (in our study neuropathological scores in the different studied regions) and identified clusters that represent different pathology deposition patterns during this agglomerative approach.
Subjects included in the study belonged to four different clinico-pathological groups: (1) dementia subjects (excluding PDD) who met neuropathological criteria for AD and had LRP that met criteria for DLB or did not have enough LRP for DLB (Dem-AD-LB group); (2) PD subjects with or without dementia who did not meet neuropathological criteria for AD (PD group); (3) PD subjects with or without dementia who met neuropathological criteria for AD (PD-AD group); 4) DLB subjects who did not meet neuropathological criteria for a concurrent AD diagnosis (DLB group).
Materials and methods
Subjects and pathological assessments
Autopsy cases were selected from the Center of Neurodegenerative Disease Research (CNDR) at the University of Pennsylvania [44] and the Arizona Study of Aging and Neurodegenerative Disorders, part of the Banner Sun Health Research Institute Brain and Body Donation Program (BBDP) [3, 4]. The protocols for brain harvesting, selection of areas to obtain tissue blocks (Supplementary Table 1) and staining and diagnostic procedures followed have been previously reported in detail [3, 4, 44]. The center-specific histochemical approaches are summarized in supplementary Table 2.
Cases with a complete neuropathological study, including α-synuclein immunohistochemistry (IHC), based on the protocols of each of the centers, were selected for the study. Four groups of subjects were included in the study: (1) dementia subjects (excluding PDD) who met neuropathological criteria for AD [13] and had LRP that met criteria for DLB or did not have enough LRP for DLB (Dem-AD-LB group) [34]; (2) PD subjects with or without dementia [15, 18, 25] who did not meet neuropathological criteria for AD (PD group); (3) PD subjects with or without dementia [15, 18, 25] who met neuropathological criteria for AD (PD-AD group); (4) DLB subjects [16, 34] who did not meet neuropathological criteria for a concurrent AD diagnosis (DLB group) (Table 1).
A neuropathological diagnosis of AD was established based on the presence of plaques and tangles using the National Institute of Aging Reagan criteria [13, 27]. We classified subjects as having AD neuropathological diagnosis based on combined results from the CERAD scoring system for neocortical neuritic plaque density (B or C) [35] and a distribution of NFT that corresponded to a Braak NFT stage of III–VI [7, 8], consistent with an intermediate or high probability of dementia being due to AD [13]. Due to differences in staining techniques applied in the CNDR (PHF-1) and the BBDP (Gallias) cohort (Supplementary Table 2) different versions of the Braak staging criteria were applied [7, 8]. Both staining approaches are still recognized by the most current AD neuropathological diagnostic criteria [26, 36]. Current AD neuropathological AD diagnostic criteria [26, 36] were not used due to the retrospective nature and large number of cases included in the study.
The distinction between PDD and DLB was made based on the clinical presentation [20, 32, 34]; if dementia preceded or appeared within 1 year after the onset of the parkinsonian motor signs, the current convention based on consensus criteria is that subjects are diagnosed clinically as DLB while when the onset of dementia is more than 1 year after the onset of motor signs of PD, the clinical diagnosis is PDD.
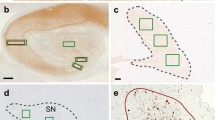
Substantia nigra (SN) slides were scanned using the Aperio Leica Biosystems microscope and nigra pigmented cell density was counted manually using 2-mm-wide boxes centered in the SN in transversal midbrain sections at the level of the red nucleus (Supplementary Figure 1). For dopamine active transporter (DAT) IHC, formalin-fixed paraffin-embedded sections of the post-commissural putamen were pretreated with citrate-based antigen retrieval unmasking solution. The primary antibody, rat anti-dopamine active transporter monoclonal antibody (EMD Millipore, MAB369, rat IgG2A kappa), was then added to slides at a 1:500 dilution in the CNDR cohort. Slides were scanned using the Aperio Leica Biosystems microscope and putamen percent-area DAT IHC staining was quantified using HALO software. In cases with severe dopaminergic denervation of the putamen that prevented the delineation of the total putaminal area in the DAT stained slide, we used slides stained with Syn303 to quantify the total area of the putamen and assess that the putamen was present.
Statistical analysis
For the comparison of the groups in the demographic table, ANOVA, Kruskall–Wallis and Fisher exact tests were applied to analyze differences between quantitative normally and non-normally distributed variables and quantitative variables, respectively. For the comparison of the pathological scores, a semi-quantitative ordinal grading scale (the Bruner, Dette Munk test) was applied to handle tied values (which are not the case of the Kruskall–Wallis test). The p values were adjusted for multiple comparisons using the Bonferroni correction for the non-hypothesis-based comparisons. For the analysis of categorical variables, Fisher exact test and multinomial logistic regression were applied.
A hierarchical clustering method was used to identify different patterns of α-synuclein distribution including all the subjects in each cohort together. This approach successively groups subjects based on the similarity of the values across the studied variables (here LRP scores in studied areas) which can be seen in the dendrograms (Supplementary Figures 2–4). In each step differences (the dissimilarity) between all subjects is calculated and then the subjects/groups showing the smallest difference/dissimilarity are merged. Number of clusters was selected based on the presence of a “knee” in the Hubert index and a peak in the Hubert index and D-index second differences plots to detect groups of subjects that represent common patterns of LRP deposition based on two large cohorts that represent the whole spectrum of LRP. Two-tailed p values below 0.05 were considered statistically significant.
Results
Subjects
A total of 308 Dem-AD-LB, 141 PD, 80 PD-AD and 13 DLB cases were included in the study (Table 1). Dem-AD-LB presented the lowest percentage of male subjects and highest percentage of APOE ε4 carriers. When we evaluated the last clinical diagnosis of the studied subjects, only four PD cases were clinically misdiagnosed. On the other hand most of the Dem-AD-LB subjects were clinically diagnosed as AD dementia.
Unsupervised clustering of α-synuclein pathology in the CNDR cohort
For the initial clustering in the CNDR cohort, we included two limbic (amygdala and cingulate cortex), two subcortical (putamen and thalamus), two brainstem (medulla and SN), a combined medial temporal score (hippocampus and entorhinal cortex) and two neocortical areas (frontal and angular cortex) to get a balanced representation of different systems. The clustering analysis identified a four-cluster solution in the CNDR cohort (Fig. 1 and Supplementary Figure 2): limbic transitional cluster 1, neocortical cluster 2, amygdala cluster 3 and limbic predominant cluster 4.

CNDR clusters heatmaps (top) and brain maps showing LRP pathology distribution in the clusters (bottom). First colored column on the left of the heatmaps represents cluster identity and second column represents clinico-pathological diagnosis with LRP-only groups in green colors (PD and DLB) and groups with coincident LRP and AD in magenta colors (AD-LB and PD-AD). Color scale of the brain maps below represents the mean semi-quantitative scores in each cluster
The clinico-pathologically defined groups were unevenly distributed among the clusters (p < 0.0001) (Table 3): the amygdala and limbic predominant clusters 3 and 4 were only present in the Dem-AD-LB group, whereas the limbic transitional and the neocortical clusters 1 and 2 were present in all the clinico-pathologically defined groups.
The amygdala cluster 3 showed the lowest burden of pathology and was characterized by LRP in the amygdala. Limbic LRP was shared by the limbic transitional and limbic predominant clusters 1 and 4, although both clusters showed significant differences (Table 3). LRP was absent in the SN and very infrequent in thalamus, medulla and putamen in the limbic-predominant cluster 4, which only consisted of Dem-AD-LB cases, whereas the aforementioned areas showed a moderate-to-severe pathology burden in the limbic transitional cluster 1. Finally, the neocortical cluster 2 showed the highest burden of LRP, extending to frontal and parietal cortices.
The neocortical cluster 2 was more frequent in the PD-AD than the PD group (OR = 1.7, p = 0.004), whereas all DLB cases were included in the neocortical cluster 2, which was also the largest, encompassing 48.3 % of the subjects. We therefore performed an additional clustering analysis, excluding areas that showed no variance in this cluster and including an additional neocortical area which is affected in later stages (occipital cortex).
With this approach, we found three new clusters (Fig. 1 and Supplementary Figure 3) with frequencies that differed across the three clinico-pathological groups (p < 0.0001) (Table 2). The neocortical occipital cluster 2B was mainly present in the Dem-AD-LB group and PD-AD groups and the main differences when compared with the low burden neocortical cluster 2A and moderate burden neocortical cluster 2C was the burden of LRP pathology in the occipital cortex (Table 3). The low burden neocortical showed lower burden of pathology in all neocortical areas and the cingulate cortex compared to the other two neocortical clusters. Again, the PD group was most frequently in the cluster with the lowest amount of pathology (low burden neocortical cluster 2A) and the PD-AD group was associated with a higher odds of being include in the occipital cluster 2B (OR = 3.2, p = 0.002) and moderate burden neocortical cluster 2C (OR = 2.8, p = 0.005).
When we tested if APOE ε4 carrier status was associated with any specific cluster, we discovered that in Dem-AD-LB subjects it increased the odds of being classified in cluster 2B (adjusting for NFT Braak stage). Similarly APOE ε4 carrier status was associated with increased odds of being classified in clusters 2B and 2C in the PD and PD-AD groups (adjusting for coincident AD diagnosis). The CERAD score and NFT Braak stages for the different clusters are summarized in supplementary Table 3.
Unsupervised clustering of α-synuclein pathology in the BBDP cohort
Olfactory bulb sections were available for 121 Dem-AD-LB, 63 PD-AD, 5 DLB and 44 PD subjects in the BBDP cohort. Because of differences in sampling criteria (Supplementary Table 1), there was no scoring available for lentiform, thalamus and hippocampus. Five clusters were found in the BBDP cohort (Fig. 2 and Supplementary Figure 3) with different frequencies across the different clinico-pathological groups (p < 0.0001, Table 2). The clusters were generally similar to those in the CNDR analysis (Supplementary Table 5). Again two clusters were only present in the Dem-AD-LB group (clusters 3 and 4). The olfactory bulb cluster 4 presented the lowest burden of pathology. The limbic predominant cluster 3 presented greater LRP pathology in the amygdala and transentorhinal cortex compared to the olfactory bulb cluster 4 (Table 4) and, like the olfactory bulb cluster, lacked pathology in the SN and showed similarly low LRP pathology in the dorsal motor nucleus of the vagus. The limbic transitional cluster 5 had a higher burden of pathology in the brainstem and cingulate cortex compared to limbic predominant cluster 3. The moderate burden neocortical cluster 1 had an overall higher burden of LRP pathology in most of the studied regions compared to previously described clusters, but was mainly characterized by greater neocortical LRP pathology.

BBDP clusters heatmaps (top) and brain maps showing LRP pathology distribution in the clusters (bottom). First colored column on the left of the heatmaps represents cluster identity and second column represents clinico-pathological diagnosis with LRP-only groups in green colors (PD and DLB) and groups with coincident LRP and AD in magenta colors (AD-LB and PD-AD). Color scale of the brain maps below represents the mean semi-quantitative scores in each cluster
PD-AD cases had higher odds of being classified into the moderate neocortical cluster 1 (OR = 3.68, p = 0.002) and the low burden neocortical cluster 2 (OR = 15.4, p = 0.001) vs. the limbic transitional cluster 5 (lower burden of LRP) compared to PD cases (as in the CNDR cohort).
Extracranial and nigrostriatal α-synuclein assessment
There were 80 PD subjects of the BBDP cohort (30 PD, 1 DLB, 14 PD-AD and 35 Dem-AD-LB) in which α-synuclein pathology was evaluated in the lumbar spinal cord, esophagus, submandibular gland or vagus nerve. Dem-AD-LB subjects showed the lowest presence of α-synuclein pathology in the four regions compared to the PD and PD-AD groups (Table 5; Fig. 3), whereas the latter groups showed no differences. For further comparisons we grouped the PD and PD-AD categories together due to their similarity and limited sample size. When compared to the lowest burden LRP PD/PD-AD cluster (limbic transitional cluster 5), the two BBDP Dem-AD-LB specific clusters (the olfactory bulb cluster 3 and the limbic predominant cluster 4) showed a lower burden of LRP pathology in all extracranial areas. On the other hand, Dem-AD-LB low and moderate neocortical burden clusters (clusters 1 and 2) showed a higher frequency of extracranial LRP pathology than Dem-AD-LB olfactory bulb cluster 3 and limbic predominant cluster 4. Finally, when comparing the PD/PD-AD and Dem-AD-LB clusters with highest LRP pathology (clusters 1 and 2), the esophagus and submandibular gland still showed lower LRP frequency in the Dem-AD-LB group.

LRP presence in the esophagus, lumbar spinal cord, submandibular gland and vagus nerve. Bar plots represent percentage of cases with LRP based on neuropathological and cluster groups. Measurements were performed in the BBDP cohort
In the CNDR cohort, the PD-AD (n = 10, p = 0 < 0.0001), DLB (n = 4, p = 0.003) and PD (n = 14, p = <0.0001) groups showed a lower SN pigmented neuron density compared to the Dem-AD-LB (n = 35) group, whereas none of the statistical clusters showed any associations with SN pigmented neuron density (Fig. 4). When compared to the Dem-AD-LB group (n = 29), the PD (n = 11, p < 0.0001) and PD-AD (n = 6, p < 0.0001) groups showed a lower percentage of DAT IHC positive area, whereas the unremarkable brain subjects (UB) showed a higher percentage of DAT IHC positive area (n = 6, p = 0.030) (Fig. 4 and Supplementary Figure 5). When clusters were compared within the Dem-AD-LB group, the limbic transitional cluster 1 (n = 9, p = 0.011), the olfactory bulb cluster 3 (n = 8, p = 0.0002) and the limbic predominant cluster 4 (n = 4, p = 0.0001) showed a higher percentage of DAT IHC positive area than the neocortical cluster 2 (n = 8). When evaluating clinical associations within the Dem-AD-LB group, we found that those with a DLB clinical diagnosis (n = 4) showed a lower nigra cell count than those with and AD (n = 22) clinical diagnosis (p = 0.010).

SN cell density (first row) and percentage of DAT IHC area in the putamen (second row). Measurements were performed in the CNDR cohort
Clinical differences between clusters in the CNDR cohort
There was a large variability in the clinical diagnoses in the CNDR Dem-AD-LB group that differed across the distinct clusters defined here (Supplementary Table 4). When compared to the limbic transitional cluster 1, only the neocortical cluster 2 was associated with a higher frequency of DLB/corticobasal syndrome (CBS) diagnosis (OR = 1.96, p value = 0.050). This association was mainly due to subjects who were further subclassified into the occipital neocortical cluster 2B and the moderate burden neocortical cluster 2C. In the PD and PD-AD groups the neocortical cluster 2 was associated with increased odds of being demented at time of death (OR = 1.6, p = 0.0003), which was mainly driven by the occipital neocortical cluster 2B.
Finally, we evaluated the association of pathology with clinical progression in the CNDR cohort. When all the clinico-pathological groups were studied, PD (OR = 0.29, p = 0.0006) and PD-AD (OR = 0.45, p = 0.003) groups had longer disease duration than the Dem-AD-LB, but there was no association between the clusters and disease duration (Fig. 5). When we evaluated progression to dementia in the PD and PD-AD groups (Dem-AD-LB was excluded due to having dementia present from the onset), we found that the occipital neocortical cluster 2B and the moderate burden neocortical cluster 2C (compared to the limbic transitional cluster 1) and PD-AD clinico-pathological diagnosis predicted an increased progression to dementia (Table 6).

Disease duration based on neuropathological diagnosis (top row left) and cluster classification (top row right). Conversion from PD to PDD based on neuropathological diagnosis (bottom row left) and cluster classification (bottom row right)
Discussion
Using an unsupervised data-driven clustering approach independently in two large cohorts encompassing a wide spectrum of LRP pathology with and without coincident AD pathology, we found that (1) in Dem-AD-LB LRP pathology seems to start in the olfactory bulb progressing to the amygdala and limbic system with overall sparing of the brainstem; (2) in the amygdala and limbic predominant Dem-AD-LB clusters there is a relative sparing of LRP in the spinal cord, enteric nervous system and submandibular gland; (3) PD-AD subjects were more frequently present in clusters with greater LRP burden than PD subjects; (4) the nigrostriatal pathway was preserved in Dem-AD-LB compared to PD and PD-AD subjects in matched clusters; (5) the occipital neocortical cluster 2B presented most frequently as DLB/CBS and was associated with a shorter motor–dementia interval in PD and PD-AD subjects and APOE ε4 carrier status.
Unsupervised LRP clusters point to different spreading patterns
In this study, we identified two LRP distribution clusters that were only present in the Dem-AD-LB cases and not in the PD-AD and PD cases in both cohorts. These clusters mainly overlapped with the amygdala-predominant and olfactory bulb-only stages of LRP. Neither of these stages was originally included in the Braak PD or DLB Consortium staging systems [10, 34].
The consensus DLB diagnostic criteria originally included brainstem, limbic and neocortical stages but not an amygdala predominant or olfactory bulb-only stage of LRP [34]. Later reports included the amygdala-predominant LRP category (amygdala cluster 3 in the CNDR cohort) [31].
Our analysis identified, using a data-driven approach, the importance of pathology originated in the limbic system that appeared in cases with significant burdens of concomitant AD pathology as suggested by recent studies [17, 29, 30, 38, 47]. Amygdala and limbic LRP only appeared in the presence of AD pathology in the same locations. These findings emphasize that the classically used transitional and the more recently defined limbic-predominant stages are not interchangeable and our results, based on data-driven and not a priori based classification, confirm that there may be at least two patterns of LRP spreading into two different limbic stages that are mainly characterized by differential LRP burdens in the brainstem [2]. For cases following the classical PD spreading pattern, by the time there is significant hippocampal and amygdala LRP there is a similar degree of LRP deposition in the cingulate cortex and the basal ganglia. On the other hand, in cases extending from the amygdala to the hippocampus, for a similar LRP burden in the amygdala and adjacent medial temporal regions, there was a more limited burden in the brainstem and cingulate cortex.
Previously, the Unified Staging System for LB disease (USSLB) specifically differentiated between two limbic stages that differed based on the presence (Stage III: brainstem and limbic) or absence or low involvement of the brainstem (Stage IIb: limbic predominant) [2]. Our data-driven findings strongly support this staging system, based on the independent analyses performed in two different cohorts. These analyses describe the presence of two clusters in each cohort that were characterized by a high burden of LRP limited to the olfactory bulb, or olfactory bulb, amygdala and medial temporal lobe, in the absence of or with a minimal burden of brainstem LRP. Interestingly, the clustering approach indicated that most Dem-AD-LB cases without significant neocortical LRP were classified into clusters without any SN LRP in both cohorts (81.0 % of 105 Dem-AD-LB subjects in the CNDR Dem-AD-LB groups and 77.5 % of 71 AD-LB subjects in the BBDP cohort), which is strikingly different from the spreading pattern observed in PD [10, 11] while AD cases with coincident LRP are mainly but not exclusively associated with an olfactory bulb-limbic pattern.
Due to the relative infrequency with which early stage PD cases come to autopsy, there were very few short duration PD cases in either cohorts, and thus brainstem only stage cases were underrepresented in our study. However, several studies have described these cases as having significant brainstem pathology in the absence of limbic and neocortical LRP [10, 15, 17]. When the burden of LRP in the Dem-AD-LB specific limbic predominant clusters (4 in CNDR and 3 in BBDP) was compared to the transitional limbic clusters (1 in CNDR and 5 in BBDP), which was predominantly present in PD and PD-AD groups and less frequent in the Dem-AD-LB group, there were no differences in LRP burden within the amygdala, hippocampus and transentorhinal cortex, whereas the brainstem and limbic clusters showed higher LRP in the cingulate and basal ganglia in addition to the brainstem.
The PD-AD group is associated with increased LRP, APOE ε4 carriers status and is more frequently present in Dem-AD-LB clusters
Notably, our study suggests that the presence of AD pathology and APOE ε4 carriers status are associated with, and hence may modulate, the progression of LRP. Previously, we had shown a correlation between LRP and tau NFT semi-quantitative scores [28]. Our new results show, in two independent cohorts and three different cluster analyses, that PD-AD subjects were more frequently classified into clusters that showed a higher burden of LRP and that the LRP frequency distribution for PD-AD subjects was between the one observed for PD and Dem-AD-LB subjects. This would indicate that the presence of AD pathology may facilitate the spreading of LRP and lead to an overall higher burden of LRP in subjects who present clinically as PD and have coincident AD pathology. Increasing evidence points to the fact that neurodegenerative disease proteins such as tau, Aβ and α-synuclein exist in different strain-like conformations or post-translationally modified species [12, 22, 24, 33, 39], while tau and α-synuclein were shown to cross-fibrillize with one another but not with Aβ [19]. One study described that one strain of α-synuclein (strain A) preformed fibrils (PFFs) could induce only α-synuclein pathology, while a second strain of α-synuclein PFFs (strain B) induced more tau than α-synuclein pathology in cultured neurons and in tau transgenic mice [22]. Thus, the effects seen here might reflect the interactions of different strains of tau and/or Aβ that differentially induce formation of α-synuclein LRP in a region-specific manner or facilitate the spread of α-synuclein along with the spread of tau and Aβ pathology. These preliminary results need to be validated evaluating characteristics of brain extracts of deceased patients representing different groups of the Lewy body disease spectrum and their correlates in different cell and animal disease models. Thus, this is an area requiring further study to understand if strains of AD and PD-associated proteins account for the findings here, but it also is essential to develop reliable biomarkers indicative of the presence of LRP in addition to the existing AD biomarkers which correlate with plaques and tangles [42] to improve efforts for predicting the neurodegenerative pathologies underlying dementia and parkinsonism in living patients.
Extracranial LRP pathology differs across clusters and clinico-pathological diagnoses
Differences between the LRP clusters described here that would represent the olfactory bulb and limbic predominant stages (Dem-AD-LB cluster 3 and 4 in BBDP) and the cluster that would represent the limbic transitional stage in PD and PD-AD (cluster 5) also extended to extracranial areas beyond the brain. The Dem-AD-LB specific clusters (BBDP olfactory bulb cluster 3 and limbic predominant cluster 4) did not show any LRP in the esophagus, lumbar spinal cord and vagus nerve and only a single case showed LRP in the submandibular gland. This was in contrast with the PD limbic transitional cluster 5 which showed LRP in the same extracranial regions in approximately 80 % of the cases. Interestingly, there were some specific differences in the involvement of extracranial regions in the Dem-AD-LB vs. the PD and PD-AD groups even in the clusters that presented more widespread pathology. In the more advanced stages, these groups did not differ in the involvement of the spinal cord or the vagus nerve, but the Dem-AD-LB subjects with neocortical involvement still showed a lower frequency of LRP in the submandibular gland and esophagus, indicating that these areas are affected in later stages in Dem-AD-LB or LRP in these cases have a decreased tropism for these organs. Alternatively, it is possible that in advanced stages the cell loss is accompanied by an absence of LRP, although the finding of lesser involvement in early stages would not favor this hypothesis).
The lack of extracranial pathology in Dem-AD-LB cases is the opposite to what is described in the classical PD pattern of spreading, that would start in the peripheral nervous system around the viscera and brainstem leading to a variety of non-motor systems that precede PD’s motor symptoms [23]. In line with our results, a recent large study described the absence of LRP in the spinal cord in cases with LRP circumscribed to the olfactory bulb and amygdala, whereas all subjects with LRP in the spinal cord had LRP in the brainstem [40], which would descend from the supraspinal medullary reticular formation [14].
Therefore, we propose that in most subjects with a dementia clinical presentation and coincident AD and LB pathology, with either a primary neuropathological diagnosis of AD or DLB, the origin of the pathology is the olfactory bulb and the amygdala and its spread might be facilitated by the AD pathology, as we did not find any cases with vagal, submandibular gland or esophageal LRP in the absence of brain LRP. On the other hand, in cases who present initially with a PD motor phenotype (PD and PD-AD cases), there was a higher frequency of LRP in submandibular gland and esophagus compared to AD-LB, which would indicate a greater tropism for these areas in these groups, but still with an origin in the olfactory bulb and caudal brainstem regions followed by a simultaneous rostral and caudal spread, or alternatively, a centripetal spread from the enteric system periphery as proposed by some groups. However, none of the over 100 PD and control cases in the BBDP cohort had LRP identified in the submandibular gland, esophagus and stomach in the absence of LRP in the central nervous system (unpublished data), which would not favor the latter hypothesis. We summarize these spreading patterns across the clinico-pathological groups in Fig. 6.

LRP spreading patterns across the different clinico-pathological groups
Relative integrity of the nigrostriatal pathway in Dem-AD-LB compared to PD/PD-AD
Another critical finding is the preservation of SN neurons and the nigrostriatal pathway in Dem-AD-LB cases as measured by SN pigmented neuron density counts and DAT immunohistochemistry in the striatum (although subjects with a DLB clinical diagnosis showed significantly lower SN cell density). This explains the lack of motor symptoms in many cases, which makes cases less likely to fit the clinical DLB diagnostic criteria. It has been described that approximately 70 % of DLB subjects have parkinsonism which contrasts with the pathological findings and indicates that either limbic predominant cases or neocortical cases with low degree of SN cell loss have a similar presentation as AD cases or that the clinical presentation has not been adequately characterized. Studies of longitudinally followed subjects with neuropathological, clinical and multi-modal biomarkers will be needed to characterize the heterogeneity of DLB [43]. Furthermore, PET or SPECT scans using ligands to assess the nigrostriatal pathway might not be sensitive biomarkers for LRP in the frequent AD-LB subjects without parkinsonism. Because even some cases with neocortical Lewy body pathology showed a low degree of SN cell loss it would be important to assess SN cell loss as part of the DLB neuropathological evaluation as this might have significant implications for the clinical presentation and characterization. Alternatively, specific FDG PET patterns of hypometabolism might prove to be sensitive and specific biomarkers for coincident LRP in AD [21, 43]. A previous study of the BBDP showed that AD-LB cases had striatal tyrosine hydroxylase (measured using ELISA) in the same range as control subjects as opposed to PD cases, although this study only included two cases in the AD-LB group with moderate or high LRP [2]. The same study showed in a large number of cases a lack of correlation between total LRP and semi-quantitatively rated SN cell loss although several cases presented moderate or severe SN neuronal loss and another study using semi-quantitative rating described lower SN neuron loss in DLB subjects compared to PD subjects [45].
Association of LRP with clinical diagnosis and motor–dementia interval
In addition, we were able to evaluate the clinical associations in the CNDR cohort. Interestingly, the neocortical cluster 2 was associated with increased odds of dementia in the PD and PD-AD cases and with a higher frequency of non-Alzheimer type dementia, mainly driven by combined motor and dementia clinical presentations, namely DLB and CBS. These clinical associations were mainly driven by the occipital neocortical cluster 2B and the moderate burden neocortical cluster 2C. This would indicate that neocortical distributions represented in these two clusters are the ones that show the highest association with the referred clinical phenotypes.
Interestingly, the LRP clusters were not associated with the disease duration and it was the clinico-pathological diagnostic group that predicted disease duration in age, gender and APOE ε4 adjusted models. AD-LB cases showed the shortest disease duration as compared to PD. However, the distribution of LRP pathology was the strongest predictor of the onset of dementia even adjusting for coincident AD pathology (that was also associated with the dementia progression rate). Specifically, cluster 2B was the one with a 1.7 higher HR than coincident AD pathology and cluster 2C (cluster 2C only showing a lower burden of LRP in the occipital lobe). These result`s expand on our previous finding of differences of neocortical LRP burden in faster and slower progressors [28], as we now define specific distributions based on LRP in the whole brain, include the important occipital region, which was not considered preciously, and performed a more continuous assessment of progression using a Cox hazard model.
Previous studies have described olfactory dysfunction in PD, DLB and AD and hyposmia has been associated with the risk of progressing to mild cognitive impairment during follow-up [37, 46]. Hyposmia has been associated with limbic pathology [6]. Therefore, it is possible that hyposmia in cases with AD clinical diagnosis might be related to cases with limbic or diffuse LRP pathology.
Conclusion
Our study identified clusters of LRP in distinct forms of Lewy body disease which indicate that there are at least two LRP distribution patterns showing different frequencies across the Dem-AD-LB, PD and PD-AD groups and that coincident AD pathology may alter the spread of LRP in PD. Although the number of DLB cases without AD pathology was limited, the distribution of LRP and dopaminergic loss in these cases was similar to the one in PD cases. Differences between clusters and clinico-pathological groups extended to extracranial regions and to differences in the integrity of the nigrostriatal pathway which have important clinical implications for the selection of biomarkers. In addition, the cluster-defined groups were associated with different clinical diagnoses in the Dem-AD-LB group and rate of progression to dementia in PD and PD-AD cases.
References
Aarsland D, Ballard CG, Halliday G (2004) Are Parkinson’s disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol 17(3):137–145. doi:10.1177/0891988704267470
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J et al (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117(6):613–634. doi:10.1007/s00401-009-0538-8
Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG et al (2015) Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology 35(4):354–389. doi:10.1111/neup.12189
Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L et al (2008) the sun health research institute brain donation program: description and experience, 1987–2007. Cell Tissue Bank 9(3):229–245. doi:10.1007/s10561-008-9067-2
Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T et al (2014) Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 29(4):454–462. doi:10.1002/mds.25844
Bohnen NI, Muller ML, Kotagal V, Koeppe RA, Kilbourn MA, Albin RL et al (2010) Olfactory dysfunction, central cholinergic integrity and cognitive impairment in Parkinson’s disease. Brain 133(Pt 6):1747–1754. doi:10.1093/brain/awq079
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112(4):389–404. doi:10.1007/s00401-006-0127-z
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Braak H, Del Tredici K (2008) Invited article: nervous system pathology in sporadic Parkinson disease. Neurology 70(20):1916–1925. doi:10.1212/01.wnl.0000312279.49272.9f
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211 (pii: S0197458002000659)
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318(1):121–134. doi:10.1007/s00441-004-0956-9
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11(7):909–913. doi:10.1038/ncb1901
Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (1997). Neurobiol Aging 18 (4 Suppl):S1-2
Del Tredici K, Braak H (2012) Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol 124(5):643–664. doi:10.1007/s00401-012-1028-y
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM et al (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8(12):1150–1157
Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA, Frigerio R et al (2009) Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord 15(Suppl 3):S1–S5. doi:10.1016/S1353-8020(09)70769-2
Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y (2010) Evidence in favor of braak staging of Parkinson’s disease. Mov Disord 25(Suppl 1):S78–S82. doi:10.1002/mds.22637
Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y et al (2007) Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 22(12):1689–1707. doi:10.1002/mds.21507 (quiz 1837)
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT et al (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300(5619):636–640. doi:10.1126/science.1082324
Goetz C, Emre M, Dubois B (2008) Parkinson’s disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol 64(S2):S81–S92
Graff-Radford J, Murray ME, Lowe VJ, Boeve BF, Ferman TJ, Przybelski SA et al (2014) Dementia with Lewy bodies: basis of cingulate island sign. Neurology 83(9):801–809. doi:10.1212/WNL.0000000000000734
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B et al (2013) Distinct alpha-synuclein strains differentially promote tau Inclusions in neurons. Cell 154(1):103–117. doi:10.1016/j.cell.2013.05.057
Hawkes CH, Del Tredici K, Braak H (2010) A timeline for Parkinson’s disease. Parkinsonism Relat Disord 16(2):79–84. doi:10.1016/j.parkreldis.2009.08.007
Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A et al (2013) Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep 14(11):1017–1022. doi:10.1038/embor.2013.137
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosur Psychiatry 55:181–184. doi:10.1136/jnnp.55.3.181
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al (2012) National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8(1):1–13. doi:10.1016/j.jalz.2011.10.007
Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56(10):1095–1097
Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V et al (2012) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72(4):587–598. doi:10.1002/ana.23659
Jellinger KA (2009) A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792(7):730–740. doi:10.1016/j.bbadis.2008.07.006
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) Controversies over the staging of alpha-synuclein pathology in Parkinson’s disease. Acta Neuropathol 116(1):125–128. doi:10.1007/s00401-008-0381-3 (author reply 129–131)
Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB et al (2008) Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 18(2):220–224. doi:10.1111/j.1750-3639.2007.00117.x
Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF et al (2007) DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 68(11):812–819. doi:10.1212/01.wnl.0000256715.13907.d3
Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R (2013) Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154(6):1257–1268. doi:10.1016/j.cell.2013.08.035
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65(12):1863–1872. doi:10.1212/01.wnl.0000187889.17253.b1
Mirra SS (1997) The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging 18(4 Suppl):S91–S94
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123(1):1–11. doi:10.1007/s00401-011-0910-3
Olichney JM, Murphy C, Hofstetter CR, Foster K, Hansen LA, Thal LJ et al (2005) Anosmia is very common in the Lewy body variant of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 76(10):1342–1347. doi:10.1136/jnnp.2003.032003
Parkkinen L, Pirttila T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol 115(4):399–407. doi:10.1007/s00401-008-0346-6
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. doi:10.1016/j.neuron.2014.04.047
Sumikura H, Takao M, Hatsuta H, Ito S, Nakano Y, Uchino A et al (2015) Distribution of alpha-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol Commun 3:57. doi:10.1186/s40478-015-0236-9
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58(12):1791–1800
Toledo JB, Brettschneider J, Grossman M, Arnold SE, Hu WT, Xie SX et al (2012) CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol 124(1):23–35. doi:10.1007/s00401-012-0983-7
Toledo JB, Cairns NJ, Da X, Chen K, Carter D, Fleisher A et al (2013) Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun 1(1):65. doi:10.1186/2051-5960-1-65
Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL et al (2014) A platform for discovery: the university of pennsylvania integrated neurodegenerative disease biobank. Alzheimers dement 10(4):477–484. doi:10.1016/j.jalz.2013.06.003 (e471)
Tsuboi Y, Dickson DW (2005) Dementia with Lewy bodies and Parkinson’s disease with dementia: are they different? Parkinsonism Relat Disord 11(Suppl 1):S47–S51. doi:10.1016/j.parkreldis.2004.10.014
Wilson RS, Arnold SE, Schneider JA, Boyle PA, Buchman AS, Bennett DA (2009) Olfactory impairment in presymptomatic Alzheimer’s disease. Ann N Y Acad Sci 1170:730–735. doi:10.1111/j.1749-6632.2009.04013.x
Zaccai J, Brayne C, McKeith I, Matthews F, Ince PG (2008) Patterns and stages of alpha-synucleinopathy: relevance in a population-based cohort. Neurology 70(13):1042–1048. doi:10.1212/01.wnl.0000306697.48738.b6
Acknowledgments
This study was supported by NIH grants: AG05136, AG006781 and Morris K Udall Center for Parkinson’s disease Research Core grants P50 NS053488 and NS062684. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. DJI is supported by NS088341. Finally, we thank our patients and their families for making this research possible.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Toledo, J.B., Gopal, P., Raible, K. et al. Pathological α-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol 131, 393–409 (2016). https://doi.org/10.1007/s00401-015-1526-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1526-9