
Abstract
Microglia are phagocytic cells that survey the brain and perform neuroprotective functions in response to tissue damage, but their activating receptors are largely unknown. Triggering receptor expressed on myeloid cells 2 (TREM2) is a microglial immunoreceptor whose loss-of-function mutations in humans cause presenile dementia, while genetic variants are associated with increased risk of neurodegenerative diseases. In myeloid cells, TREM2 has been involved in the regulation of phagocytosis, cell proliferation and inflammatory responses in vitro. However, it is unknown how TREM2 contributes to microglia function in vivo. Here, we identify a critical role for TREM2 in the activation and function of microglia during cuprizone (CPZ)-induced demyelination. TREM2-deficient (TREM2−/−) mice had defective clearance of myelin debris and more axonal pathology, resulting in impaired clinical performances compared to wild-type (WT) mice. TREM2−/− microglia proliferated less in areas of demyelination and were less activated, displaying a more resting morphology and decreased expression of the activation markers MHC II and inducible nitric oxide synthase as compared to WT. Mechanistically, gene expression and ultrastructural analysis of microglia suggested a defect in myelin degradation and phagosome processing during CPZ intoxication in TREM2−/− microglia. These findings place TREM2 as a key regulator of microglia activation in vivo in response to tissue damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microglia are unique CNS resident myeloid cells that originate from yolk-sac progenitors during embryogenesis and self-renew as an autonomous population throughout life [12, 21]. Microglia are motile cells that constantly survey the central nervous system (CNS) parenchyma [10]. They are important in CNS homeostatic functions as well as for the progression and resolution of diseases. Microglia have been proposed to support tissue homeostasis by playing important roles in the clearance of apoptotic bodies and debris, secretion of neurotrophic factors and remodeling neural connections. Upon tissue injury, microglia rapidly respond by undergoing a rapid transformation from resting, ramified forms to an amoeboid morphology [66]. However, the receptors involved and the molecular changes that occur in microglia during their activation are incompletely understood.
Triggering receptor expressed on myeloid cells 2 (TREM2) belongs to the TREM family of immunoreceptors. The TREM2 cytoplasmic domain has no intrinsic signaling capacity, but associates with the adaptor molecule DAP12, which can signal through an immunoreceptor tyrosine activation motif (ITAM) and is required for TREM2 surface expression [9, 16]. Most studies have shown that TREM2 is selectively expressed in microglia in the CNS and in osteoclasts in bone, but not in peripheral blood leukocytes in human and mouse in the steady-state or in inflammatory conditions [7, 28, 53]. TREM2 ligands are poorly characterized and include poly-anionic compounds such as bacterial products and neuronal debris, although signaling upon receptor engagement by these compounds has not been proven [11, 30, 50]. Moreover, undefined TREM2 ligands are expressed by astrocytes and neurons [11, 30, 63] and by in vitro-generated macrophages and dendritic cells [24, 32].
An important role for TREM2 in modulating microglia functions in the CNS is evinced by genetic human studies. Homozygous, loss-of-function mutations in TREM2 or DAP12 genes cause polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL, also known as Nasu-Hakola disease) [39, 61]. Furthermore, TREM2 mutations are associated with cases of frontotemporal dementia (FTD)-like syndrome without bone pathology [23]. More recently, studies have demonstrated that rare TREM2 genetic variants significantly increase the risk for Alzheimer’s disease (AD) [22, 34, 35, 64, 71]. Intriguingly, recent reports have also shown an association of TREM2 variants with FTD, Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) [3, 5, 8, 67]. These findings suggest an important role for TREM2 and microglia in neurodegenerative diseases.
TREM2 can promote phagocytosis of apoptotic material and inhibit inflammatory cytokine production in response to apoptotic material and TLR agonists [24, 80, 81]. Moreover, TREM2 and DAP12 promote cell proliferation and survival in response to macrophage colony-stimulating factor (CSF-1) in vitro, the ligand for CSF-1 receptor (CSF-1R) [59, 60] which is required for the development and maintenance of microglia [13, 14]. DAP12 deficiency results in fewer microglial cells in certain areas of the CNS [36, 60]. These findings have led to the hypothesis that TREM2 may function as a CSF-1R co-receptor in microglia.
A strong limitation of previous TREM2 functional studies is the use of cell lines or primary microglial cells derived from newborn mice that are not bona fide microglia [7, 72]. Thus, in vivo studies are needed to assess TREM2 function in the CNS. Here, we have explored the role for TREM2 in microglia activation and function in the cuprizone (CPZ)-induced demyelination model. This is a well-characterized model in which oligodendrocyte degeneration in the brain is followed by a strong microglial response consisting in rapid activation, proliferation and clearance of damaged myelin debris with an intact blood–brain barrier. The CPZ model is a suitable tool to study specifically microglia responses because of minimal CNS infiltration of peripheral inflammatory cells [29, 33]. We provide evidence that TREM2-deficient mice have a broad defect in microglia response to myelin damage including defective activation, proliferation and lipid degradation within the cells, resulting in more profound CNS demyelination and clinical impairment. These findings suggest that similar mechanisms may be impaired in TREM2-associated human neurodegenerative disease, possibly rendering microglia less efficient at clearance of cells and toxic debris.
Materials and methods
Mice
TREM2−/− and littermate control WT mice (backcrossed 12 generations to the C57BL/6 background) were obtained from Marco Colonna. The two strains were bred in parallel. Animal experiments were approved by the Animal Study Committee (ASC) at Washington University in St. Louis.
Mouse model of CPZ-induced demyelination and tissue processing
Six- to eight-week-old TREM2−/− and WT mice were fed a standard diet (Harlan) containing 0.2 % CPZ [finely powdered oxalic bis(cyclohexylidenehydrazide); Sigma–Aldrich] for 4, 6 or 12 weeks. Brains were removed after mouse perfusion with 4 % paraformaldehyde (PFA), fixed in 4 % PFA for 24 h, followed by immersion in 30 % sucrose for 24–48 h. Forty-five WT and 48 TREM2−/− mice were used in cuprizone feeding studies. A total of nine WT and ten TREM2−/− naïve mice were compared in the analyses.
Behavioral testing
The following behavioral tests were performed on age-matched TREM2−/− and WT mice after 12 weeks of CPZ:
1-h Locomotor activity and sensorimotor battery
Locomotor activity was evaluated using transparent (47.6 × 25.4 × 20.6 cm high) polystyrene enclosures and computerized photobeam instrumentation [84]. General activity variables (total ambulations, vertical rearings) along with indices of emotionality including time spent, distance traveled and entries made in a 33 × 11 cm central zone were analyzed. A battery of sensorimotor tests was performed to assess balance (ledge and platform), strength (inverted screen), coordination (pole and inclined screens) and initiation of movement (walking initiation), as previously described [20, 84].
Rotarod
Motor co-ordination and balance were studied as published [6, 20]. The rotarod protocol consisted of three training sessions with each session including one stationary rod trial, two constant speed (2.5 rpm for 60 s max) and two accelerating speed rotarod trials (2.5–10.5 rpm over 1–180 s max). Sessions were separated by 4 days to minimize motor learning, and time spent on the rod was used as the dependent variable to assess performance.
Statistical analyses for behavioral data
Repeated measures (rm) ANOVA models containing two between-subjects variables (Genotype and Sex) and one within-subjects (repeated measures) variable (i.e., Time Blocks) were used to analyze the general ambulatory and rearing frequency data, as well as the constant speed and accelerating rotarod data. One-way ANOVA models were used to analyze differences between groups for the other activity variables and measures in the sensorimotor battery and stationary rod data. Pairwise comparisons were conducted following relevant, significant overall ANOVA effects, which were subjected to Bonferroni correction when appropriate.
Histological and immunohistochemical analyses
Solochrome cyanine was used to stain myelin as described [37]. The following primary antibodies were used: anti-MBP (1:100; Abcam, ab7349), -dMBP (1:2000; Millipore, ab5864) [31], -βAPP (1:100; Invitrogen, 51-2700), -SMI-31 (1:1000; Covance, smi-31R), -Iba1 (1:600; Wako, 019-19741), -BrdU (1:250; Abcam, ab1893), -GFAP (1:200; Invitrogen, 13-0300), -iNOS (1:100; BD Pharmingen, 610329), -LPL (1:400, from Dr. G. Olivecrona) [57] and -MHC II (1:100; BD Pharmingen, 553549). Images were acquired using a Nikon Eclipse 90i fluorescent and bright field microscope and analyzed for quantitation with the Metamorph 7.7 software. Iba1+ and bromodeoxyuridine (BrdU) positive microglia were manually counted using two 10× and four 60× images, respectively, on two non-adjacent sections. Colocalization of BrdU with the DAPI staining in Iba+ cell nuclei determined which cells were marked as proliferating. Total number of cells was divided by the total area (# of cells/mm2). GFAP, dMBP, βAPP and SMI-31 staining were analyzed as the percentage area of positive staining (number of positive pixels/1 mm2) within the area of interest. LPL, iNOS and MHC II were analyzed for the percentage of colocalization with Iba1 using Metamorph software. Quantitative evaluation of microglial cell morphology in tissue sections and in vitro was performed as described in the literature using ramification index (RI) calculated by the following equation: 4π × cell area/(cell perimeter)2 [26]. The RI of perfectly round cells is 1; if morphology deviates from perfectly circular form, RI is smaller than 1; and when the cell is highly ramified the RI is close to zero. Cell area and perimeter were calculated with the assistance of the Metamorph software.
Analysis of microglia proliferation in vivo
Microglia proliferation in vivo was measured during CPZ feeding by BrdU incorporation. BrdU was administered (25 mg/kg body weight, Sigma) by intraperitoneal injection once every 8 h for 4 days. 8 h after the last injection, brains were removed for immunohistochemistry (IHC) or flow cytometry analyses. For flow cytometry studies, cells were labeled using antibodies to CD11b (BD Pharmingen, 553311), CD45 (Biolegend, 101245). BrdU staining was performed using a commercial kit (BD Biosciences). Cells were analyzed on a FACS Gallios (Beckman Coulter Inc.).
Microarray processing and analysis
Microarrays were performed by the Genome Technology Access Center (GTAC) at Washington University. Total RNA was isolated from the caudal corpus callosum of WT and TREM2−/− mice obtained at 6 (4WT and 3 TREM2−/− mice) and 12 weeks (5WT and 6 TREM2−/− mice) after CPZ treatment using the RNeasy mini kit (Qiagen). RNA quality and integrity were assessed using the Bioanalyzer (Agilent Technologies) with all samples being of high quality (RNA integrity number = 7.9–8.9). Hybridization was performed with Affimetrix GeneChip Mouse Exon 1.0 ST arrays. Biotinylated cDNA was prepared according to the standard NuGen WT-Ovation Pico RNA Amplification system from 500 pg to 50 ng total RNA. Probe summarization (Robust Multichip Analysis, RMA), quality control analysis, and probe annotation were performed according to recommended guidelines (Expression Console Software, Affymetrix, Inc.). Statistical analyses of the 6- and 12 weeks time point experiments were done separately. Microarray data produced were quality checked [70] under R version 3.1.0 Bioconductor software [18]; undetectable and low detection value probes were removed. To identify differentially expressed genes, based on a moderate t test, the Limma package [83] was used, and genes were selected based on a P value cut-off <0.05 (after Benjamini and Hochberg adjustment) and absolute logarithmic fold change >1.
IPA (Ingenuity) analysis
Microarray data were analyzed by IPA (Ingenuity System, www.ingenuity.com). Differentially expressed genes were incorporated in canonical pathways and bio-functions and were used to generate biological networks. Uploaded data sets for analysis were filtered using the following cutoff definitions: one log fold change, adjusted P value <0.05. Core analysis was performed using the following settings: Mouse Gene 1.0 ST Array. Relationship to include: direct and indirect does not include endogenous chemicals. Filter summary: consider only relationships where confidence = High (predicted) or experimentally observed. The network Score was based on the hypergeometric distribution and was calculated with the right-tailed Fisher’s Exact Test. Downstream Effect Analysis P value of overlap was calculated by the Fisher’s Exact Test.
Real-time PCR on microglia
RNA was purified from FACS sorted microglia using the RNeasy Micro Kit (Qiagen), converted into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and used at 50 ng/μl in quantitative real-time PCR (qRT-PCR) analysis (ABI 7000 Real-Time PCR System, Applied Biosystems). The ΔΔC t method was applied to determine differences in gene expression levels after normalization to the arithmetic mean of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal standard. The TaqMan probes (Life technologies) were the following: SiglecH (Mm00618627_m1); Tmem119 (Mm00525305_m1); P2ry12 (Mm00446026_m1); P2ry13 (Mm01951265_s1); Gpr34 (Mm02620221_s1); Fcrls (Mm00472833_m1); Olfml3 (Mm00513567_m1); Slc2a5 (Mm00600311_m1); LPL (Mm01345523_m1).
In vitro adult microglia culture
TREM2−/− and WT mice were deeply anesthetized, perfused with PBS and the brains were aseptically removed. Mononuclear cells isolated from the brains as previously described [1] were stained with anti-CD11b phycoerythrin (BD Pharmingen, 553311) and anti-CD45 AF700 (Biolegend, 103128) and sorted on a FACSAria (BD Biosciences). CD11b+CD45low microglial cells were collected and cultured for 72 h in DMEM complete medium (Gibco) in the presence of 100 ng/ml CSF-1 and 50 ng/ml TGFβ with or without human myelin (100 μg/ml).
Pre-embedding immuno-electron microscopy
Mice were perfused with 4 % PFA, brains were removed and immersion fixed. Brain sections were cut using a vibratome (100 μm) and incubated in 0.5 % gelatin, 5 % horse serum and 0.01 % saponin in PBS for 5 h. Microglia staining was performed using a Rb anti-Iba1 (1:600; Wako), a secondary biotinylated anti-Rb (1:500; Rockland), streptavidin-HRP (1:1000; Rockland, S000-03) for 3 h and visualized using DAB Substrate Kit (Cell Marque, 957D). Tissue was then fixed with 2 % PFA, 2.5 % GA and sections were processed using routine EM protocols. Grids were not counterstained to preserve immunostaining. Finished blocks were sectioned using a DiATOME ultra 45° diamond knife and a LEICA Ultracut UC7. Sections (90 nm) were cut and picked up onto 200 hex mesh, formvar-carbon coated copper grids (Ted Pella, 01800-F). Images were captured using a JEOL 1200 EX II Transmission Electron Microscope with AMT digital camera.
Statistical analyses
IHC results and in vitro assay analyses were expressed as mean value ± SD or ± SEM. Medians are reported when data were not parametric. Data were analyzed using Mann–Whitney U tests (GraphPad Prism, GraphPad Software). Statistical analyses of behavioral testing and microarray results were performed as described in “Materials and methods”.
Results
Lack of TREM2 leads to defective clearance of myelin debris in the CPZ model
CPZ is a copper-chelating toxic agent that, when administered in the diet, causes oligodendrocyte death and consequent demyelination in the CNS. Mice fed CPZ for 3 weeks exhibited extensive reactive gliosis accompanied by oligodendrocyte apoptosis. Demyelination was evident after 4 weeks of intoxication in multiple structures, including the cortex, hippocampus, cerebellum, striatum and most notably the corpus callosum [45, 75]. After 12 weeks on CPZ, demyelination is more profound. The effects of 4, 6 and 12 weeks CPZ feeding on CNS myelin were compared in TREM2-deficient (TREM2−/−) versus wild-type (WT) mice. We focused our analysis on the corpus callosum (CC) because this large tract of white matter exhibits the greatest and most consistent pathological changes in this model. Myelin integrity in the CC of TREM2−/− and WT mice was assessed histologically by solochrome cyanine staining. Intact myelin (stained in dark blue) was observed in the CC of naïve TREM2−/− and WT mice without any differences between the two groups (Fig. 1a, top row). Consistent with previous reports, after 4 weeks on CPZ, WT mice showed loss of myelin involving the hippocampal commissure, lying beneath the CC, which extended progressively into the CC at 6 and 12 weeks (Fig. 1a). At higher magnification in areas of demyelination in WT mice no myelin staining was detected suggesting that they had cleared myelin debris at 4, 6 and 12 weeks as observed histologically. In contrast, in the TREM2−/− mice on CPZ, myelin staining was still detectable in most areas of the CC, but was noted to have a lighter intensity of stain. At higher magnification the myelin staining in these areas suggested loss of myelin integrity (Fig. 1a, 60× images). Therefore, myelin integrity was assessed by immunostaining using antibodies specific for intact or degraded myelin basic protein (MBP and dMBP, respectively). The dMBP antibody has been previously shown to bind a MBP epitope that is accessible only in areas of myelin degradation [44]. The amount of MBP and dMBP was assessed and quantified in the CC (Fig. 1b, c). In naïve TREM2−/− and WT mice, normal MBP staining was observed without labeling of dMBP. In WT mice at 4, 6 and 12 weeks on CPZ a limited amount of dMBP staining was detected in demyelinated areas of the CC. In contrast, more extensive accumulation of dMBP was observed in TREM2−/− mice at all time points (Fig. 1b). The quantity of dMBP in the CC was significantly more in TREM2−/− than in WT mice at all time points (Fig. 1c, left panel). dMBP was also quantified with similar results in the CA1 region of the hippocampus which also undergoes demyelination after CPZ treatment [56] (Fig. 1c, right panel). Additionally, loosened and uncompacted myelin was readily demonstrated in the CC of TREM2−/− mice by electron microscopy (EM) at 4 and 12 weeks on cuprizone (Supplementary Fig. 1). Thus, we observed more extensive accumulation of degraded myelin staining in TREM2−/− vs. WT mice, which could be due to defective clearance of myelin debris.

Defective clearance of myelin debris in TREM2−/− mice during CPZ-induced demyelination. a Myelin was studied histologically by solochrome cyanine staining (intact myelin stained in dark blue) in WT and TREM2−/− naïve mice and mice fed CPZ for 4, 6 and 12 weeks. The coronal section of the mouse brain on the top shows in the boxed area the region of the caudal corpus callosum (CC) that was analyzed (modified from Sidman, High Resolution Mouse Brain Atlas, http://www.hms.harvard.edu/research/brain/index.html). The images on the left for each WT and TREM2−/− panel (first and third columns) depict the CC (×4 magnification). A higher magnification (×60) of the boxed area is shown on the right of each image. b Myelin integrity was assessed in the CC of WT and TREM2−/− naïve and CPZ-fed mice by immunostaining using antibodies specific for intact or degraded myelin basic protein (MBP in red and dMBP in green, respectively; ×20). c dMBP accumulation was quantified in the CC and in the CA1 region of the hippocampus at the different time points. Data are from one experiment of two independent experiments performed with similar results. In the presented experiment for week 0 n = 3 each for naive WT and TREM2−/− mice; for week 4 n = 7 WT and n = 5 TREM2−/− mice; for week 6 n = 9 for both groups; for week 12 n = 4 WT and n = 6 TREM2−/− mice. Values in the graphs are mean ± SD. *P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.0001 using Mann–Whitney test
TREM2−/− mice have more axonal pathology
Axons in the CC of TREM2−/− and WT mice at 4, 6 and 12 weeks of CPZ treatment were assessed by staining for beta-amyloid precursor protein (APP) (Fig. 2a). APP accumulation reflects impaired axonal transport in injured axons [15], a change that can be reversible if the causative insult does not persist. At all time-points we observed a significantly greater extent of APP staining in the CC of TREM2−/− compared to WT, with more diffuse axonal beading (Fig. 2b, left panel). It has been reported that axonal APP staining in CPZ treatment decreases between 4 and 12 weeks, without substantial loss of axons [85]. In accord with this, we observed diminishing APP staining from 4 to 12 weeks of CPZ intoxication in both TREM2−/− and WT mice. To compare the fate of axons in the CC in the two strains with CPZ treatment, we used SMI-31 immunostaining (for intact phosphorylated neurofilaments) to quantify non-injured axons. Significantly less SMI-31 staining was observed in TREM2−/− compared to WT mice CC at 12 weeks of CPZ treatment, but not at 4 weeks (Fig. 2b, right panel). The reduced SMI-31-stained axons at 12 weeks suggested that axonal swelling in TREM2−/− mice is followed by axonal degeneration to a greater extent than in WT mice. To confirm this, we performed EM studies of the CC obtained at 4 and 6 weeks on CPZ in each strain (Fig. 2c). Axonal damage was identified based on the typical morphology of damaged axons containing numerous autophagocytic vesicles. At 4 weeks on CPZ we observed a higher number of dystrophic axons in TREM2−/− than in WT mice, but this was not statistically significant. At 12 weeks compared to 4 weeks on CPZ, the number of dystrophic axons was increased in TREM2−/− mice and was significantly higher than in WT mice, where the degree of axonal damage diminished compared to the 4-week time-point (Fig. 2d). Overall, at all time-points we observed more extensive axonal damage in TREM2−/− than in WT mice.

TREM2−/− mice show more severe axonal pathology after CPZ. a Immunofluorescent APP (in green) and SMI-31 (in red) staining of WT and TREM2−/− naive mice and mice after 4, 6 and 12 weeks of CPZ treatment. The analysis was performed in the caudal CC area (×10 magnification). b Quantification of axonal beading (depicted by APP fluorescent staining), and of healthy, phosphorylated axons (measured by SMI-31 positivity) in the CC at the different time points. Data are from one experiment of two independent experiments performed with similar results. In the presented experiment for week 0 n = 3 naive WT and n = 5 naïve TREM2−/− mice; for week 4 n = 5 WT and n = 3 TREM2−/− mice; for week 6 n = 6 WT and n = 8 TREM2−/− mice; for week 12 n = 4 WT and n = 6 TREM2−/− mice. Values in the graphs are mean ± SD. c EM images of WT and TREM2−/− at 12 weeks of CPZ treatment. Black arrows indicate dystrophic autophagocytic axons and asterisks indicate Iba1+ immunolabeled microglia. d Quantification of the number of dystrophic axons at 4 and 12 weeks of CPZ treatment counted per field at ×3,000 magnification (area = 381 μm2; n = 2 mice/group). The horizontal lines in the graph represent medians. *P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.0001 using Mann–Whitney test
Clinical deficits in TREM2−/− mice after CPZ treatment relative to WT mice
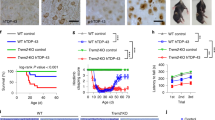
To evaluate possible functional deficits related to lack of TREM2 in mice treated with CPZ, behavioral testing was performed after 12 weeks of CPZ, a time-point at which axonal damage was markedly different between the two groups. We assessed neurologic functions by conducting a 1-h locomotor activity test, a battery of sensorimotor measures and the rotarod test. Locomotor activity data showed no differences between groups with regard to general ambulatory activity, frequency of vertical rearings or in terms of the entries made, time spent, or distance traveled in the center (data not shown). However, sensorimotor tests revealed that TREM2−/− mice were significantly impaired on 3/7 measures. Specifically, the TREM2−/− mice took significantly longer time to climb down a vertical pole without falling and longer to climb to the top of 60° or 90° screens compared to the WT (Fig. 3a). No differences were observed with regard to initiation of movement out of a small, circumscribed area (walking initiation test), or on tests of balance (ledge and platform), or in grip strength (inverted screen) (data not shown). The TREM2−/− mice also exhibited performance deficits on the two more difficult components of the rotarod test, in which they spent significantly less time on the constant speed and accelerating rotarod conditions compared to WT (Fig. 3d, e), while times on the stationary rod were not different. In general, the behavioral findings suggest that CPZ treatment resulted in coordination deficits in TREM2−/− mice as documented by the rotarod rod results, and this interpretation is consistent with the slowed responses of the TREM2−/− mice on the pole and inclined screen tests which require coordination between the forelimbs and hindlimbs. The lack of differences between the groups on the general activity variables and other sensorimotor tests suggest that the observed behavioral deficits were not due to general malaise or torpor produced by the CPZ treatment and that the impairment was relatively selective since functions like balance or strength did not appear to be affected in TREM2−/− mice.

Clinical deficits in TREM2−/− mice after CPZ treatment. Behavioral testing in WT and TREM2−/− mice was performed after 12 weeks of CPZ treatment (n = 10/group). TREM2−/− mice showed impaired motor coordination compared to WT as measured by (a) time to climb down a pole and (b, c) time to reach the top of a 60° and 90° inclined screen. d, e TREM2−/− mice spent significantly less time on the constant speed (d Genotype effect: P = 0.038) and accelerating (e Genotype effect: P = 0.00016 and Genotype × Trials interaction: P = 0.013) rotarod trials compared to WT, indicating impaired performance on this test. Subsequent pairwise comparisons conducted on the accelerating rotarod data showed significant differences between groups (beyond Bonferroni correction; P = 0.05/6 = 0.0083) for Trial 2 during Sessions 2 and 3 (P = 0.006 and 0.001, respectively), while large differences were also found during Trial 1 for Sessions 1 and 2 (P = 0.040 and 0.011, respectively). Pairwise comparisons also showed that large differences occurred during Trial 2, Session 2 and Trial 1, Session 3 (P = 0.048 and 0.043, respectively) for the constant speed rotarod condition. *P ≤ 0.05; **P ≤ 0.005 using ANOVA
Decreased number of microglia in TREM2−/− mice relative to WT during acute demyelination
In the CNS, TREM2 is specifically expressed by microglial cells, which are believed to be critical for the clearance of tissue debris in homeostatic conditions and during neurologic diseases [51, 52]. We reasoned that the lack of clearance of myelin debris in the CC of TREM2−/− mice in the CPZ model, associated with worse pathology and clinical impairment, could be related to a defect in microglia number and/or functions. The number of microglial cells was quantified in the CC of TREM2−/− and WT mice in naïve mice and after 4, 6 and 12 weeks of CPZ treatment by immunostaining for ionized calcium-binding adapter molecule 1 (Iba1), a microglial cell marker (Fig. 4a). No significant differences were noted in the number of microglia in the CC of naïve TREM2−/− and WT mice (age 6–8 months old), with just a trend toward fewer microglia in TREM2−/− mice. At 4 and 6 weeks on CPZ treatment, accumulation of Iba1+ cells compared to naive was observed in the CC of WT and TREM2−/− mice, but the number of Iba1+ cells was significantly lower in TREM2−/− mice compared to WT (Fig. 4a, b). At 12 weeks on CPZ, the number of Iba1+ cells in WT and TREM2−/− CC was greatly reduced compared to 4 and 6 weeks and no significant differences were noted between the two groups, but a trend toward more Iba1+ cells in the CC TREM2−/− mice.

Decreased number of microglia in TREM2−/− mice during acute demyelination. a The number of microglial cells was evaluated in the CC of TREM2−/− and WT mice in naïve mice and after 4, 6 and 12 weeks of CPZ treatment by immunostaining for Iba1. Images were acquired in the CC of WT and TREM2−/− mice (×10 and ×60 magnification images are presented for WT and TREM2−/− panels). b The number of Iba1+ microglial cells in the CC was quantified. Data are from one experiment of three independent experiments performed with similar results. In the presented experiment for week 0 n = 3 each for naive WT and TREM2−/− mice; for week 4 n = 7 WT and n = 5 TREM2−/− mice; for week 6 n = 9 WT and n = 9 TREM2−/− mice; for week 12 n = 5 WT and n = 9 TREM2−/− mice. Values are mean ± SD. c Microglia morphometric analysis at the different time points was done using a ramification index [RI = 4π × cell area/(cell perimeter)2] that describes microglia cell shape. RI of 1 is a perfectly round cell. RI is smaller than 1 if morphology deviates from perfectly circular and RI is close to zero when the cell is ramified (10 cells/group were analyzed in naïve mice and a total of 40 cells/group were analyzed from 4 to 9 independent mice/group at all other time points). Horizontal lines are means. d MHC II e iNOS and f Mac-3 expression was evaluated by IHC on Iba1+ cells in the CC of TREM2−/− and WT mice at the different time points (n = 4/group at all time points). Horizontal lines in the graphs represent medians. *P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.0001, using Mann–Whitney test
Notably, we also observed differences in microglia morphology in the two groups over time. Microglia in a resting state display a ramified morphology and once activated acquire an amoeboid shape [29]. We quantified microglial cell morphology at the different time points during CPZ treatment using a ramification index (as defined in methods). In naïve WT and TREM2−/− mice, microglia displayed a ramified morphology characteristic of a resting state (Fig. 4a, 60× images) and the RI was close to zero for both groups (Fig. 4c). At 4 weeks on CPZ during acute demyelination, while WT microglia acquired a typical activated amoeboid shape, TREM2−/− microglia retained a ramified morphology (Fig. 4a, 60× images). At this time, the RI for WT microglia was significantly higher than the RI of TREM2−/− mice (0.6 ± 0.1 and 0.1 ± 0.1, respectively, P < 0.0001). At 6 weeks on CPZ most of WT microglia in the CC appeared to return to a ramified shape, as the RI went down compared to that at 4 weeks, at which point no significant differences in RI were noted between WT and TREM2−/− microglia. At 12 weeks on CPZ most WT microglia resumed a ramified morphology characteristic of a resting state, with RI close to zero. Notably, at this time point TREM2−/− microglia displayed a RI that was slightly but significantly higher than WT (Fig. 4c).
It was previously reported that astrogliosis accompanies microglia accumulation in areas of demyelination due to CPZ feeding [29, 45]. We investigated astrocyte number and distribution in the CC in WT and TREM2−/− naïve mice at 4, 6 and 12 weeks on CPZ by immunostaining for GFAP. As expected, GFAP expression was nil in both WT and TREM2−/− naïve mice. In WT mice fed CPZ, GFAP staining progressively increased in the CC from 4 to 12 weeks (mean ± SD for % of GFAP staining was 16 ± 3, 39 ± 10 and 41 ± 18 at 4, 6 and 12 weeks, respectively). In contrast, in TREM2−/− mice GFAP staining increased at 4 weeks compared to naïve mice but then remained stable at 6 and 12 weeks (GFAP staining in TREM2−/− CC was 14 % ± 3, 11 % ± 4 and 20 % ± 7 at 4, 6 and 12 weeks, with P values vs. WT not significant, <0.0001 and <0.01, at the respective time-points).
Defective activation of TREM2−/− microglia during acute demyelination
Our morphometric analysis comparing TREM2−/− and WT microglia prompted us to postulate that activation of TREM2−/− microglia is defective. To test this hypothesis, we compared microglial expression of MHC II, which increases upon microglial activation in this model [82]. As expected, microglia in the CC of naïve TREM2−/− and WT mice did not express MHC class II. At 4 weeks on CPZ MHC II expression increased 60-fold in WT microglia. In contrast, at 4 weeks MHC II expression was still minimal in TREM2−/− microglia. At 6 weeks on CPZ, MHC II expression by WT microglia decreased but remained significantly higher than baseline and higher compared to TREM2−/− microglia. At 12 weeks, levels of MHC II expression declined almost to naive levels, and no significant differences were noted between the two groups (Fig. 4d). Next, we evaluated the expression of another marker of microglial activation, inducible nitric oxide synthase (iNOS). iNOS was upregulated 50-fold in WT microglia at 4 weeks compared to resting conditions. In contrast, iNOS upregulation at 4 weeks after CPZ on TREM2−/− microglia was about 20-fold, significantly lower than that in WT mice. At 6 weeks after CPZ, iNOS expression declined in both WT and TREM2−/− microglia, but remained significantly lower in the latter. At 12 weeks, iNOS expression returned to baseline in WT but not in TREM2−/− microglia (Fig. 4e). At that time, TREM2−/− expressed significantly more iNOS than WT. Expression of Mac-3, which increases on microglia upon activation, was also evaluated by IHC. At 4 weeks, Mac-3 was upregulated sixfold in WT microglia compared to resting conditions. In contrast, Mac-3 upregulation at 4 weeks after CPZ on TREM2−/− microglia was about threefold, significantly lower than in WT mice (Fig. 4f). In summary, TREM2−/− microglia demonstrate less activation than WT microglia during CPZ feeding but have a delayed return toward baseline.
Defect of microglia proliferation in TREM2−/− mice
In the CPZ model, the increased number of microglia in the CC is mainly due to local microglial proliferation [47, 82]. We compared microglial proliferation in the CC in TREM2−/− and WT mice at 4 weeks on CPZ by BrdU incorporation, using IHC (Fig. 5a). The percentage of BrdU+Iba1+ microglia was significantly decreased in TREM2−/− compared to WT mice (Fig. 5b). The Iba1 marker does not reliably discriminate between microglia and macrophages. To specifically assess proliferation of microglia versus macrophages by BrdU incorporation, we isolated mononuclear cells from whole brains of TREM2−/− and WT mice after 4 weeks of CPZ treatment. Microglia and macrophages were discriminated by flow cytometry using the markers CD45 and CD11b with microglia being CD11b+CD45low and macrophages CD11b+CD45high. In the CPZ model, microglia activation is not accompanied by significantly increased expression of CD45 and CD11b [82]. Proliferation of TREM2−/− microglia was significantly less than WT microglia (Fig. 5c). In contrast, no differences were noted in macrophage proliferation between the two groups (Fig. 5d). Thus, lack of TREM2 was associated with reduced proliferation by microglia which contributed to the reduced number of Iba1+ cells in the TREM2−/− CNS in response to CPZ intoxication.

Defect of microglia proliferation in TREM2−/− mice. a Proliferation of microglia in the CC in TREM2−/− and WT mice at 4 weeks on CPZ after BrdU incorporation was evaluated by IHC (×60 images). White arrows indicate the nuclei of BrdU+/Iba+ proliferating microglia. b The percentage of BrdU+/Iba+ microglia relative to the total number of Iba+ microglia was quantified in the CC of TREM2−/− and WT mice after 4 weeks on cuprizione (n = 4 mice/group). This is one representative experiment out of two independent experiments performed with similar results. Values are median ± range. c Brain mononuclear cells were isolated from TREM2−/− and WT mice at 4 weeks on CPZ and after BrdU+ treatment. Microglia and macrophages were distinguished by flow cytometry using the CD45 and CD11b markers. BrdU incorporation was quantified by flow cytometry in CD11b+CD45low microglia and CD11b+CD45high macrophages (n = 5/group). This is one representative experiment out of two performed with similar results. Values are mean ± SD. *P ≤ 0.05; **P ≤ 0.01 using Mann–Whitney test
Defect in lipid metabolism in TREM2−/− microglia
To identify the mechanisms underlying the lack of activation of TREM2-deficient microglia in the CPZ-induced model, we compared gene expression profiling of the CC from TREM2−/− vs. WT mice at 6 and 12 weeks. Gene arrays identified 13 and 62 genes that were differentially expressed using pre-specified cut-off values between WT and TREM2−/− mice at 6 and 12 weeks, respectively (Supplementary Table S1). Most of the differentially expressed genes were changing in the same direction at the two time points (although statistical significance may not have been reached at both time points); For TREM2−/− mice, five genes had lower expression and 50 had higher expression compared to WT at each time point, whereas 7 genes changed in opposite directions at the two time points (Fig. 6b). We used two published datasets that define a naïve adult microglia gene expression signature [7, 28] to analyze our list of differentially expressed genes. Based on the published microglia-unique genes the TREM2−/− and WT expression arrays were clearly different, with the hierarchical analysis based on one published data set shown in Fig. 6c [28]. No major differences in WT vs TREM2−/− hierarchical gene expression for astrocyte- or oligodendrocyte-associated genes [28] were observed in our dataset (data not shown). Notably, many of those gene transcripts recently defined as naïve adult microglia-unique genes were expressed at higher levels in the CC of TREM2−/− mice compared to WT after CPZ treatment, including P2ry12, P2ry13, Fcrls, Tmem119, Gpr34, Siglech, Olfml3 and Slc2a5 (Fig. 6b, c). Increased expression of these genes in TREM2−/− compared to WT microglia purified from the brain after 4 weeks on CPZ was validated by qRT-PCR (Fig. 6d).

Microarray analysis in the TREM2−/− and WT corpus callosum at 6 and 12 weeks on CPZ. Gene expression array analysis was conducted on the CC isolated from WT and TREM2−/− mice at 6 (4 WT and 3 TREM2−/− mice) and 12 weeks (5 WT and 6 TREM2−/− mice) after CPZ treatment. a Venn diagram showing the number of genes that were differentially expressed (cut-off values: logarithmic fold change 1 and adjusted P value <0.05) between TREM2−/− and WT at 6 and 12 weeks. b List of all differentially expressed genes between the two groups for both time points using the predefined cut-off values (TREM2−/− vs. WT mice). c Hierarchical clustering analysis based on the published data set of microglia-unique gene expression signature [28]. d Gene expression quantification of some microglia-unique genes by qRT-PCR in purified WT and TREM2−/− microglia after 4 weeks on CPZ (from n = 6 WT and 8 TREM2−/− mice). Values are mean ± SD. *P ≤ 0.05; **P ≤ 0.01 using Mann–Whitney test
Next, we used Ingenuity IPA Network Generation software to identify the major cellular functions implicated by the differentially expressed genes in TREM2−/− vs. WT CC. Based on the prediction algorithm, the network called “lipid metabolism, molecular transport, small molecule biochemistry” was the top altered in the TREM2−/− mice at 12 weeks (score of 46) (Supplementary Fig. 2a). Nineteen genes in that pathway had higher expression and one, lipoprotein lipase (LPL), had lower expression in the TREM2−/− mice compared to WT. Analysis with the Ingenuity downstream effect analysis tool identified the cellular concentration of fatty acids as a biological function that would be affected (P = 6.29 × 10−5 by Fisher’s Exact Test) given the observed gene expression changes in our dataset. Seven genes are normally implicated in regulating cellular fatty acid concentration and they were all listed among those differentially expressed in TREM2−/− mice compared to WT (Fig. 6b). Five of these pathway activator genes (Alox5AP, PTGS1, LTC4S, HSD11B1, P2RY13) were induced in TREM2−/− mice consistently with that predicted by the analysis tool in respect of the downstream effects on fatty acid concentration. In contrast, two genes had expression levels that were opposite to what predicted: phosphodiesterase 3B (PDE3B-a pathway inhibitor), which was induced and LPL (a pathway activator) that was abnormally decreased in TREM2−/− mice compared to what expected by downstream effects (Supplementary Fig. 2b).
LPL was among the top genes whose expression was significantly reduced in TREM2−/− mice compared to WT after CPZ treatment at both 6 (−2 log fold change, adjusted P value of 3.8 × 10−2) and 12 weeks (−1.3 log fold change, adjusted P value of 4.3 × 10−2) (Fig. 6b). LPL is an enzyme implicated in the degradation of lipids that is expressed in microglia in the CNS [19, 43]. We evaluated LPL expression by qRT-PCR in TREM2−/− and WT microglia isolated from the brain of naïve TREM2−/− and WT mice and at 4 weeks on CPZ. LPL transcripts were greatly induced in WT but not by TREM2−/− microglia after CPZ (Supplementary Fig. 2c). LPL protein expression was evaluated in the CC by IHC in naïve mice and after 4 weeks on CPZ, with significant increase of LPL observed at 4 weeks in WT but not in TREM2−/− mice (Supplementary Fig. 2d).
Defective myelin degradation by microglia in TREM2−/− mice
Defective clearance of myelin debris in TREM2−/− mice could be due in part to the lower number of microglial cells in TREM2−/− mice. However, the defective microglial activation in these mice suggested that there might also be a defect in myelin uptake and/or degradation by TREM2−/− microglia. To directly assess myelin uptake, WT and TREM2−/− adult microglia were isolated and cultured with myelin and then analyzed for MBP content by IHC as a measure of myelin uptake. No differences were noted between the two groups (Fig. 7a). In this in vitro assay, TREM2−/− microglia had a lower RI compared to WT, similar to our in vivo observations (Fig. 7b)

Defect in myelin degradation in TREM2−/− microglia. a Microglia were purified by sorting based on CD11b and CD45 expression from the brain of TREM2−/− and WT naïve mice and cultured in vitro for 24 h in presence of myelin. Myelin uptake was quantified by immunofluorescence staining for MBP detected in Iba1+ microglia cells. Mean ± SEM are shown. b Microglia morphology was quantified using the RI as expression of activation. RI equals 1 for a round cell and below 1 if the morphology deviated from round. Mean ± SEM are shown. c Immuno-EM images of TREM2−/− and WT microglia stained with Iba1 in the CC at 4 and 12 weeks on CPZ treatment. Images on the left in WT and TREM2−/−panels at week 4 and 12 (3,000× magnification) depict Iba+ microglial cells (asterisks). A higher magnification (15,000×) for the boxed area is shown on the right of each image. Black arrows indicate phagosomes containing myelin debris. White arrows indicate pi granules. d Number of phagosomes containing myelin. e The diameter of phagosomes and f the number of pi granules were quantified per field at 3,000× magnification (area = 381 μm2) (n = 2 mice/group). Horizontal lines are medians. *P ≤ 0.05; **P ≤ 0.0005; ***P ≤ 0.0001 using Mann–Whitney test. NA not applicable
To investigate myelin processing by microglia in vivo, we performed immuno-EM of Iba1 labeled microglia in WT and TREM2−/− mice after 4 and 12 weeks on CPZ (Fig. 7c). We quantified the number of phagosomes containing myelin figures (lamellate structures) in Iba1+ microglia in the CC of WT and TREM2−/− mice (Fig. 7d). At 4 weeks, the number of phagosomes was significantly higher in TREM2−/− than in WT CC. At 12 weeks no phagosomes containing myelin were detected in WT mice, while phagosomes filled with myelin figures were amply present in TREM2−/− CC (Fig. 7d). TREM2−/− phagosomes also had significantly larger diameters at 4 weeks (Fig. 7e). At 12 weeks in WT mice, microglial cells displayed extensive presence of lipopigments by EM, called pi granules, which are considered to be residue of myelin breakdown [40]. Pi granules were absent at 4 weeks during acute demyelination in CC of both WT and TREM2−/− mice. At 12 weeks, we observed significantly more pi granules in WT microglia than in TREM2−/− CC (Fig. 7f). These results suggest that TREM2−/− microglia have a defect in lipid metabolism and myelin breakdown, but do not support a defect in myelin uptake.
Discussion
More than 10 years ago it was described that human genetic deficiency of the microglial receptor TREM2 leads to a neurodegenerative disease, PLOSL, characterized by severe brain atrophy with major white matter involvement and diffuse neuronal and axonal loss [61]. Moreover, recent reports have demonstrated that rare variants of TREM2 confer an increased risk to develop common neurodegenerative diseases including AD, FTD, PD and ALS [3, 5, 8, 22, 34, 35, 64, 67, 71, 77]. TREM2 has been reported to exert a variety of functions in different myeloid cell types, such as to positively regulate phagocytosis in microglia in vitro [80], to decrease pro-inflammatory responses of mouse macrophages and microglia in vitro [79, 80] and to promote proliferation of immature bone marrow-derived macrophages [59]. These results have been extrapolated to postulate that TREM2 expressed in resident microglia would have a key role in CNS homeostasis, in the clearance of cell debris and metabolic waste and in sustaining an anti-inflammatory state [53]. However, the relevance of these studies to microglia function is largely speculative since the functions of TREM2 in microglia in vivo have not been investigated.
In this study, we define a role for TREM2 as a critical regulator of microglia activation in the CNS in vivo in response to oligodendrocyte damage using the well-characterized CPZ-induced demyelination model. This model allows the study of brain-resident microglia function. Microglia are the first responders to CPZ-induced injury and their proliferation is largely responsible for Iba1+ cell accumulation with very limited infiltration of monocyte/macrophages from the periphery [29, 47, 69]. TREM2 is markedly upregulated on microglia during CPZ treatment [82]. Herein, we provide in vivo evidence that lack of TREM2 in the mouse results in defective microglia activation in response to myelin damage and defective clearance of myelin debris. TREM2 deficiency affected multiple microglial functions including proliferation and the capacity to degrade myelin and was associated with altered morphology and cell marker profiles suggestive of a broad defect in microglia activation. The result of the microglial dysfunction was the accumulation of myelin debris in the CNS, with more axonal damage and clinical impairments in TREM2−/− mice. These findings indicate that TREM2 promotes neuroprotective functions of resident microglia in the CNS in response to local cell damage. Most importantly, our results support the notion that TREM2 engagement is essential for microglia activation in response to tissue injury.
The function of TREM2 in neurological diseases has been evaluated in just a few studies. We and others have previously shown a protective role for TREM2 in an animal model for multiple sclerosis, experimental autoimmune encephalomyelitis, in which TREM2 expression was preferentially expressed by resident microglia [63, 78]. In autopsied multiple sclerosis subjects TREM2 was highly expressed on macrophages/microglia that had engulfed myelin debris [62]. More recently, TREM2 genetic variants have been associated with susceptibility to AD and other neurodegenerative diseases [68]. In transgenic mouse models of AD, TREM2 expression is increased in amyloid plaque-associated microglia, particularly in the outer zone of amyloid plaques [17, 48]. Since TREM2 may affect the uptake of aggregated proteins in vitro [38], and clearance of these products is postulated to play a key role in protection from AD and other neurodegenerative proteinopathies [2, 46], it is tempting to speculate that TREM2 mutations could favor neurodegeneration by impairing microglia function to clear cell debris and metabolic waste in the CNS. Notably, treatment with CSF-1 or IL-34, which increases microglia numbers and internalization of β-amyloid, leads to reduction of amyloid deposition and amelioration of memory deficits in transgenic AD mouse models [4, 42, 49]. Loss-of-function mutations in the gene coding for CSF-1 and IL-34 receptor, CSF-1R, cause hereditary diffuse leukoencephalopathy with axonal spheroids and pigmented orthochromatic leukodystrophy, two genetic diseases with striking similarities in terms of clinical phenotype and pathological findings with PLOSL, due to TREM2/DAP12 mutations [54, 65]. Since the TREM2/DAP12 complex cooperates with CSF-1R [59, 60, 76], this could be a mechanism through which TREM2 regulates microglia activities.
Most neurological diseases display activated microglia in areas of tissue damage. In response to tissue injury, microglia undergo a rapid transition from resting, ramified forms to amoeboid morphologies [66]. TREM2 deficiency increases the expression of pro-inflammatory products in mouse macrophages and microglial cell lines in response to TLR ligands and neuronal debris [24, 79, 80]. Thus, it has been hypothesized that loss-of-function mutations in TREM2 may promote neurotoxic microglial activation [27]. However, in our model, TREM2-deficient mice showed increased axonal damage as a result of impaired microglia activation and function, with no evidence of higher pro-inflammatory cytokine production based on transcriptome analysis. Interestingly, many gene transcripts recently defined as unique to naïve adult microglia were found at higher levels in TREM2−/− mice compared to WT (Fig. 6b-c), including the purinergic receptor P2ry12, Tmem119, Gpr34, Fcrls+ and SiglecH. These genes are expressed in naïve microglia, but their expression is dramatically reduced after microglia activation [25, 41]. These results further support a less activated phenotype of TREM2−/− compared to WT microglia during CPZ intoxication. This is consistent with reduced transcription of pro-inflammatory cytokines and chemokines described in TREM2−/− mice subjected to an inflammatory brain ischemia model in its sub-acute phase [73]. Another study in an AD mouse model showed a decrease of plaque-associated microglia in mice with only one copy of TREM2 [81]. Our study expands on these previous findings by providing evidence that lack of TREM2 leads to a much broader defect in microglia activation, including proliferation and degradation of cell debris. As an aside, we observed much less astrogliosis in TREM2−/− compared to WT, suggesting that dysfunctional microglial activation and proliferation affect astrocyte functions. Interestingly, Skripuletz et al. [74] showed that depletion of astrocytes in vivo results in a decrease in microglia accumulation, suggesting that astrocytes can also provide signals that promote microglia recruitment and activation to clear debris. TREM2 could directly mediate this interaction as astrocytes have been shown to express binding sites for TREM2 [11, 63].
The human disease PLOSL usually presents with CNS symptoms in the third–fourth decades of life, suggesting that TREM2 deficiency in CNS microglia requires accumulation of damage to become clinically evident. Naïve TREM2-deficient mice do not show a phenotype resembling human PLOSL (our unpublished results), which may be due to the short lifespan of mice as compared to humans. However, upon prolonged exposure to CPZ in our studies, TREM2-deficient mice were significantly impaired on measures of motor coordination compared to WT mice. These differences only became apparent in the more difficult components of the behavioral testing. Interestingly, prolonged CPZ exposure (12 weeks in our system) in our studies was associated with a slightly but significantly less ramified morphology of TREM2−/− microglia and with a significant higher iNOS expression compared to WT. At higher magnification by fluorescence microscopy, TREM2−/− microglia had a vacuolated appearance, which was further demonstrated by EM to derive from the persistent presence of cell vacuoles filled with undigested myelin. Together, these findings suggest that lack of TREM2 on microglia in the presence of a prolonged insult leads to a dysfunctional microglia phenotype. This aberrant microglia state could contribute to the accumulation of tissue damage observed in the mouse model and in humans, but this hypothesis needs to be investigated.
Herein we show that lack of TREM2 is associated with a defect in myelin degradation. Several pieces of evidence support this: first, the continued presence of phagosomes containing myelin in TREM2−/− microglia after 12 weeks on CPZ, a time point when most myelin have been degraded in WT microglia. Second, gene expression analysis suggested that absence of TREM2 is associated with a defect in lipid metabolism. We identified decreased LPL as one potential factor leading to defective myelin degradation in the absence of TREM2. We showed that LPL expression is robustly induced both transcriptionally and at the protein level in WT microglia in areas of demyelination after 4 weeks on CPZ. This was in accord with a prior report that proposed that LPL could be involved in recycling of lipids from degenerating myelin for use by remyelinating oligodendrocytes [58]. LPL induction after CPZ intoxication was not observed in TREM2−/− microglia. However, we cannot conclude if the reduction of LPL is a downstream effect or a direct consequence of lack of TREM2. Interestingly, LPL accumulates in senile plaques of AD brains [55], and microglial LPL has been suggested to play a role in amyloid β uptake and degradation in vitro [43].
In summary, our findings reveal a key role for the receptor TREM2 in promoting resident microglia activation and response to tissue injury caused by CPZ. Our data support the hypothesis that TREM2 facilitates multiple neuroprotective functions of microglia in response to CNS tissue damage, functions that could relate to many different neurological diseases. The broad defects we have observed in TREM2−/− microglia suggest that TREM2 engagement on the cell surface could be one of the first events in microglia activation. Alternatively, TREM2 may function downstream of this pathway as an essential molecule to control several different cell functions (proliferation, phagocytosis and lipid degradation) independently. Further clarifying the role of TREM2 may open new avenues for therapeutic intervention in neurodegenerative diseases.
References
Archambault AS, Sim J, McCandless EE, Klein RS, Russell JH (2006) Region-specific regulation of inflammation and pathogenesis in experimental autoimmune encephalomyelitis. J Neuroimmunol 181:122–132. doi:10.1016/j.jneuroim.2006.08.012
Bayer TA (2013) Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2013.03.007
Benitez BA, Cruchaga C (2013) TREM2 and neurodegenerative disease. N Engl J Med 369:1567–1568. doi:10.1056/NEJMc1306509#SA4
Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S (2009) Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain 132:1078–1092. doi:10.1093/brain/awn331
Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, Fenoglio C, Piaceri I, Archetti S, Bonvicini C et al (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35(934):e937. doi:10.1016/j.neurobiolaging.2013.09.017
Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM, Bayly PV (2007) Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma 24:657–673. doi:10.1089/neu.2006.0011
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE et al (2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143. doi:10.1038/nn.3599
Cady J, Koval ED, Benitez BA, Zaidman C, Jockel-Balsarotti J, Allred P, Baloh RH, Ravits J, Simpson E, Appel SH et al (2014) TREM2 variant p. R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol 71:449–453. doi:10.1001/jamaneurol.2013.6237
Colonna M (2003) TREMs in the immune system and beyond. Nat Rev Immunol 3:445–453. doi:10.1038/nri1106
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nature Neurosci 8(6):752–758
Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE (2003) Pattern recognition by TREM-2: binding of anionic ligands. J Immunol 171:594–599
Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP (2012) Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 60:717–727. doi:10.1002/glia.22298
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL et al (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. doi:10.1016/j.neuron.2014.02.040
Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW (2011) Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 6:e26317. doi:10.1371/journal.pone.0026317
Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3):393–399
Ford JW, McVicar DW (2009) TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol 21:38–46. doi:10.1016/j.coi.2009.01.009
Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T (2008) TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56:1438–1447. doi:10.1002/glia.20710
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Geno Biol 5:R80. doi:10.1186/gb-2004-5-10-r80
Gong H, Dong W, Rostad SW, Marcovina SM, Albers JJ, Brunzell JD, Vuletic S (2013) Lipoprotein lipase (LPL) is associated with neurite pathology and its levels are markedly reduced in the dentate gyrus of Alzheimer’s disease brains. J Histochem Cytochem 61:857–868. doi:10.1369/0022155413505601
Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR (2006) Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J Neurosci 26:2841–2851. doi:10.1523/JNEUROSCI.4823-05.2006
Greter M, Merad M (2013) Regulation of microglia development and homeostasis. Glia 61:121–127. doi:10.1002/glia.22408
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S et al (2012) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127. doi:10.1056/NEJMoa1211851
Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H et al (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol 70:78–84
Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 177:2051–2055
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9:1512–1519. doi:10.1038/nn1805
Heppner FL, Roth K, Nitsch R, Hailer NP (1998) Vitamin E induces ramification and downregulation of adhesion molecules in cultured microglial cells. Glia 22:180–188
Hickman SE, El Khoury J (2014) TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem Pharmacol 88:495–498. doi:10.1016/j.bcp.2013.11.021
Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. doi:10.1038/nn.3554
Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK (1998) Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol 92:38–49
Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109(4): 1144–1156
Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, Oakley AE, Englund E, O’Brien JT, Ince PG, Kalaria RN (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 119:579–589. doi:10.1007/s00401-009-0635-8
Ito H, Hamerman JA (2012) TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol 42:176–185. doi:10.1002/eji.201141679
Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP (2010) The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci 30:15811–15820. doi:10.1523/JNEUROSCI.4088-10.2010
Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, Norton JB, Hsu S, Harari O, Cai Y et al (2014) Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. doi:10.1093/hmg/ddu277
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ et al (2012) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–116. doi:10.1056/NEJMoa1211103
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C et al (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16:273–280. doi:10.1038/nn.3318
Kiernan JA (1984) Chromoxane cyanine R. II. Staining of animal tissues by the dye and its iron complexes. J Microsc 134:25–39
Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri Fet al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Trans Med 6: 243–286
Klunemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE et al (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64:1502–1507
Lassmann H, Ammerer HP, Jurecka W, Kulnig W (1978) Ultrastructural sequence of myelin degradation. II. Wallerian degeneration in the rat femoral nerve. Acta Neuropathol 44:103–109
Lewis ND, Hill JD, Juchem KW, Stefanopoulos DE, Modis LK (2014) RNA sequencing of microglia and monocyte-derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J Neuroimmunol 277: 26–38
Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, Zhu L, Alabsi H, Getachew R, Narasimhan R et al (2013) Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med 210:157–172. doi:10.1084/jem.20120412
Ma Y, Bao J, Zhao X, Shen H, Lv J, Ma S, Zhang X, Li Z, Wang S, Wang Q et al (2013) Activated cyclin-dependent kinase 5 promotes microglial phagocytosis of fibrillar beta-amyloid by up-regulating lipoprotein lipase expression. Mol Cell Proteomics 12:2833–2844. doi:10.1074/mcp.M112.026864
Matsuo A, Lee GC, Terai K, Takami K, Hickey WF, McGeer EG, McGeer PL (1997) Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: a potential mechanism for the generation of autoantigens. Am J Pathol 150:1253–1266
Matsushima GK, Morell P (2001) The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 11:107–116
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774. doi:10.1126/science.1197623
McMahon EJ, Suzuki K, Matsushima GK (2002) Peripheral macrophage recruitment in cuprizone-induced CNS demyelination despite an intact blood-brain barrier. J Neuroimmunol 130:32–45
Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, Stalder AK, Staufenbiel M, Neumann H, Carson MJ (2010) Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro 2:e00037. doi:10.1042/AN20100010
Mizuno T, Doi Y, Mizoguchi H, Jin S, Noda M, Sonobe Y, Takeuchi H, Suzumura A (2011) Interleukin-34 selectively enhances the neuroprotective effects of microglia to attenuate oligomeric amyloid-beta neurotoxicity. Am J Pathol 179: 2016–2027
N’Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, Hamerman JA, Seaman WE (2009) TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol 184(2): 215–223
Nayak D, Roth TL, McGavern DB (2014) Microglia development and function. Annu Rev Immunol 32:367–402. doi:10.1146/annurev-immunol-032713-120240
Neumann H, Kotter MR, Franklin RJ (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132: 288–295
Neumann H, Takahashi K (2007) Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 184: 92–9
Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, Nelson PT, Clark HB, Wszolek ZK, Dickson DW et al (2013) CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 80:1033–1040. doi:10.1212/WNL.0b013e31828726a7
Nishitsuji K, Hosono T, Uchimura K, Michikawa M (2011) Lipoprotein lipase is a novel amyloid beta (Abeta)-binding protein that promotes glycosaminoglycan-dependent cellular uptake of Abeta in astrocytes. J Biol Chem 286:6393–6401. doi:10.1074/jbc.M110.172106
Norkute A, Hieble A, Braun A, Johann S, Clarner T, Baumgartner W, Beyer C, Kipp M (2009) Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. J Neurosci Res 87:1343–1355. doi:10.1002/jnr.21946
Nyren R, Chang CL, Lindstrom P, Barmina A, Vorrsjo E, Ali Y, Juntti-Berggren L, Bensadoun A, Young SG, Olivecrona T et al (2012) Localization of lipoprotein lipase and GPIHBP1 in mouse pancreas: effects of diet and leptin deficiency. BMC Physiol 12:14. doi:10.1186/1472-6793-12-14
Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke HW (2012) Identification of a microglia phenotype supportive of remyelination. Glia 60:306–321. doi:10.1002/glia.21266
Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, Faccio R, Ross FP, Teitelbaum SL, Takayanagi H et al (2012) TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol 188:2612–2621. doi:10.4049/jimmunol.1102836
Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, Tassi I, Takai T, Stanley SL, Miller M et al (2009) Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol 10:734–743. doi:10.1038/ni.1744
Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A et al (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 71:656–662
Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J, Naismith RT, Panina-Bordignon P, Passini N (2008) Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131: 3081–3091
Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, Panina-Bordignon P (2007) Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol 37:1290–1301
Pottier C, Wallon D, Rousseau S, Rovelet-Lecrux A, Richard AC, Rollin-Sillaire A, Frebourg T, Campion D, Hannequin D (2013) TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J Alzheimers Dis 35:45–49. doi:10.3233/JAD-122311
Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, De Jesus-Hernandez M et al (2012) Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 44:200–205. doi:10.1038/ng.1027
Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145. doi:10.1146/annurev.immunol.021908.132528
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH et al (2013) TREM2 in neurodegeneration: evidence for association of the p. R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener 8:19. doi:10.1186/1750-1326-8-19
Reitz C, Mayeux R (2013) TREM2 and neurodegenerative disease. N Engl J Med 369:1564–1565. doi:10.1056/NEJMc1306509#SA1
Remington LT, Babcock AA, Zehntner SP, Owens T (2007) Microglial recruitment, activation, and proliferation in response to primary demyelination. Am J Pathol 170:1713–1724. doi:10.2353/ajpath.2007.060783
Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, Smyth GK (2006) Empirical array quality weights in the analysis of microarray data. BMC bioinformatics 7:261. doi:10.1186/1471-2105-7-261
Ruiz A, Dols-Icardo O, Bullido MJ, Pastor P, Rodriguez-Rodriguez E, de Lopez Munain A, de Pancorbo MM, Perez-Tur J, Alvarez V, Antonell A et al (2014) Assessing the role of the TREM2 p. R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging 35(444):e441–e444. doi:10.1016/j.neurobiolaging.2013.08.011
Schmid CD, Melchior B, Masek K, Puntambekar SS, Danielson PE, Lo DD, Sutcliffe JG, Carson MJ (2009) Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo. J Neurochem 109 (Suppl 1): 117-125
Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C (2013) Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS ONE 8:e52982. doi:10.1371/journal.pone.0052982
Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, Berger K, Kipp M, Baumgartner W, Stangel M (2013) Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 136:147–167. doi:10.1093/brain/aws262
Skripuletz T, Lindner M, Kotsiari A, Garde N, Fokuhl J, Linsmeier F, Trebst C, Stangel M (2008) Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am J Pathol 172:1053–1061. doi:10.2353/ajpath.2008.070850
Stanley ER, Chitu V (2014) CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect 6(6): a021857
Streit WJ, Xue QS (2014) Human CNS immune senescence and neurodegeneration. Curr Opin Immunol 29C:93–96. doi:10.1016/j.coi.2014.05.005
Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H (2007) TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med 4:e124
Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201:647–657
Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177:3520–3524
Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM (2014) Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener 9:20. doi:10.1186/1750-1326-9-20
Voss EV, Skuljec J, Gudi V, Skripuletz T, Pul R, Trebst C, Stangel M (2012) Characterisation of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol Dis 45:519–528. doi:10.1016/j.nbd.2011.09.008
Wettenhall JM, Simpson KM, Satterley K, Smyth GK (2006) affylmGUI: a graphical user interface for linear modeling of single channel microarray data. Bioinformatics 22:897–899. doi:10.1093/bioinformatics/btl025
Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ (2004) Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis 17:403–414. doi:10.1016/j.nbd.2004.08.006
Xie M, Tobin JE, Budde MD, Chen CI, Trinkaus K, Cross AH, McDaniel DP, Song SK, Armstrong RC (2010) Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol 69:704–716. doi:10.1097/NEN.0b013e3181e3de90
Acknowledgments
The authors thank Drs. Gregory F. Wu, Jessica Williams and Erika D. Koval for critical discussions, advice and/or for reading the manuscript; Dr. Marco Colonna for providing TREM2−/− mice; Mike Ramsbottom for technical assistance. This work was supported by grants from Fondazione Italiana Sclerosi Multipla (FISM) [2009/R/33 to L.P.], the National Multiple Sclerosis Society (NMSS, JF 2144A2/1 to L.P.) and the Dana Foundation “Program in the Neuroimmunology and Brain Infections and Cancer” (to L.P.). C.C. is a FISM fellow (2012/B/1). Histological support came from NIH PO1 NS059560 Core B to RSK. L.P. is a Harry Weaver Neuroscience Scholar of the NMSS. A.H·C. was supported by the Manny and Rosalyn Rosenthal Dr. John L. Trotter Chair in Neuroimmunology from Barnes-Jewish Hospital Foundation. GTAC is supported by NCI Cancer Center Support Grant #P30 CA91842, ICTS/CTSA Grant# UL1 TR000448 from the National Center for Research Resources (NCRR), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
Conflict of interest
The authors have no conflicting financial interests.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2015_1388_MOESM1_ESM.tif
Supplementary Fig. 1 Immuno-EM images indicating the presence of compacted or non-compacted myelin debris in WT and TREM2−/− mice at 3,000× (left column) and 15,000× (right column). Boxes indicate region of interest shown in higher magnification images on the right. Microglia were labeled with Iba and are marked with asterisks. WT mice at 4 weeks of cuprizone treatment contain small amounts of non-compacted myelin outside of microglia (white arrows) and inside microglia (black arrows). After 12 weeks of cuprizone treatment, the non-compacted myelin has been cleared, and remyelination can be seen by the presence of axons wrapped by compacted myelin (Ax). TREM2−/− mice at 4 weeks of treatment contain large amounts of non-compacted myelin outside of microglia (white arrows). After 12 weeks of treatment, TREM2−/− mice still contain significant amounts of non-compacted myelin outside of microglia (white arrows) and inside of microglia (black arrows). (TIFF 38348 kb)
401_2015_1388_MOESM2_ESM.tif
Supplementary Fig. 2 Defect in lipid metabolism in TREM2−/− mice. (a) Gene expression data for the 12 weeks on CPZ were uploaded to IPA for network analysis. Genes with a logarithmic fold change (TREM2−/− vs. WT mice) ≥ 1 and adjusted P value ≤ 0.05 were included in the analysis. The “Lipid Metabolism, Molecular Transport, Small Molecule Biochemistry” was the top network that was significantly altered in TREM2−/− compared to WT (score of 46) and it is presented in a. Red objects denote proteins that had increased gene expression, green objects denote proteins that had decreased gene expression in TREM2−/− compared to WT mice. Solid connecting lines imply direct relationship between proteins; dotted lines imply indirect interactions. (b) Analysis with the Ingenuity downstream effect analysis identified the cellular concentration of fatty acid as the biological function that is affected given the observed gene expression change. Red objects denote proteins with increased gene expression, green objects denote proteins with decreased gene expression in TREM2−/− vs. WT. Orange or yellow arrows denote when the change is consistent or inconsistent with the downstream effect, respectively. (c) LPL gene expression quantification by qRT-PCR in purified WT and TREM2−/− microglia from naïve mice (n = 3/group) or mice after 4 weeks on CPZ (from n = 6 WT and 8 TREM2−/− mice). (d) LPL protein expression quantification by IHC in the CC of naïve WT and TREM2−/− mice or after 4 weeks on CPZ (n = 4/group). Values are given as mean ± SD. * P ≤ 0.05; **P ≤ 0.001 using Mann–Whitney test (TIFF 23230 kb)
Rights and permissions
About this article

Cite this article
Cantoni, C., Bollman, B., Licastro, D. et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 129, 429–447 (2015). https://doi.org/10.1007/s00401-015-1388-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1388-1