
Abstract
Autophagic vacuolar myopathies (AVMs) are a group of disorders united by shared histopathological features on muscle biopsy that include the aberrant accumulation of autophagic vacuoles. The classic conditions that compose the AVMs include Pompe Disease, Danon Disease and X-linked myopathy with excessive autophagy (XMEA). Other disorders, including acquired myopathies like chloroquine toxicity, also have features of an autophagic myopathy. This review is focused on XMEA, a myopathy with onset of slowly progressive proximal weakness and elevated serum creatine kinase (2× to 20× normal) typically in the first decade of life. However, both late-adult onset and severe, sometimes lethal, neonatal cases also occur. Skeletal muscle pathology is characterized by numerous cytoplasmic autophagic vacuoles, complex muscle fiber splitting with internalization of capillaries, and complement C5b-9 deposition within vacuoles and along the sarcolemma. The autophagic vacuoles have sarcolemmal features. Mutations in the VMA21 gene at Xq28 cause XMEA by reducing the activity of lysosomal hydrolases. The VMA21 protein regulates the assembly of the V-ATPase required to acidify the lysosome. Increased lysosomal pH and poor degradation of cellular debris may secondarily induce autophagy, the net effect being accumulation of autophagolysosomes. The relationship of XMEA to other lysosomal disorders of muscle and potential therapeutic interventions for XMEA are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term autophagy derives from the Greek words auto (“self”) and phagein (“to eat”). It refers to the catabolism of damaged or unnecessary cellular components, including damaged proteins and organelles, and to the machinery required for this breakdown. Autophagy is a normal homeostatic cellular process employed for detoxification and for recycling of proteins and membranes [9, 41]. It is also utilized and activated under conditions of cellular and organismal stress (particularly starvation), where it serves as a rich source for nutrients. Macroautophagy is the most common form of autophagy and is the process that will be discussed henceforth in this review. Macroautophagy proceeds through the orderly development of the autophagosome from the phagophore [15, 30]. The autophagosome is a double-membrane compartment that envelopes the material to be degraded. It fuses with the lysosome to form the autophagolysosome, where hydrolases activated in the acidic environment contributed by the lysosome degrade the encased cellular components, which can then be reused/recycled as sources of cellular energy or building blocks for intracellular structures. A general detailed review of autophagy is presented in this issue [5].
Abnormalities in autophagy are an increasingly recognized cause of and contributor to human disease [2]. A subset of disorders related to aberrant autophagy is caused by genetic mutations in either the machinery that regulates autophagy or that participates in the degradation of material within the lysosome. Cardiac and skeletal muscle appear particularly susceptible to injury or damage in the setting of impaired autophagy, a fact not entirely surprising given the high energy needs of muscle, the fact that skeletal muscle is a reservoir of amino acids in starvation, and the fact that muscle is an organ system with relatively little cellular turnover and thus at risk for accumulating damaged proteins and organelles. In some myopathies, particularly the autophagic vacuolar myopathies (see next paragraph), disease is thought to result from primary defects in an aspect or aspect(s) of the autophagic pathway. In other muscle diseases, impaired autophagy is considered a secondary contributor to disease pathogenesis, potentially accounting for features seen in these disorders such as muscle atrophy and fatigue [22]. Examples where abnormalities of autophagic flux have been reported, at least at the pre-clinical model system level, include Duchenne muscular dystrophy [6], Ullrich congenital muscular dystrophy [8], and hereditary inclusion body myopathy [19].
Autophagic vacuolar myopathies are a subgroup of muscle conditions with aberrant autophagy [29]. They are unified by shared features on muscle biopsy, and particularly by the presence of abundant accumulation of autophagic vacuoles. They are the myopathies where disease is thought to result from a primary defect in autophagy and/or lysosomal function [20]. The three best-defined conditions within this subgroup are Pompe Disease, Danon Disease, and X-linked myopathy with excessive autophagy (XMEA). Pompe Disease (glycogen storage disease type II, also known as acid maltase deficiency) is caused by mutations in the acid alpha glucosidase (GAA) gene, encoding a lysosomal enzyme that hydrolyses glycogen to glucose. It is a progressive multi-system disorder (affecting muscle, heart and brain in the infantile form) with widely varying clinical forms determined by the level of enzyme activity [14]. Pompe is also one of the first muscle diseases with a significant disease-modifying therapy (enzyme replacement) that has dramatically improved clinical outcomes. Danon Disease (discussed in detail in its own review in this issue [7]) is caused by mutations in lysosomal-associated membrane protein 2 (LAMP2). It is an X-linked disorder characterized by the triad of cognitive impairment, skeletal myopathy, and cardiomyopathy. XMEA is a skeletal myopathy caused by mutations in VMA21. It is the focus of the remainder of this review.
Clinical features
The first description of XMEA was by Kalimo et al. [13]. They provided a comprehensive examination of five affected males from three generations with disease inherited in an X-linked pattern. The clinical features were relatively uniform. Onset of symptoms was approximately age 5–6, with difficulties in arising from a sitting position and impaired ankle dorsiflexion. The disease was very slowly progressive in these individuals, and all retained the ability to ambulate. Muscle groups affected included the shoulder (with mild scapular winging) and hip girdles, the anterior thigh, and ankle dorsiflexors, all with mild to moderate weakness (4/5 on MRC grading scale). There was no involvement of other organ systems; in particular, cardiac evaluation was normal.
Villanova et al. described a second family in 1995. There were five affected males in three generations [39]. Onset of disease appeared slightly later than in the initial report (symptoms most consistently noted after onset of puberty). All cases were characterized by proximal muscle weakness, more notable in the legs than shoulders, that was slowly progressive. Thigh atrophy was prominently noted. Two individuals developed progressive and significant hip weakness (MRC 1/5) and eventually required a cane for ambulation (ages 48 and 51). Ambulation, however, was preserved in all five individuals, and death (while premature in two of the cases) could not be immediately ascribed to the myopathy.
A third report describing five families and eight affected individuals was published by Chabrol et al. [1]. The general clinical description was similar to that of the first two studies. Age of onset was around 5 years in three cases and 12 years in another. Two of the individuals had yet to develop symptoms and were identified based on an affected sibling and elevated CPK levels. It is not known if these children ever developed symptoms.
Since then, additional cases have been described and a relatively uniform clinical picture has emerged [3, 12, 16]. It includes onset usually in the first decade with abnormal gait and/or difficulties running. The primary muscles affected are in the shoulder and hip girdles and in the thighs, with some additional evidence of ankle dorsiflexion weakness in some individuals. Thigh atrophy can become prominent. There is a relatively static course to the disease, though some mild progressive in adulthood is noted, and individuals where clinical features have been analyzed later in life eventually require assistance with ambulation. There appears to be no cardiac or central nervous system involvement, respiratory function is unaffected (though not closely examined in terms of nocturnal hypoventilation), and survival is not compromised.
A dramatic expansion of the clinical spectrum of XMEA has been reported since 2013. First, a case was described with first symptoms late in adulthood [4]. The affected individual had onset after age 50 and mildly impaired ambulation at age 71. A mutation in VMA21 (c.164-7T>G) was identified in this individual, confirming that he represents a case of XMEA. This same mutation had been seen previously in several cases with the classical childhood-onset phenotype [16, 32], indicating interplay of the causative mutation with substantial, likely genetic, modifiers. On the other end of the clinical spectrum, two cases were described with neonatal–infantile onset. These children had neonatal hypotonia and poor suck, early involvement of upper extremity muscles, rapid progression and wasting, loss of ambulation in the early twenties in one (the other is younger but seems to be heading in the same direction), and involvement of extraocular eye muscles. Their muscle pathology was typical of XMEA and their VMA21 mutations novel [34]. Most recently, the mutation in an even more severe XMEA variant was identified in a family first described by Nishino et al. a decade ago, including seven boys with congenital disease, of whom only two could be saved through intubation/ventilation and tube-feeding. Again, muscle pathology was typical of XMEA [42]. The surviving children walked between ages 2 and 5 and then no longer. Presently aged 18 and 20, they exhibit extreme generalized muscle wasting and require non-invasive ventilation during sleep. Their mutation has also not been seen in cases with childhood-onset classical XMEA [27].
Diagnostic considerations
Standard neuromuscular diagnostic evaluations have been performed in many of the individuals with XMEA. The muscle biopsy characteristics that define the disorder are presented in the next section. Serum CPK levels are available for most of the published cases [4, 13, 23, 39]. The majority have elevated CPKs, ranging from 400 to >4,000. A few individuals, mainly those described in the study by Villanova et al. [39], have normal CPK values. In another French study by Chabrol et al. [1], two individuals had elevated CPK without apparent weakness, though they were still quite young at the time of the published description.
Neurodiagnostic features have also been described in depth. Nerve conduction studies are uniformly normal. EMG features are variable, though typically abnormal, and essentially all affected individuals have myopathic appearing units [12, 25]. In several individuals, abnormal spontaneous activity, including myotonia and high frequency discharges, has been described, despite a lack of clinical myotonia on physical examination. Interestingly, Munteanu et al. [25] described two adult cases where EMG abnormalities including myotonia were observed in the absence of any overt weakness. Linkage studies in these cases confirmed genetic association with XMEA, and subsequent mutation analysis showed them to carry the same mutation as their clinically affected siblings (unpublished observation), in combination with the children with elevated CPK and no symptoms [1]; this suggests that some individuals with XMEA are clinically unaffected.
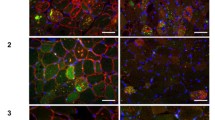
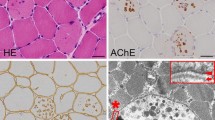
The muscle biopsy histopathology of XMEA is consistent across patients and is the defining and uniting feature of the disease (see Fig. 1) [29]. The core pathologic features of the disorder seen by light microscopy are as follows [13]: (1) numerous cytoplasmic vacuoles, (2) complex muscle fiber splitting with internalization of capillaries, and (3) deposition of complement C5b-9 [17]. Small cytoplasmic vacuoles are evident as basophilic stippling with hematoxylin and eosin (H&E) stains (Fig. 1a, b) and dark green to purple stippling with Gomori trichrome (not shown). Their true vacuolar nature is clarified by enzyme histochemistry for acetylcholinesterase (Fig. 1c) or immunostaining for sarcolemma-associated proteins [37]. This includes all members of the dystrophin–glycoprotein complex, caveolin-3, dysferlin, and spectrin (dystrophin shown in Fig. 1d). The basal lamina proteins merosin and perlecan, but not collagen VI, are also often detectable rimming cytoplasmic vacuoles, but with less intense staining than the sarcolemmal surface (Fig. 1e, f). Unlike collagen VI, both merosin and perlecan are produced by skeletal muscle. Collagen VI produced by fibroblasts does not have access to the cytoplasmic vacuoles. Immunostains for all three extracellular matrix proteins highlight the complex splits and internalized capillaries. Increased numbers of cytoplasmic lysosomes may be demonstrated by acid phosphatase enzyme histochemistry or by immunostaining for lysosomal proteins such as LAMP2 (Fig. 1g). The presence of this latter protein distinguishes XMEA from Danon disease. Complement activation with deposition of C5b-9 (the membrane attack complex) may be seen within cytoplasmic vacuoles and at the muscle fiber surface membrane (Fig. 1h) [17, 39]. Another inflammatory marker, MHC class I, can be expressed at the same sites (Fig. 1i). Calcium deposits, both along the sarcolemmal membrane and within vacuoles, have been described [1, 18]. Nonspecific myopathic features include increased fiber size variation, with both hypertrophic and atrophic fibres, and increased internal nuclei (see H&E images in Fig. 1). Importantly, there is a minimal degree or absence of myonecrosis, lymphocytic inflammation, and glycogen accumulation.

Light microscopic histopathology of XMEA. Characteristic features of XMEA that are readily observable by light microscopic analysis of muscle biopsy samples include numerous cytoplasmic autophagic vacuoles, complex splitting with internalization of nuclei and capillaries, irregularly thickened basal laminas, deposition of complement C5b-9, and expression of MHC class I. Examples from muscle biopsy cryosections that illustrate these features include H&E staining (a, b), acetylcholinesterase (AChE) enzyme histochemistry (c), and immunofluorescence for dystrophin (d), merosin (e), dual label collagen VI (green) and perlecan (red) (f), LAMP2 (g), complement C5b-9 (h), and MHC class I(i). Nuclei are stained with DAPI in panels e and f. The size bar is 100 µm for panels a, c, h, and i, and 50 µm for panels b, d, e, f, and g
Electron microscopy reveals additional characteristic features (see Fig. 2) [13]. These include the presence of abundant clustered lysosomes that are most often clearly membrane bound. Vacuoles can be found in the subsarcolemmal and intermyofibrillar spaces, as well as occasionally in contact with the muscle fiber surface membrane. This latter finding is a manifestation of autophagosome extrusion; the vacuoles are often seen adjacent to or in communication with exocytosed material. The basal lamina in these regions may be thickened or duplicated. The intracellular autophagocytic vacuoles surrounded by a discernable limiting membrane may or may not have distinct basal lamina immediately internal to the membrane. These pathologic basal lamina features may also be appreciated by light microscopy using immunostains for basal lamina proteins (see merosin and perlecan stains in Fig. 1). Other structures associated with the lysosomal clusters include membrane whorls, myelin figures and T-system proliferations.

Ultrastructural pathology of XMEA. Muscle biopsy electron microscopy shows large and small autophagic vacuoles interspersed between myofibrils (thick black arrows in a, b) or near the sarcolemma (thick white arrows in c). An example of exocytosis (extrusion) of autophagolysosome cargo is illustrated in panel d (thick white arrow). Exocytosis (extrusion) results in a serrated surface contour to some muscle fibers (c) and lysosomal debris in the extracellular space (c, d, small white asterisks) is seen between multiple layers of duplicated basal lamina (d, thin black arrows). Extensive redundant membranes (large black asterisks) and Z-band streaming (thin white arrows) are sometimes also present (b). The distance between Z-lines in this figure is approximately 3 µm
The unusual presence of sarcolemmal membrane proteins and extracellular matrix components rimming the vacuoles, a unique feature of XMEA and of Danon disease, led Nishino to introduce the term autophagic vacuoles with sarcolemmal features (AVSF) [37]. As shown by Nishino and colleagues, AVSF are distinct from other vacuolar myopathies, including myopathies with rimmed vacuoles such as sporadic inclusion body myositis and hereditary inclusion body myopathies. Immunopositivity of vacuoles for LAMP2 in XMEA provide pathologic distinction between XMEA and Danon Disease.
Genetics: linkage to Xq28 and mutations in VMA21
It was clear from the first description of XMEA that it was genetically based and that it likely followed an X-linked inheritance pattern. Linkage performed on the index family using restriction fragment length polymorphism analysis excluded dystrophin (DMD) and suggested localization to distal Xq [35]. Additional linkage analysis was performed on a second family to refine the locus to Xq28 and exclude emerin (EMD) [40]. The causative genetic locus was further refined in two subsequent mapping studies to a small region (0.5 Mb) of Xq28 [23, 26]. Finally, Ramchandran et al. [32] showed that mutations in the VMA21 gene cause the disease.
In the initial study, VMA21 mutations were identified in 14 families with XMEA [32]. Six different single-nucleotide substitutions, most intronic, were described in these 14 families. All 6 are predicted to alter gene splicing, reduce splicing efficiency, and result in reduced amount of XMEA transcript and protein. This was demonstrated by qRT-PCR (for RNA levels) and Western blot analysis (for protein levels). One of these mutations, c.164-7T>G, was separately reported in another case with classical XMEA [16]. As mentioned, this same mutation was shown to also cause a late-adulthood onset variant of the disease [4]. Of the recently described two neonatal–infantile onset cases with a more diffuse and severe phenotype than typical XMEA, one had an intronic microdeletion (VMA21 c.54-16_54-8del) and the other a 3′ untranslated region deletion (VMA21 c.*13_*104del) [34]. Finally, the most severe congenital cases with neonatal lethality (except where saved by ventilation and nasogastric feeding) had a single-nucleotide intronic mutation, VMA21 c.164-6T>G [27]. As can been seen, this mutation is one nucleotide closer to the exon than the c.164-7T>G mutation that causes both classical childhood onset slowly progressive XMEA and late-adult onset very slowly progressive XMEA. Studies of the relative effects of these two mutations on VMA21 expression and vacuolar-ATPase (V-ATPase) activity (see below for the role of VMA21 on the V-ATPase) showed that c.164-6T>G leads to deeper reductions of both VMA21 transcript and protein than does c.164-7T>G [27].
It has yet to be established whether all previously described cases clinically and pathologically compatible with XMEA are due to VMA21 mutations. Thus, it is not clear if there is genetic heterogeneity in this condition. One indication that additional genes may be found is the report of twin female siblings with myopathy and histopathology similar to XMEA [10]. Another is the failure to identify VMA21 mutations in several individuals with muscle biopsy evidence of LAMP2-positive AVM mimicking XMEA (unpublished observations, Minassian and Moore).
VMA21 function and pathogenesis of abnormal autophagy
The primary function of VMA21 is to regulate the assembly of the vacuolar ATPase (V-ATPase), though it may have additional chaperone activity. The V-ATPase is a multi-component enzyme that couples ATP hydrolysis with proton transport. V-ATPase is a ubiquitous proton pump that functions in many cellular contexts [38]; within the lysosome, its main function is to generate and maintain lysosome acidity, which in turn is required to create the acidic environment necessary for the activity of several lysosomal hydrolases [24]. The mutations of VMA21 found in patients reduce V-ATPase activity and cause a rise in mean lysosomal pH from 4.7 to 5.2 [32].
Based on knowledge of V-ATPase function and on several observations from XMEA patient cells and in vitro models of VMA21 knockdown, the following model for XMEA pathogenesis has been developed [32]: (1) mutations in VMA21 cause reduced levels of VMA21 protein; (2) reduced levels of VMA21 impair V-ATPase assembly, resulting in less V-ATPase complex and diminished V-ATPase activity; (3) diminished V-ATPase activity alters lysosomal pH; (4) Increased lysosomal pH reduces the activity of lysosomal hydrolases; (5) impaired hydrolase activity partially blocks the terminal step(s) of autophagy; (6) impaired V-ATPase activity and hydrolase function also potently induce autophagy, likely via inhibition of the mTORC1 pathway; (7) induction of autophagic flux in the setting of impaired autophagy causes a “feed forward” pathogenic loop, resulting in the accumulation of autophagolysosomes with incompletely digested contents.
Several interesting considerations emerge from this model. One is how, ultimately, the processes interrupted by loss of VMA21 result in muscle weakness and disease. It is interesting to note that mouse knockout models of several autophagy components (such as, for example, Atg7, which is required for initiation of autophagy) do not result in an XMEA-like pathologic picture [21]. Thus, disease is likely not as simple as an overall impairment in autophagy. One potential explanation for this discrepancy is the place within the autophagic pathway that the abnormality is located. The lysosome receives proteins and membranes from multiple sources and not solely from the phagophore. Thus, a primary lysosomal defect may result in a more significant accumulation of toxic/breakdown products (and of autophagic vacuoles) than an autophagy initiation problem. Another consideration is that the lysosome performs functions other than breakdown of long-lived proteins and membranes. Relevant for muscle is the growing evidence that lysosomes participate in membrane repair [11]. Perhaps aspects of the XMEA phenotype are due to interruption of sarcolemmal membrane homeostasis; this would be in keeping with the elevated CPK levels of patients with XMEA, and also with the observation of vacuoles accumulating at the sarcolemma and extruding their cargo into the extracellular space.
Another key question is what effect a more significant reduction in VMA21 levels would have in terms of human disease phenotype. V-ATPase activity is required in many cell types, and impaired lysosomal function in general is often more typically associated with central nervous system disease [31]. Perhaps complete loss of VMA21 would cause more wide spread disease that resembles a more typical lysosomal storage disease instead of purely a skeletal myopathy. This is suggested by the observation that cultured fibroblasts from XMEA patients also develop vacuoles, indicating the potential for disturbed lysosomal function in non-muscle cell types. Conversely, it may be that VMA21 in vivo is required/necessary primarily in muscle, and more deleterious mutations may manifest only as earlier onset and more severe muscle disease.
Future work related to these and other questions concerning the function of VMA21 and disease pathogenesis will be greatly aided by the development of in vivo model systems. At present, there are no vertebrate models with VMA21 loss of function, a clear barrier to understanding the protein and the disease.
Treatment considerations
Currently, there are no specific therapies for XMEA; management is symptomatic and based on maximization of mobility and reduction of joint contractures. As mentioned above, pre-clinical development of disease-modifying therapies is hindered due to the lack of animal model(s) of the disease. Potential therapeutic strategies must be extrapolated from other contexts.
One clear question is whether modification of autophagic flux would be expected to modify disease. This is a complicated issue, as reduced levels of VMA21 appear to both impair the completion of autophagy and excessively activate autophagic flux [32]. Thus, a simple approach of either activating autophagy (such as with a drug like rapamycin) or inhibiting autophagic initiation (like with 3-methyladenine) would be of uncertain benefit. Of the two, inhibition of autophagic flux may seem the better alternative, as it might prevent accumulation of autophagolysosomes. However, if disease is instead more related to lysosome function, particularly non-autophagy-related roles, then this would not help and would potentially worsen the disease process.
An intriguing potential treatment strategy was described by Nemazanyy et al. [28]. They examined the function in skeletal muscle of Vps15, a regulatory subunit of the class III phosphoinositide kinase PIK3C3 (also called Vps34), and a key participant in autophagy and endolysosomal maturation. Knockout of Vps15, or of the kinase Pik3c3 [33], results in muscle pathology very similar to that seen in XMEA, including the presence of AVSF. Of most relevance, overexpression of Vps15 + PIK3C3 in myoblast cell lines from patients with Danon disease resulted in a decrease in aberrant autophagy (as determined by LC3 levels, see [5]) and a reduction in the accumulation of glycogen. Given that there appears to be significant overlap in the pathogenesis of Danon disease and XMEA, a similar upregulation of Vps15/PIK3C3 might improve disease pathology in XMEA as well. Further studies will obviously be necessary to investigate this possibility, as will pre-clinical testing of chemical activators of PIK3C3.
Another exciting and potentially relevant observation is the discovery that TFEB overexpression can improve aspects of Pompe disease pathology [36]. TFEB is a transcription factor that drives lysosome biogenesis [5]. It also stimulates autophagy and enhances lysosomal exocytosis. Viral-mediated TFEB expression in fibroblasts from patients with Pompe disease and myofibers from GAA knockout mice reduced glycogen accumulation and promoted the clearance via exocytosis of enlarged lysosomes. It will be of interest to see if the beneficial effects of TFEB might also improve aspects of the pathologic changes in Danon disease and XMEA. The fact that TFEB enhances exocytosis of aberrant material trapped in defective lysosomes suggests that this approach may have merit in XMEA, as of the subsarcolemmal accumulation of autophagic vacuoles in some XMEA muscle fibers suggests that exocytosis may be impaired.
Summary
Remarkable progress regarding XMEA has been made in the past several years. First described in 1988, XMEA is now a cohesive clinical and pathologic entity distinct from other muscle diseases. The discovery of mutations in VMA21 in all genetically “solved” cases of XMEA support that this disorder is a unique condition caused by impaired autophagy and lysosomal function. Further study of VMA21 should improve our understanding of disease pathogenesis and the role of the lysosomes in muscle homeostasis and disease. This in turn may lead to rational development of therapies for the condition. It will be of interest to see if mutations in VMA21 are the only cause of this type of autophagic myopathy, or whether mutations in additional genes (and additional phenotypes) will be discovered in conditions that share the unique XMEA histopathology.
References
Chabrol B, Figarella-Branger D, Coquet M et al (2001) X-linked myopathy with excessive autophagy: a clinicopathological study of five new families. Neuromuscul Disord 11:376–388
Choi AMK, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:1845–1846. doi:10.1056/NEJMc1303158
Chow G, Beesley CE, Robson K et al (2006) Case of X-linked myopathy with excessive autophagy. J Child Neurol 21:431–433
Crockett CD, Ruggieri A, Gujrati M et al (2014) Late-adult onset of X-linked myopathy with excessive autophagy (XMEA). Muscle Nerve 50(1):138–144. doi:10.1002/mus.24197
Damme M, Suntio T, Saftig P, Eskelinen E-L (2014) Autophagy in neuronal cells: general principles and physiological and pathological functions. Acta Neuropathol. doi:10.1007/s00401-014-1361-4
De Palma C, Morisi F, Cheli S et al (2012) Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 3:e418. doi:10.1038/cddis.2012.159
Endo Y, Furuta A, Nishino I (2015) Danon disease: a phenotypic expression of LAMP2-deficiency. Acta Neuropathol [Epub ahead of print]. doi:10.1007/s00401-015-1385-4
Grumati P, Coletto L, Sabatelli P et al (2010) Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16:1313–1320. doi:10.1038/nm.2247
Hansen TE, Johansen T (2011) Following autophagy step by step. BMC Biol 9:39. doi:10.1186/1741-7007-9-39
Holton JL, Beesley C, Jackson M et al (2006) Autophagic vacuolar myopathy in twin girls. Neuropathol Appl Neurobiol 32:253–259. doi:10.1111/j.1365-2990.2006.00691.x
Huynh C, Roth D, Ward DM et al (2004) Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc Natl Acad Sci 101:16795–16800. doi:10.1073/pnas.0405905101
Jääskeläinen SK, Juel VC, Udd B et al (2002) Electrophysiological findings in X-linked myopathy with excessive autophagy. Ann Neurol 51:648–652. doi:10.1002/ana.10173
Kalimo H, Savontaus ML, Lang H et al (1988) X-linked myopathy with excessive autophagy: a new hereditary muscle disease. Ann Neurol 23:258–265. doi:10.1002/ana.410230308
Katzin LW, Amato AA (2008) Pompe disease: a review of the current diagnosis and treatment recommendations in the era of enzyme replacement therapy. J Clin Neuromuscul Dis 9:421–431. doi:10.1097/CND.0b013e318176dbe4
Klionsky DJ, Codogno P (2013) The mechanism and physiological function of macroautophagy. J Innate Immun 5:427–433. doi:10.1159/000351979
Kurashige T, Takahashi T, Yamazaki Y et al (2013) Elevated urinary β2 microglobulin in the first identified Japanese family afflicted by X-linked myopathy with excessive autophagy. Neuromuscul Disord 23:911–916. doi:10.1016/j.nmd.2013.06.003
Louboutin JP, Villanova M, Ulrich G et al (1996) Elevated levels of complement components C5 and C9 and decreased antitrypsin activity in the serum of patients with X-linked vacuolated myopathy. Muscle Nerve 19:1144–1147. doi:10.1002/(SICI)1097-4598(199609)19:9<1144:AID-MUS10>3.0.CO;2-V
Louboutin JP, Villanova M, Lucas-Héron B, Fardeau M (1997) X-linked vacuolated myopathy: membrane attack complex deposition on muscle fiber membranes with calcium accumulation on sarcolemma. Ann Neurol 41:117–120. doi:10.1002/ana.410410121
Malicdan MCV, Noguchi S, Nishino I (2007) Autophagy in a mouse model of distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Autophagy 3:396–398
Malicdan MC, Noguchi S, Nonaka I et al (2008) Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul Disord 18:521–529. doi:10.1016/j.nmd.2008.04.010
Masiero E, Agatea L, Mammucari C et al (2009) Autophagy is required to maintain muscle mass. Cell Metab 10:507–515. doi:10.1016/j.cmet.2009.10.008
Merlini L, Nishino I, Consortium for Autophagy in Muscular Dystrophies (2014) 201st ENMC International Workshop: autophagy in muscular dystrophies—Translational approach, 1–3 November 2013, Bussum, The Netherlands. Neuromuscul Disord. doi:10.1016/j.nmd.2014.03.009
Minassian BA, Aiyar R, Alic S et al (2002) Narrowing in on the causative defect of an intriguing X-linked myopathy with excessive autophagy. Neurology 59:596–601
Mindell JA (2012) Lysosomal acidification mechanisms. Annu Rev Physiol 74:69–86. doi:10.1146/annurev-physiol-012110-142317
Munteanu I, Ackerley CA, Mnatzakanian GN et al (2005) Electrophysiology extends the phenotypic spectrum of X-linked myopathy with excessive autophagy. Neurology 64:927–928. doi:10.1212/01.WNL.0000152884.11980.53
Munteanu I, Ramachandran N, Mnatzakanian GN et al (2008) Fine-mapping the gene for X-linked myopathy with excessive autophagy. Neurology 71:951–953. doi:10.1212/01.wnl.0000325991.01899.35
Munteanu I, Ramachandran N, Ruggieri A et al (2015) Congenital autophagic vacuolar myopathy is allelic to X-linked myopathy with excessive autophagy. Neurology (in press)
Nemazanyy I, Blaauw B, Paolini C et al (2013) Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med 5:870–890. doi:10.1002/emmm.201202057
Nishino I (2006) Autophagic vacuolar myopathy. Semin Pediatr Neurol 13:90–95. doi:10.1016/j.spen.2006.06.004
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20:460–473. doi:10.1089/ars.2013.5371
Platt FM, Boland B, van der Spoel AC (2012) The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 199:723–734. doi:10.1083/jcb.201208152
Ramachandran N, Munteanu I, Wang P et al (2013) VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol 125:439–457. doi:10.1007/s00401-012-1073-6
Reifler A, Li X, Archambeau AJ et al (2014) Conditional knockout of Pik3c3 causes a murine muscular dystrophy. Am J Pathol. doi:10.1016/j.ajpath.2014.02.012
Ruggieri A, Ramachandran N, Wang P et al (2015) Non-coding VMA21 deletions cause X-linked myopathy with excessive autophagy. Neuromusc Dis (in press)
Saviranta P, Lindlöf M, Lehesjoki AE et al (1988) Linkage studies in a new X-linked myopathy, suggesting exclusion of DMD locus and tentative assignment to distal Xq. Am J Hum Genet 42:84–88
Spampanato C, Feeney E, Li L et al (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 5:691–706. doi:10.1002/emmm.201202176
Sugie K, Noguchi S, Kozuka Y et al (2005) Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J Neuropathol Exp Neurol 64:513–522
Sun-Wada G-H, Wada Y (2013) Vacuolar-type proton pump ATPases: acidification and pathological relationships. Histol Histopathol 28:805–815
Villanova M, Louboutin JP, Chateau D et al (1995) X-linked vacuolated myopathy: complement membrane attack complex on surface membrane of injured muscle fibers. Ann Neurol 37:637–645. doi:10.1002/ana.410370514
Villard L, Portes des V, Levy N et al (2000) Linkage of X-linked myopathy with excessive autophagy (XMEA) to Xq28. Eur J Hum Genet 8:125–129. doi:10.1038/sj.ejhg.5200432
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109. doi:10.1038/ncb1007-1102
Yan C, Tanaka M, Sugie K et al (2005) A new congenital form of X-linked autophagic vacuolar myopathy. Neurology 65:1132–1134. doi:10.1212/01.wnl.0000178979.19887.f5
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dowling, J.J., Moore, S.A., Kalimo, H. et al. X-linked myopathy with excessive autophagy: a failure of self-eating. Acta Neuropathol 129, 383–390 (2015). https://doi.org/10.1007/s00401-015-1393-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1393-4