
Abstract
Interfacial rheology has become a powerful tool to study the viscoelastic properties of interfaces in several multiphase polymer-based systems such as multilayer liquids containing surfactants, proteins or solid particles, and also in polymer blends, 3D printed multimaterials and coextruded multilayer films. During all these manufacturing processes, the elongational flow at interface is predominant. Nevertheless, direct interfacial rheological measurements in extension devoted to such polymer systems are not plentiful and are often based on indirect modelling methods. In the present work, interfacial dilational rheology testing based on the rising oscillating drop method was used to probe surface (and interfacial) properties of model Newtonian polymer melts: polydimethylsiloxane (PDMS)/polyisobutylene (PIB) systems. The interfacial properties in both oscillatory and static drop experiments were carefully corrected, considering the inertia and the contribution of the coexisting phase viscosities during the processing of the numerical data. The influence of molecular weight and temperature on the interfacial rheological responses was particularly examined. A new approach was developed to determine the dilational relaxation times (τ) of the studied polymer systems using a square pulse relaxation test. It was found that the evolution of τ with the temperature followed an Arrhenius behaviour. A comparison with capillary breakup extensional rheometry revealed similar overall values to those obtained with the pulse method. Finally, using interfacial shear rheology, we focused on the Trouton correlation between shear and dilational surface rheology, and a direct link between shear surface viscosities and elongational relaxation times was evidenced for the first time and over the entire viscosity range studied.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Multiphase systems constitute a topic of capital importance in biology, chemistry and material sciences. The interfacial or surface tension is the first criterion that one looks to evaluate when studying these systems. However, it is not the only surface or interfacial property worth considering. The tension gradient can provide information about the deformation resistance properties at the interface. For example, when surfactants or nanoparticles adsorb at liquid interfaces, they not only reduce the surface tension, but also confer intrinsic rheological properties to the corresponding interfaces.
When discussing the deformation of the interface (surface), we refer to the interfacial rheology in shear (Slattery et al. 2007; Vandebril et al. 2010; Renggli et al. 2020) as well as in dilatation/compression (Derkach et al., 2009).
Interfacial or surface dilational rheology, which is the main subject of this article, has become a powerful tool to study the static and dynamic properties of surfaces and interfaces such as interfacial layers containing surfactants (Lai et al. 2017), polymers (Sun et al. 2011), proteins (Erik M Freer et al. 2004) or solid particles (Noskov and Bykov 2018). Understanding the properties of these complex interfaces (surfaces) is the main challenge faced when aiming to control many technological and natural processes involving multiphase systems such as polymer blends (Dadouche et al. 2021), multilayer coextruded polymers (Zhang et al. 2015), overmolded parts, additively manufactured multimaterials, emulsions (Lei et al. 2019) or foams (Hetang Wang et al. 2019).
Furthermore, the close link that exists between the dynamic properties of complex interfaces as well as their adsorption mechanism makes dilational rheology one of the key tools for accessing the transport and kinetic processes involved in the physicochemical properties of a particular fluid/fluid system.
There are many experimental methods for studying interfacial dilational rheology, with all of them based on a disruption of the mechanical equilibrium at the interface and the analytical measurement of the interfacial response. Two methods are used to investigate dilational properties. The first one relies on the use of a Langmuir balance (Murray and Nelson 1996), which enables the measurement of the surface pressure by means of a tank topped by a pressure sensor and enclosed by two barriers that permit surface compression. This technique uses a Wilhelmy plate to measure the variation of the surface tension. The second method is the oscillating bubble/drop technique (Lunkenheimer and Kretzschmar 1975; Del Rıo and Neumann 1997), which consists in deforming a pendant drop (or a rising drop) at the end of a capillary in another liquid or gaseous phase.
Interfacial rheology in situations of dilatation/compression evaluates the change occurring in the interfacial tension with respect to the variation of the area or the volume of the interface (Lucassen and Van den Tempel 1972), whereas interfacial shear rheology measurements are performed using a constant interfacial area (Fig. 1) (Derkach et al. 2009).

Different methods for deforming an interface
In other words, the test consists in measuring the surface or interfacial tension γ when the area A of the interface changes (Derkach et al. 2009; Sun et al. 2011). The dilatational modulus Ed and the dilatational viscosity ηd are defined as:
In the case of oscillatory measurements (harmonic testing of the interface at different frequencies) where a surface at equilibrium is subjected to small periodic disturbances, a sinusoidal change in the surface tension with a certain phase shift is observed. This can be explained by the viscoelastic nature of the interface (Lucassen and Van den Tempel 1972).
Here we define the dilatational complex modulus \({E}_{d}^{*}\), including an elastic part Eʹ and a viscous part Eʺ,
where δ is the phase angle, A0 is the initial interfacial area, γ0 is the surface (or interfacial) tension at equilibrium, and ΔA and Δ \(\gamma\) are the amplitude of the interfacial area and surface tension, respectively (Lucassen and Van den Tempel 1972).
Oscillating bubble/drop method
The oscillating bubble/drop method is an important technique for studying interfacial dilational rheology. It allows the interfacial tension to be evaluated in either of two different ways. One is based on the analysis of the drop/bubble profile (Fig. 2) (axisymmetric drop shape analysis (ADSA)) (Ravera et al. 2010), while the other technique is based on the capillary pressure measurement (Mobius and Miller 1998). Both techniques exploit the relationship between the interface curvature, the pressure difference across the two phases and their interfacial tension, as described by the Laplace formula (Eq. (7)) (Young 1805; Laplace 1805).

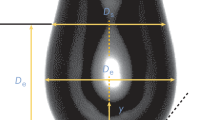
A pendant drop image indicating the coordinate system used for determining the surface tension. The drop shape is fit to a solution of the Young–Laplace equation
where γ is the surface/interfacial tension,
- R1 and R2:
-
are the first and second principal radii of curvature,
- ∆P = Pin − Pout:
-
is the Laplace pressure, which is the pressure difference across the interface,
- ∆P0:
-
is a reference pressure at z = 0,
- ∆ρgz:
-
is the hydrostatic pressure, and
- ∆ρ = ρd − ρc:
-
is the density difference. ρd and ρc are respectively the density of the drop phase and the density of the continuous phase.
Given the drop axisymmetry of the pendant drop, this relationship cannot be solved analytically but instead must be solved mostly numerically (the interfacial tension γ tends to reach an infinite value). The Young–Laplace law can be expressed in terms of cylindrical coordinates z and x, according to differential equations in terms of arc length s measured from the apex of the drop and also depending on the Bond number Bd.
This relation can also be expressed as a function of the tangent angle \(\varphi\) in a curvilinear coordinate system x(s) and z(s), which corresponds to a smoothed solution of the Laplace equation.
From the arc length s measured from the drop apex, the first and second principal radii of curvature are given by:
Because of the nature of the drop, the curvature at the apex is constant in all directions:
The reference pressure could be expressed as:
Therefore Eq. (7) can be obtained as a coupled set of differential equations in terms of the arc length x measured from the drop apex:
where Rapex is the radius of curvature of the surface.
Bd is the Bond number, a dimensionless quantity that represents the ratio between the gravitational forces and the surface tension on an interface between two fluids. The shape of the drop depends on this dimensionless number.
According to the Bond number expression, it is interesting to note that as the diameter of the needle decreases, the minimum Bond number value needed for an accurate calculation decreases. Berry et al. (2015) introduced a universal solution that is independent of the needle diameter by using a non-dimensional number called the Worthington number, Wo, which scales from 0 to 1.
Wo is defined as the ratio of the drop volume V to the theoretical maximum drop volume Vmax and expressed as:
where Vmax is expressed as:
and Dc is the diameter of the needle.
A low Bond number means that the drop is more spherical. However, the more the drop takes on a spherical shape, the greater the relative error of the measurement becomes. Furthermore, when the two liquids are of the same density, the measurement will be delicate (Berry et al. 2015). For high accuracy, a high Worthington number value is needed. To do this, it is necessary to increase the radius of curvature at the apex, which in turn leads to a higher drop volume.
After all of these precautions are taken, the drop is subjected to a change in volume or area (through a piezoelectric or mechanical motor) (Fig. 3) with a sinusoidal deformation profile, and the shape of the drop at each oscillation allows the dilatational viscoelastic modulus Ed to be calculated.

Diagram of the experimental setup for the oscillating drop/bubble method
Inertial and viscosity effects versus interfacial effects
In static drop measurements, the Young–Laplace law is valid. In other words, the gravitational forces acting on the drop are in equilibrium with the interfacial forces. However, in the case of oscillatory measurements, the interfacial area changes during the time of the experiment. In this instance, inertial forces and viscous forces appear and modify the shape of the drop/bubble. Therefore, the Young–Laplace law becomes invalid, thereby restricting the range of frequencies that can be employed in the measurements (the highest frequency is limited) (E. M. Freer et al. 2005; Santini et al. 2007).
To quantify the inertial and viscous forces, one must calculate both the capillary number Ca, the ratio of viscous to capillary forces, and the Weber number We, the ratio of inertial to capillary forces, as defined below (E. M. Freer et al. 2005):
where Δη and \(\Delta\) ρ are the difference between the Newtonian viscosities and densities of the drop and the continuous fluid, ω is the oscillation frequency of the drop, ΔV is the amplitude of the volume oscillation, \({\varvec{\gamma}}\) is the equilibrium interfacial tension and Rc is the radius of the capillary.
In the limits of Ca < < 1 and We < < 1, the drop shape is that of a static drop and is well described by the Young–Laplace equation. To obtain low capillary and Weber numbers, we must take certain experimental precautions, such as limiting the applied amplitudes and frequencies and using a needle with high radius (Rc).
When studying the dilatational interfacial properties of a viscous system, the measured loss modulus is the sum of the interfacial loss modulus (Eʺinterface) and the bulk loss modulus (Eʺvisc) (Alexandrov et al. 2009; Russev et al. 2008):
where
Rs,eq is the initial radius (apex) before the oscillation begins (i.e. the equilibrium value).
χ = Hs,eq/Rc and \({\text{H}}_{\text{s,eq}}\) is the initial drop height before starting the oscillation.
If the interfacial modulus is evaluated using pressure measurement, the applied pressure needs to form the drop or bubble, and contains the capillary pressure (Pc) (Alexandrov et al. 2009) in addition to other contributions. In this case, for a Newtonian fluid, Eq. (22) will be written as follows:
where L is the capillary length.
Various techniques and methods are currently available for measuring the shear interfacial rheological properties of liquid systems, the most common of these being interfacial rheometers equipped with bicone or double wall ring (DWR) and oscillating drop instruments (Jaensson et al. 2021). These instruments are most often applicable to systems with low viscosity, typically above a few mPa.s.
In this work, we use a new type of interfacial rheological setup that enables the interfacial dilational rheology measurements to be extended to high viscosity polymer systems. Special emphasis has been placed on the effect of the temperature and the molecular weight of the two coexisting phases (PDMS and PIB) on the interfacial elastic modulus, particularly in the case of interfaces formed from polymer/air or polymer/polymer interfaces with an asymmetry in molecular weight.
Note that the main advantage of these polymers is that they are in the molten state at room temperature, and their refractive indices are sufficiently different (1.41 for PIB and 1.5 for PDMS) for one to observe PIB in PDMS and vice versa using dilational tensiometry. On the other hand, the higher density component (PDMS) forming the matrix is highly incompatible with the rising droplet phase (PIB).
Furthermore, we have developed a new approach to access the characteristic elongational rheological times of air-polymer surfaces and polymer–polymer interfaces via a simple test called a ‘pulse’ using the rising oscillating drop/bubble method. Additionally, the surface extensional properties measured with the capillary breakup extensional rheometer (CaBER) were investigated for comparison. Experimental difficulties encountered, possible artefacts and precautions necessary for reliable measurements are highlighted particularly in the presence of high molecular weight polymers.
Finally, we aim to see if there is a correlation between the interfacial dilatational properties (via measured interfacial dilational relaxation times) and the interfacial shear properties (measured interfacial shear viscosities) in the air-PDMS and air-PIB interfaces following Trouton’s relationship. Furthermore, PDMS/PIB combinations were chosen in such a way that they exhibit similar viscosities and close polydispersity indices. Therefore, the effect of the viscosity and polydispersity will not be discussed extensively hereafter.
Experimental
Materials and methods
Materials
The model fluids chosen were PDMS (polydimethylsiloxane) trimethylsiloxy terminated supplied from abcr and Alfa Aesar, and PIB (polyisobutene) supplied by INEOS. The PDMS and PIB used in this work presented different molecular weights. Table 1 shows the composition of each material.
The above materials were used to prepare three PDMS and three PIB materials with a low viscosity difference (Δη tending to zero) at 25 °C using the mixing law (Grizzuti et al. 2000). All of these materials were prepared by using an overhead stirrer with a wide speed range. Table 2 summarises the composition of each PDMS and PIB. Here, lv, mv and hv respectively denote low, medium and high viscosity systems.
It is interesting to indicate that all PIB and PDMS melts investigated in this study are entangled polymers since they have an average weight molecular weight (Mw) higher than the critical molecular weight Mc. Indeed, the Mc is 28000 g/mol and 1060 g/mol for PDMS and PIB, respectively (El Omari et al. 2021).
The different polymer surfaces and polymer/polymer interfaces investigated in the present work are summarised in Table 3.
Characterisation methods
Bulk rheological measurements
To extract the rheological behaviour of the studied model fluids, a controlled-stress rheometer (DHR-2 Rheometer from TA Instruments, USA) with cone-plate geometry (diameter 40 mm, angle 1.994°) was used. The dynamic oscillatory measurements were performed using a frequency sweep (100 to 0.1 rad/s). All complex viscosity |η*| measurements were carried out at constant temperatures of 25, 45 and 60 °C.
Interfacial tension measurements
The surface and interfacial tension measurements were performed at different temperatures using an automatic drop tensiometer (TRACKER-H from TECLIS Scientific, France). A drop of PIB was formed inside the PDMS (ρPDMS > ρPIB) using the rising drop configuration (Fig. 4). The detailed experimental protocol for this measurement was reported in a recent paper from our team (El Omari et al. 2021).

Rising drop configuration. Sectional view of the cell (left) and cell in closed position (right)
Interfacial dilational rheology
The interfacial dilational measurement was performed using the oscillating drop/bubble method on the TRACKER-H tensiometer from TECLIS Scientific (France). The drop/bubble was subjected to sinusoidal compression/dilation cycles using a mechanical motor. The viscoelastic modulus was determined at each frequency by analysing the shape of the drop as described previously. For the dynamic drop experiment, we started by defining the linear region with an amplitude sweep test (Becker et al. 1991), and then, the frequency sweep was performed to determine the elastic and loss dilational moduli. The first step consisted in forming a drop of PIB (rising drop) inside the PDMS (receiving phase) and then waiting for the stationary state to be achieved (plateau γ(t) = constant). The next step was the amplitude sweep at the highest frequency (applied in the frequency sweep test). To minimise the capillary and Weber numbers, a syringe with a small volume (10 µL) was used to enable the application of a small amplitude ΔV, in addition to a needle with a high diameter (1 mm).
Surface and interfacial stress relaxation measurements
There are many experimental techniques for studying interfacial relaxations in air-polymer and polymer–polymer interfaces (Serrien et al. 1992). The oldest relaxation technique is wave damping (R Miller et al. 1991, 1993). Miller et al. were the first to use the pendant drop method to initiate transient relaxation of aqueous solutions of surfactants and proteins. Here, we applied the ‘pulse’ technique to study interfaces and surfaces of molten polymer systems. First, we began by reaching the equilibrium of the surface (or interfacial) tension, and then, a constant amplitude (T) was applied during a time ∆t = t2 − t1. The evolution of the surface (or interfacial) tension makes it possible to characterise the relaxation of each surface or interface as a function of the temperature (Fig. 5).

Interfacial relaxation method or ‘pulse’ technique used in this study
The equation of this square variation of the drop volume noted V(t) is described by a Fourier series as expressed below:
where \({\varvec{\Delta}}{\varvec{V}}\) is the applied amplitude, ω is the frequency (ω = 2π/T) and n is the number of the squares.
During one cycle (compression/dilatation), the applied square pulse disturbance is an expansion-relaxation-compression-relaxation cycle with amplitude ΔV applied to the surface or interface with initial volume V0 as presented in Fig. 5. In the actual experiment, the dilatation and compression ramps were done at low flow rate (low frequencies); thus, the intrinsic viscous contribution to the dilational properties can be neglected. Using the square pulse perturbations, an initial increase of the surface (or interfacial) tension was first observed, followed by a relaxation towards the stationary state value \({{\varvec{\gamma}}}_{0}\). In the compression stage, the surface (or interfacial) tension \({\varvec{\gamma}}\) decreased below its stationary value, and then, at the end of the compression, the relaxation of \({\varvec{\gamma}}\) towards the stationary value was noticed.
Capillary breakup extensional rheometry (CaBER)
A capillary breakup extensional rheometer (CaBER) is conceptually based on the design of Bazilevsky et al. (1990). It is a rheological device that can quantify the elongational properties of low to medium viscosity fluids. A capillary bridge (a volume of fluid with a free surface, held by surface tension between two solid surfaces) is placed in an unstable situation that leads to its rupture into two distinct volumes of fluid (Fig. 6). This rupture is an elongational flow whose dynamics depend solely on the surface tension and the viscosity of the fluid. The recording of these dynamics (which can be very fast, in the order of few milliseconds) allows the extraction of the apparent elongational viscosity and the elongational relaxation time of the fluid.

Schematic representation of the CaBER instrument
In the present work, a Thermo Scientific™ HAAKE™ CaBER™ 1 capillary breakup extensional rheometer (USA) was used. A tiny droplet of the studied model fluid is placed between two parallel plates of diameter 4 mm, separated by an initial height H0 of 500 µm. Then, one plate is moved using an extension velocity v0 of 430 mm/s to a constant gap Hf of 40 mm. The experiment is recorded by a high-speed camera, Olympus i-SPEED 3, up to 5000 frames per second. The instrument also uses a laser micrometre to monitor the diameter at the midplane of the thinning filament, D(t).
It is essential to mention that the droplet must be homogeneous and must not contain any air bubbles; otherwise, the filament will break, and the extracted parameters will be incorrect. On the other hand, it is useful to mention that during the elongation and thinning of the filament, viscous forces tend to stabilise the cylindrical shape of the filament, whereas gravitational forces can drag the fluid below the mid-filament. The competition between viscous and gravitational forces can be estimated by the ratio of the Bond number to the capillary number, \(\frac{{B}_{0}}{{C}_{a}} = \frac{\rho g {D}_{0}}{2 {\eta }_{0} \dot{\varepsilon }}\), where ρ is the density of the fluid, g is the gravity constant and \(\dot{\varepsilon }\) is the strain rate. In the present study, the \(\frac{{B}_{0}}{{C}_{a}}\) ratio was calculated and found to be lower than 0.5, ensuring a viscous-dominated thinning mechanism (McKinley and Tripathi 2000).
The diameter versus time D(t) data, which represents the raw output of the CaBER, is then used to determine extensional rheological parameters. For a viscoelastic system, Anna and McKinley (2001) and Naillon et al. (2019) proposed the following fitting function:
where α, β and δ are the fitting parameters that have physical relevance, and τ is the elongational relaxation time.
If the surface tension γ of the system is known, the evolution of an apparent extensional viscosity \({\eta }_{E}\) can easily be calculated (Eq. (27)). The thinning of the filament is driven by the competition between capillarity and elasticity (Erik Miller et al. 2009; Anna and McKinley 2001; van Berlo et al. 2021).
where R(t) and \(\dot{\varepsilon }(t)\) are, respectively, the filament radius and extension rate during time evolution.
The elongational relaxation time and the apparent elongational viscosity of the model fluids investigated (PDMS and PIB) were extracted with Eq. (23) and compared to the dilatational relaxation time obtained by the pulse method (dilational tensiometry).
Surface shear measurements
The interfacial shear properties of the surfaces were evaluated using a novel titanium biconical geometry designed by Anton Paar Research and Development Service (Stuttgart, Germany) (D: 68.28 mm, angle 5°). This new Ti-bicone was attached to an MCR 302 rheometer in combination with a newly designed interfacial cell that was used for the first time in a previous work (El Omari et al. 2021). All details about the theoretical and experimental considerations are described in the reference (El Omari et al. 2021). The home-made lightweight Ti-bicone setup has low inertia (0.022733 mN.m.s2) in comparison to the commercial stainless-steel one (0.01433 mN.m.s2). The low inertia of the Ti-bicone geometry allows for reliable measurements even on high molecular weight PDMS/PIB polymer systems with sub-phase viscosities up to 300 Pa.s (El Omari et al. 2021). In addition, to carry out the ISR measurements on the Ti-bicone geometry, and in order to obtain the absolute values of surface shear viscosity, we take into account in each new experiment the height (H1), the density (ρ1) and the viscosity (η1) of the polymer of the denser phase as well as the height (H2), the density (ρ2) and the viscosity (η2) of the fluid of the less dense phase (Fig. 7). These data will allow corrections for the effects of the sub-phases based on the Oh and Slattery algorithm integrated in the RheoCompass software of the rheometer, which deduced an exact solution to the velocity distribution in the two sub-phases and the interface (Oh and Slattery 1978). The calculation of these parameters was always performed the same way, regardless of the Boussinesq number.

Schematic overview of the biconical interfacial rheometer (El Omari et al. 2021)
All steady surface viscosity ηs (\(\dot{\gamma })\) measurements were carried out from 0.1 and 10 s−1 at constant temperatures of 25, 45 and 60 °C.
Results
Bulk rheological measurements
In Fig. 8, the master curves of PDMS and PIB melts are depicted. It has been observed that all of the PDMS and PIB melt polymers present Newtonian behaviour for a frequency range lower than 1.6 Hz (10 rad.s−1). The latter is greater than the maximum frequency (0.4 Hz) used during interfacial dilational measurements. On the other hand, the vertical (y-axis) shift factor (bT) gives information about the temperature dependency of their densities. Indeed, the bT values clearly show the high temperature sensitivity of PIB compared to PDMS. Therefore, the two phases have very similar viscosities at 25 °C, but they do not exhibit the same tendency when the temperature is increased. The zero-shear viscosity and shift factor values of the different investigated PDMS and PIB are shown in Tables 4 and 5. It would be useful to recall that the main objective is to ensure a difference in viscosity of the coexisting phases that tends to low values during the dilational interfacial rheological experiments in order to minimise capillary and inertial effects (Δη tending to zero).

Master curves of the different model fluids at Treference = 25 °C
Interfacial tension measurements
The interfacial tension at equilibrium between PIB and PDMS was determined using the static experiment method. The volume of the drop of PIB was kept constant and the evolution of the interfacial tension was monitored until an equilibrium state (plateau) was reached. The range of Bond number values was between 0.3 and 0.5, whereas the Worthington number W0 gives values in the 0.7–0.8 range, confirming the precision of the interfacial tension measurements.
The measurement was performed at 25, 45 and 60 °C. Table 6 summarises the interfacial tension at equilibrium for the studied interfaces.
From Table 6, it can be observed that interfacial tension between PDMS and PIB decreased with decreasing molecular weight. LeGrand and Gaines Jr. (1975) noticed the same trend using similar polymer systems. The authors developed the empirical formula given in Eq. (28) in order to model the dependency of the interfacial tension on the molar mass:
where \({\gamma }_{12}^{\infty }\) is the equilibrium interfacial tension, k1 and k2 are constants and M1 and M2 are the molecular weight of the system components, respectively. Later, Vinckier et al. (1996) used LeGrand and Gaines’s relationship to calculate the interfacial tension between different PIB/PDMS systems, concluding that the interfacial tension increased when the molecular weight increased, which is once again consistent with our results.
On the other hand, from Table 6, it can be observed that for the lowest viscosity system (PIB lv/PDMS lv), the interfacial tension increased with the temperature. In the presence of higher viscosity polymers (PIB mv/PDMS mv and PIB hv/PDMS hv), however, the interfacial tension decreased with increasing temperature (Fig. 9). This phenomenon began starting from a specific molecular weight of sub-phases, as noted elsewhere (El Omari et al. 2021), using similar PIB/PDMS polymer systems. Based on the interfacial shear rheology and the solubility parameter modelling, the authors proved that the transport phenomenon was responsible for the decrease in the interfacial tension. The authors showed that the interfacial shear viscosity increased for the high viscosity polymer systems when the temperature was increased. This phenomenon that increases at high temperature was related to the short-chain components of broadly distributed samples migrating into the interface to reduce the interfacial tension and consequently the Gibbs energy of the entire system.

Diagram representing the migration of short PIB chains into the interface of PIB/PDMS polymer systems
Wagner and Wolf (1993) analysed the variation of the interfacial tension γ versus temperature (T) of different PDMS/PIB systems with comparatively narrow molecular weight distributions (MWD). They found that the evolution of γ(T) depends on molecular weight. The authors argued based on the modelling of the solubility parameters that it was not trivial to interpret their experimental results by the Flory–Huggins interaction parameter since the two polymers are not miscible. Later, various studies investigated the effect of the temperature and molecular weight on the same PDMS/PIB polymer systems (Tufano et al., 2008; Gabriele et al. 2011; Ziegler and Wolf 2004). According to Ziegler and Wolf (2004), in the case of apolar polymer systems (the case of PDMS/PIB) the interfacial properties are dependent on the average molecular weight and the polydispersity of both coexisting phases but also on the molecular weight difference, i.e. the asymmetry across the interface.
Interfacial dilational measurements
It is important to mention that dimensionless numbers (Bond, Weber and capillary numbers) were carefully checked to correctly conduct such dilatational measurements for each PIB/PDMS system. The viscosity contrast (Δη) between PIB and PDMS phases was 0.1 Pa.s, the density difference (Δρ) was 100 kg/m3, the radius of the nozzle was 0.5 mm, the interfacial tension was around 2.1 mN/m, the minimum and maximum frequencies used were 0.02 and 0.4 Hz, respectively, the initial volume (V0) of the drop was 4.8 µL and the ΔV amplitude was 0.3 µL. Table 7 displays the range values of each dimensionless number. The capillary number Ca was in the 10−4 to 10−1 range and We was around 10−8, which is still very low (< < 1). Therefore, viscous stress and inertia effects are sufficiently negligible.
On the other hand, the Bond number was calculated instantaneously by the tensiometer software according to Eq. (15) and analysed immediately before each measurement launch. Values of Bd were found in the range between 0.3 and 0.5 for all experiments carried out in this study.
Figure 10 shows the frequency sweeps at 25, 45 and 60 °C. For all PIB/PDMS systems, the absence of the loss modulus was noticed. It is useful to mention that in our case, the probed interfaces are free (no particles or surfactants present on the interface), which induces small phase angle values between interface area oscillations and interfacial tension variations. This means that the storage modulus Eʹ is much larger than the loss modulus Eʺ (Lucassen and Van den Tempel 1972).

Dilational frequency sweep experiment performed on the PIB/PDMS interfaces
From Fig. 10, it was observed that at a constant temperature, the more the viscosity of the coexisting phases increased, the more the interfacial elastic modulus Eʹ increased. For the lowest viscosity system (PIB lv/PDMS lv), the interfacial elastic modulus Eʹ increased when the temperature decreased. However, by increasing the viscosity of the coexisting phases (PIB mv/PDMS mv), the opposite tendency was observed. The interfacial elastic modulus decreased when the temperature increased, which is in agreement with our observations during the static drop measurements, indicating a migration phenomenon (El Omari et al. 2021; Tufano et al. 2008). No data are presented for the PIB hv/PDMS hv interface at 45 and 60 °C. Due to the high difference in viscosities (Δη) of PIB hv and PDMS hv when the temperature was increased (Table 5), high capillary and Weber numbers were obtained; as a result, the interfacial forces are not dominant and the measured droplets for these interfaces are not Laplacian (the drop profile does not follow the theoretical profile predicted by the Laplace equation). Therefore, no measurements could be carried out in this case, as it was not feasible to properly impose a sinusoidal volume or area variation (Fig. 11).

Dilational frequency sweep experiment conducted on a Laplacian and non-Laplacian drop (pictures taken from the WINDROP software of the TRACKER apparatus)
Interfacial relaxation of PIB/PDMS systems
In this section, the relaxation of the surfaces and interfaces of PIB/PDMS polymer systems is examined. The response of the surface or interface is related to its temporal relaxation. The surface or interfacial tension decreased when a positive amplitude + ΔV was applied to return to the equilibrium state (γ0). The temporal variation of the surface/interfacial tension was fitted using the Kohlrausch law (Anderssen et al. 2003):
where Δγ is an amplitude factor (Fig. 5), τ is the characteristic relaxation time and β is the stretching parameter, which varies between 0 and 1 and describes the width of the relaxation time distribution (Yousfi et al. 2009). In our case, the time-decay data of the relaxation processes of PIB/PDMS systems are better described in terms of a simple exponential function (β = 1) (Eq. (30)), implying that the tested polymers exhibit monodisperse relaxation characterising the rheological model of Newtonian Maxwellian behaviour.
This allows us to extract the values of the characteristic relaxation times of the surfaces and interfaces.
It is useful to mention that the Kohlrausch law has been widely used in the literature to model several time-decaying behaviours in the relaxation processes of polymers. For instance, Yousfi et al. (2009) fitted the evolution of the scattering intensity with time in dPS/PBMA (deuterated polystyrene/polybutyl methacrylate) nanoblends using small angle neutron scattering (SANS) to extract the relaxation time of the dPS droplets in a PBMA matrix. Boyd et al. (1997) fitted the dielectric data of PVAc (polyvinyl acetate) in the glass transition region using the Kohlrausch law to extract its relaxation kinetics.
Air/PDMS and air/PIB surfaces
Figures 12 and 13 show the decay of the surface tension γ with time for all PDMS and PIB surfaces at 25 °C. This decay in γ(t) may be related to a rearrangement of the macromolecules, or the end groups at the surface in contact with the air molecules, in order to decrease the surface tension and reach the equilibrium state described by γ0 (Quintero et al. 2009; Kleingartner et al. 2013). From Fig. 14, one can observe that the dilational relaxation times of the surfaces increased with the viscosity of the coexisting phases, but they decreased when the temperature increased. Thus, the higher the viscosity, the longer the time required to create the new free surface. These results could have been predicted beforehand. Indeed, recently, El Omari et al. (2021), using similar PDMS and PIB polymers, found that the surface shear viscosity increases linearly with the shear viscosity of the bulk, since the creation of the free surface starts from the bulk sub-phase.

Fitting of the variation of the surface tension as a function of time at 25 °C of air/PDMS surfaces a PDMS lv. b PDMS mv and c PDMS hv. The dots represent the experimental relaxation measurements using the ‘pulse mode’, and the continuous line is the fit using the Kohlrausch model

Fitting of the variation of the surface tension as a function of time at 25 °C of air/PIB surfaces a PIB lv, b PIB mv and c PIB hv. The dots represent the experimental relaxation measurements made using the ‘pulse mode’, and the continuous line is the fit obtained using the Kohlrausch model

Variation of the surface dilational relaxation times (in seconds) as a function of bulk viscosity at different temperatures for PDMS (a) and PIB (b)
Several studies that have investigated pure liquid surfaces or free polymer surface systems with no surface-active agent (surfactants, nanoparticles, etc.) as in the present work have confirmed the presence of variations in interfacial tension by dynamic interfacial tension measurements using the pendant drop technique (Jůza 2019; Kwok et al. 1998), by Wilhelmy plate force tensiometry (Sauer and Dipaolo 1991; Rahman et al. 2019), and interfacial properties (interfacial moduli) measured by dilational rheology (Peters et al. 2005).
In the ‘dynamic relaxation’ pulse mode, the tensiometer imposes square pulse variations of the drop volume and records the interfacial surface area variation as well as the results of the variation of the interfacial tension. Compared to other methods that impose both elongation and shear stresses on the interface (Verwijlen et al. 2012, 2013), the square pulse method has the advantage of imposing pure contractions/expansions that allow a direct measurement of the dilatational properties of liquid/liquid or air/liquid interfaces. But it is of paramount importance to point out that our measurements were performed using a slow dilatation/compression rate (\(\dot{\varepsilon }\)< 0.1 s−1). In these conditions, viscous stresses are insignificant (small capillary numbers), particularly in the case of low to medium viscosity polymers. Consequently, we expect that the characteristic elongational relaxation times deduced from the fit of the exponential decay of γ(t) are related mainly to the dilatational properties of polymer–polymer and air-polymer interfaces rather than to bulk extensional rheology.
However, one may accept that these statements can be debatable in the case of high molecular weight melt surfaces (air/PDMS hv and air/PIB hv systems) where Δη is rather significant. Afterwards, the viscous forces are no longer negligible (high capillary numbers). Consequently, we expect that the characteristic elongational relaxation times deduced from the fit of γ(t) of PDMS hv and PIB hv are the result of the balance between the relaxation of the chains on the surface and the viscous force contribution. But it is quite difficult to deconvolute the two phenomena.
Jalbert et al. (1993) measured the surface tensions of amine- and methyl-terminated poly(dimethylsiloxane) (PDMS) with molecular weights ranging from 1000 to 75 000 g/mol by pendant drop tensiometry. It has been reported that methyl chain ends of PDMS have low surface energy compared to those of the chain backbone. These low surface energy chains (‘methyl ends’) have an entropic preference and attraction to adsorb to the surface, which in turn was expected to cause the short chains of a polydisperse melt to segregate to the surface (Mahmoudi and Matsen 2017). Moreover, Mahmoudi and Matsen (2017) demonstrated that in the case of polydisperse melts, the chain-end segregation simultaneously induced an entropic enrichment of short chains at the surface, since they have more ends per unit volume.
Jalbert et al. (1993) highlighted that the chain ends at the surface of a polymer melt control the evolution of surface tension γ with molecular weight. For the methyl-terminated polymer melts, γ increased with molecular weight. The end-group effect was confirmed a year later by Elman et al. (1994) using neutron reflectivity.
In our study, we used polydisperse methylsiloxy-terminated PDMS melts. We suspect that the kinetics of transport of short chains and chain ends connected to the chain backbone into the surface of a polymer melt of high Mw will be slower and will require more time to equilibrate compared to low Mw ones. These interpretations support why high molecular weight melts lead to longer dilational characteristic relaxation times. The latter reflect the rate of the chain ends and short chains to move to the air/polymer interface.
The mobility of the macro-chains at the surface was also observed in the stability of foams using proteins (Dickinson 2001). Through the hydrophobicity of these macromolecules and their possible conformational reorganisation, rapid adsorption at the air/water interface is enabled, leading to the formation of an elastic adsorbed layer (Dickinson 2001; Foegeding et al. 2006). Some studies have also highlighted the role that polysaccharides play at the interfacial film regarding enhancement of the stability of foams, due to a thickening or gelling effect on the aqueous solution (Langevin 2001; Klitzing and Müller 2002; Schmidt et al. 2010).
We have also compared the evolution of the relaxation time of the studied surfaces with respect to temperature. For this purpose, Ln (τ) as a function of 1000/T was plotted. Figure 15 shows this evolution for PIB and PDMS surfaces.

Arrhenius plot of the relaxation time τ as obtained from fits of the surface relaxation test
The variation of relaxation time with temperature (T in Kelvin) is calculated using an Arrhenius function (Eq. 31).
where Ea is the activation energy (representing, in this case, the temperature sensitivity of the relaxation process), τ0 is the pre-exponential factor and R is the universal gas constant.
Figure 15 shows a linear evolution, demonstrating that the Arrhenius law fits these curves successfully. For all surfaces, regardless of the viscosity of the coexisting phases, we obtained an identical positive slope indicating that the activation energy is independent of the viscosity. These activation energies are higher than the bulk activation energy of PIB and PDMS (between 15 and 18 kJ/mol) (Table 8) (Roland and Santangelo 2002).
PIB/PDMS interfaces
From Fig. 16, it can be seen that the decrease during pulse mode sometimes had additional small steps before contraction. In fact, a deformation transition (between dilatation and compression steps) takes a short time since it cannot be instantaneous. Indeed, if that is the case, the drop will not immediately follow the fast decrease of the volume (because of its inertia) and it will have an additional dilational deformation (not a Laplacian drop) (Molaei and Crocker, 2020).

Interfacial relaxation measurements conducted in pulse mode of a PIB lv/PDMS lv, b PIB mv/PDMS mv and c PIB hv/PDMS hv
It has been seen that regardless of the type of PIB/PDMS investigated, the interfacial relaxation is faster at high temperature than at low temperature, indicating a thermal dependency of the interfacial tension.
The time dependency of the interfacial tension during the pulse interface perturbations can be described according to the model of Shi et al. (2004). Immediately after the formation of a new interface between two immiscible polymer systems such as PDMS and PIB, which is constituted of broad molecular weight distribution chains, the interfacial tension decreases rapidly because of the preferential migration of the shorter chains into the interface. The effect of this favoured incorporation into the interface slows down as time proceeds. Therefore, a steady state is reached (Fig. 16a); i.e. the interfacial tension becomes constant, as the rates of diffusion of short chains into the interface and out of it become identical (Shi et al. 2004). On the other hand, in the presence of high molecular weight components (i.e. at low temperatures of measurement), the transport of shorter chains into the interface takes place so slowly that the stationary state cannot be reached, since it is higher than the imposed expansion/compression period time of the measurements (Fig. 16c).
It is useful to mention that Shi et al. (2004) measured the variation of the interfacial tension γ with time using the static pendant drop technique. In their experiments, they formed a drop and then followed the decrease in interfacial tension until equilibrium (when the calculated interfacial tension does not change with time) but without imposing any variation in drop volume (pulse) as exercised in our study. They observed that γ decreased but with a large time scale (from minutes to several hours). They ascribed this drop in the interfacial tension to the molecular weight and polydispersity of the PIB and PDMS used. According to the authors, since the interfacial tension depends on the size of the chain components at the interface, the low molecular weight chains (especially PIB) can migrate into the interface, thus lowering the interfacial energy of the system. Since the PDMS/PIB systems used in our work are polydisperse, this transport effect might occur in our experiments as well.
Later, Peters et al. (2005) used confocal Raman spectroscopy for the estimation of the thickness of the diffusion interfacial zone in PDMS/PIB systems and confirmed the migration and diffusion effect observed in the pendant drop technique by Shi et al. (2004).
We note that with the increase of the average molecular weight of both phases (the case of the PDMS hv/PIB hv system), the interfacial tension and the interfacial viscosity also increase (El Omari et al. 2021), and thus substantially slow down the mobility and migration to the interface of entangled chains, as is observed from the shape of the change of γ with time (Fig. 16).
To conclude, most of these authors came to an agreement that short macromolecules of PIB can migrate from the bulk into the newly generated interface with PDMS. Since MwPIB < MwPDMS (Table 2), the chain mobility of PIB will be higher, and they could diffuse into the interface. These short chains could act as a surfactant and lower the interfacial tension.
We suspect that the relaxation times determined by the pulse method correspond to long relaxation times of macromolecular chains (a few seconds), which are much higher than stretch time relaxations at the molecular scale (Rouse time ~ a few ms).
Peters et al. (2005) assumed that the chain migration could occur also in dynamic shear experiments, but at time scales much shorter than for the pendant drop method.
According to several studies, the characteristic elongational relaxation times of interfaces are also higher than the relaxation times of droplet interfaces measured by dynamic shear rheology in the case of immiscible polymer blends (Shi et al. 2004; Peters et al. 2005).
In this study, for all PDMS/PIB interfaces, the characteristic interfacial relaxation times increased with increasing viscosity of the coexisting phases, but these times decreased with increasing temperature (Table 9). Interfacial relaxation times are higher than those of surfaces. This can be explained by the higher degrees of freedom of macromolecular chains at the surface compared to the interfaces. The relaxation shape of PIB hv/PDMS hv systems at 25 and 45 °C was slow, with the relaxation time tending to an infinite value. This phenomenon could be explained by the high viscosity of the PIB hv and PDMS hv polymers as explained above. Moreover, increasing the viscosity induced an increase in the volume of the drop (compression cycles), and a significant increase of the interfacial tension at high temperature was noticed. This is related to the significant difference in the viscosities of their coexisting phases Δη (Table 5). The drop of PIB becomes more deformed in the continuous phase (PDMS), which leads to an imbalance at the interfaces. Thus, the drop is no longer Laplacian and the viscous and inertial effects are considerably more important.
Surprisingly, the characteristic dilational relaxation times of PIB/PDMS interfaces obtained in this work using the dilational pulse mode measurements were comparable to those measured on similar polymer systems by other authors using other techniques such as spinning drop extensiometry (Joseph et al. 1992) and the retraction of deformed droplets (Siahcheshm et al. 2018).
Compared to the air/PDMS and air/PIB surfaces, the PDMS/PIB interfaces presented a relatively low activation energy value (18 kJ/mol for interfaces versus 31 kJ/mol for surfaces) (Table 10). This result suggests that chain motion at the interface between PIB and PDMS is different compared to PDMS and PIB surfaces.
The apparent activation energy varies slightly with increasing source and receiving phase viscosities. Knowing that the relaxation time of the PIB hv/PDMS hv system was obtained at 60 °C and assuming that the activation energy is very close to the other systems, the relaxation times at 25 and 45 °C can be determined by extrapolation. Table 11 shows an estimation of these relaxation times.
Capillary breakup extensional rheometry (CaBER)
There are two ways to extract the evolution of the filament diameter. The first way is by using the recording movie (Fig. 17) of the filament thinning until the breakup—in other words, by using image software to post-analyse the saved pictures in order to extract D(t).

Sequence of images of capillary breakup for a PIB filament
The second way is to use the laser micrometre to measure the diameter of a filament during the experiment. This latter method was used here to extract D(t).
Figure 18 shows the evolution of the stretched filament diameter as a function of time for PIB and PDMS at 25 °C. The diameter of the filament decreases during the experiment time. The raw diameter data were fitted using Eq. (26) to determine the elongational relaxation time of PIB and PDMS (Figs. 19 and 20).

Evolution of the stretched filament diameter as a function of time, for PIB (a) and PDMS (b) at 25 °C

Fitting of the variation of the filament diameter as a function of time of PIB at 25 °C. a PIB lv. b PIB mv. c PIB hv

Fitting of the variation of the filament diameter as a function of time of PDMS at 25 °C. a PDMS lv. b PDMS mv
It should be noted that the fit of the data did not include the initial data just after the initial step-stretch; this evolution of the diameter near t = 0 is dependent on the initial elongation rate. Miller et al. (2009) demonstrated that most of the CaBER experiments were conducted on surfactant wormlike micelle solutions and immiscible polymer blends, to obtain a relaxation time that is insensitive to step-stretch conditions.
As mentioned in Fig. 19, there is no data about the PDMS hv. For this model fluid, we failed to form a filament and to study the evolution of its diameter. The filament broke immediately after applying the initial constant step-stretch. This might be explained by the distribution of the molar weight that could be bimodal. It can be related to the presence of a narrow peak for the long macromolecules and a wide one for the short macromolecules. Molar mass measurements are in progress to validate these postulates.
Table 12 summarises the variation of the elongational relaxation times with the temperature for all the bulk viscosities of the coexisting phases. We can clearly observe that the elongational relaxation times for both PIB and PDMS increase with the molar weight of the bulk and decrease with temperature. For low viscosity fluids and at high temperatures (PDMS lv and PIB lv), the breakup of the filament is instantaneous, and the saved data are minimal (only three or four points): the data are not enough for the fit to be carried out.
Wagner et al. (2018) suspected in the case of highly entangled PS melts that premature breakage of the filament during the stretching step could happen when the elongation rate \(\dot{\varepsilon }\) is significantly larger than the inverse of the Rouse stretch time ‘τR’ of the polymer chain, i.e. an extension process time scale much faster than ‘τR’. Feng et al. (2019) demonstrated the same finding using polyisoprene (PI) melts with various weight average molecular weights (Mw). They found that in the presence of high molar mass polyisoprene melts (Mw > 430 K), high extension rates make the filament break quickly compared to PIs with low Mw. The authors suggested that chain scission due to finite extensibility effects is the cause of the filament rupture. However, elucidation of this phenomenon still represents a challenge, and there is currently no consensus for the description of the underlying physics in such melt polymer systems (Wang 2019) .
It is worth pointing out that in CaBER experiments, the normal stress (viscous force effect) happens simultaneously with the capillary forces in the visco-capillary thinning domain. Nonetheless, in all our measurements with CaBER, the relaxation times have been extracted in the elasto-capillary thinning region where the viscous effect is insignificant (Anna and McKinley 2001). Therefore, the calculated elongational relaxation times are mainly related to the capillary stress effect in analogy with tensiometry experiments. From Fig. 21, one can see that the extensional relaxation times obtained from CaBER measurements are very close to the dilational relaxation times obtained with the ‘pulse’ method (several seconds in both cases depending on the viscosity and molecular weight of the polymer). It might be probable that the preferential migration of short chains into the bulk during thinning manifests itself in the same way for the polymer filament as in the rising or pendant drop.

Elongational relaxation times measured by the pulse method and CaBER technique
This is not the case when compared to relaxation measurements carried out with other techniques. Recently, Rahman et al. (2019), using a Wilhelmy plate force tensiometer, measured the variation of the bulk relaxation time of different liquids with viscosities ranging from 1 mPa.s to 1 Pa.s. The relaxation time was defined as the time to attain the equilibrium of the surface tension γ during its decay. The latter was measured with the help of a force sensor connected to the vertical Wilhelmy plate. Interestingly, they found a power law dependency between the viscosity and the relaxation time. Highly viscous fluids take much more time to attain the equilibrium compared to low viscosity liquids. However, it should be noted that the dilatational interfacial relaxation times measured by the pulse relaxation technique of the present work are much shorter than the bulk relaxation times obtained by the Wilhelmy plate force tensiometer. For example, Rahman et al. showed a relaxation time of 54 s in the case of the liquid with 1 Pa.s, while in our case PDMS or PIB with 2.8 Pa.s gave a dilatational interfacial relaxation time of 0.3 s or 0.2 s, respectively.
We note that at a specific temperature and due to the difference in their average molecular weights, the characteristic elongational relaxation time of PIB lv is lower than that of PDMS lv, although they have the same melt viscosity since MwPIB < MwPDMS. The same trend was observed when comparing PDMS mv and PIB mv, or PDMS hv with PIB hv.
Recently, several authors (Sachsenheimer et al. 2012; Zell et al. 2010; Rodd et al. 2005) have tried to study the extensional behaviour using CaBER of PEO solutions (in solvents) with varying molecular weights in combination with time-dependent surface tension measurements. In all cases, they found that the surface tension decreases with time after a new, fresh interface is created. They explained that with time, polymer from the bulk diffused and adsorbed to the interface and reduced the surface tension. Their data showed that the higher molecular weight PEO took a longer time to approach equilibrium than the more mobile polymers. On the other hand, it was ascertained that the values of extensional relaxation times extracted from CaBER measurements for the PEO solutions are seen to increase with molecular weight, in qualitative agreement with data from the dynamic surface tension (DST). However, the authors argued that DST alone cannot explain the difference in the shape of the curves in the capillary thinning process of CaBER experiments.
The evolution of the apparent elongational viscosity as a function of time for the model fluids at 25 °C is also carried out and depicted in Fig. 22. The elongational viscosity passed through a transient stage before reaching a steady terminal regime. Using the latter, a Trouton ratio (Tr) of around 3 was found for the PDMS and PIB melted polymers, which confirms the reliability of measurements obtained with CaBER.

Evolution of the apparent elongational viscosity as a function of time, for PIB (a) and PDMS (b) at 25 °C
Surface shear rheology
In the interfacial shear measurements, the corrected surface shear viscosities were determined for low Boussinesq values (B0 < 1) to reach true surface viscosity values. In this work, the calculated Boussinesq numbers were in the range of 0.2 to 0.6.
Zero-shear surface viscosities of air/PIB and air/PDMS are summarised in Table 13.
All the surfaces were Newtonian at the studied shear rate. Moreover, the surface viscosity of air/PIB and air/PDMS increased with the viscosity of the sub-phases.
Velandia et al. (2021) found the same tendency when they studied the link between interfacial and bulk viscoelasticity in reverse Pickering emulsions. For both bulk and interfaces, they demonstrate, using the Double Wall Ring (DWR) [2], that elastic modulus increases with the silica content, according to a power law dependency.
We also compared the zero-shear surface viscosities obtained by interfacial shear rheology with the elongational relaxation times acquired by the pulse relaxation test. Figure 23 shows the variation of the relaxation times with the zero-shear surface viscosities of air/PIB and air/PDMS.

Evolution of the relaxation times with the zero-shear surface viscosities of air/PIB (a) and air/PDMS (b) systems
One can show that zero-shear surface viscosities of the studied surfaces increase with the elongational relaxation time. Furthermore, since this tendency follows a power law dependency, we suspect the existence of a direct relationship between the interfacial shear and elongational properties in our air/PIB and air/PDMS systems, described generally by an interfacial or surface Trouton ratio. Verwijlen et al. (2012) defined the interfacial Trouton ratio Trs. Similarly to bulk fluids, Trs is defined as the ratio between the surface dilational ηd and the surface shear viscosity ηs:
In our published article (El Omari et al. 2021), we have shown a linear correlation between the bulk viscosity and the surface viscosity in the case of free surfaces of PDMS and PIB, i.e. air/PIB and air/PDMS. A similar power law tendency between the viscosity and relaxation time has been noticed by other authors (Rahman et al. 2019; Zell et al. 2010). These observations require further study with the use of viscoelastic interfaces allowing reliable interfacial dilational viscosity measurements and will be the subject of an article in the near future.
Conclusions
In the present work, the rising oscillating drop method has been used to probe the dilational rheological properties of PDMS/PIB interfaces. The effects of molecular weight and temperature have been taken into account. To prevent inertial and capillary effects, a number of material and experimental precautions were carefully respected. During the static and dynamic experiments, the creation of an interphase was evidenced in the case of coexisting phases with a high molecular weight. Then, the characteristic dilational relaxation times of the studied surfaces and interfaces were extracted using the new pulse relaxation method. The empirical Kohlrausch formula was used to fit experimental data showing the evolution of surface or interfacial tension with time, and the Arrhenius law was applied to validate the temperature dependency of the relaxation time. It was observed that the surface and interfacial relaxation times increased with the melt viscosity of the polymer and decreased with temperature. For the PDMS/PIB interfaces probed at high temperatures, however, the oscillating drop/bubble method and the relaxation test did not provide exploitable experimental data due to the presence of high Weber and capillary numbers caused by the significant viscosity differences of the coexisting phases. Capillary breakup extensional rheometry (CaBER) was used to probe the elongational rheological properties of model fluids. Thanks to the fit of D(t), elongational relaxation times were determined. The latter were comparable to the characteristic dilational times obtained with the pulse relaxation method. Finally, the possibility of a Troutonian correlation between shear and dilational surface rheology was discussed, and a direct link between zero-shear surface viscosities and relaxation elongational times was highlighted.
The original method developed in this study could be applied to a wide range of polymer melts and fluids and to even more complex polymeric systems.
References
Alexandrov N, Marinova KG, Danov KD, Ivanov IB (2009) Surface dilatational rheology measurements for oil/water systems with viscous oils. J Colloid Interface Sci 339(2):545–550
Anderssen RS, Husain SA, Loy R (2003) The Kohlrausch function: properties and applications. Anziam J 45:C800–C816
Anna SL, McKinley GH (2001) Elasto-capillary thinning and breakup of model elastic liquids. J Rheol 45(1):115–138
Bazilevsky A, Entov V, Rozhkov A (1990) Liquid filament microrheometer and some of its applications. Third European rheology conference and golden jubilee meeting of the British society of rheology. Springer, Dordrecht, pp 41–43
Becker E, Hiller W, Kowalewski T (1991) Experimental and theoretical investigation of large-amplitude oscillations of liquid droplets. J Fluid Mech 231:189–210
Berry JD, Neeson MJ, Dagastine RR, Chan DY, Tabor RF (2015) Measurement of surface and interfacial tension using pendant drop tensiometry. J Colloid Interface Sci 454:226–237
Boyd RH, Liu F, Runt J, Fitzgerald J (1997) Dielectric spectroscopy of semicrystalline polymers. In: Runt JP, Fitzgerald JJ (eds) Dielectric spectroscopy of polymeric materials- fundamental and applications. ACS, Washington, DC, pp 107–136
Dadouche T, Yousfi M, Samuel C, Lacrampe MF, Soulestin J (2021) (Nano) Fibrillar morphology development in biobased poly (butylene succinate-co-adipate)/poly (amide-11) blown films. Polym Eng Sci 61(5):1324–1337
Del Rıo O, Neumann A (1997) Axisymmetric drop shape analysis: computational methods for the measurement of interfacial properties from the shape and dimensions of pendant and sessile drops. J Colloid Interface Sci 196(2):136–147
Derkach S, Krägel J, Miller R (2009) Methods of measuring rheological properties of interfacial layers (experimental methods of 2D rheology). Colloid J 71(1):1–17
Dickinson E (2001) Milk protein interfacial layers and the relationship to emulsion stability and rheology. Colloids Surf B 20(3):197–210
El Omari Y, Yousfi M, Duchet-Rumeau J, Maazouz A (2021) Interfacial rheology testing of molten polymer systems: effect of molecular weight and temperature on the interfacial properties. Polym Testing 101:107280
Elman J, Johs B, Long T, Koberstein J (1994) A neutron reflectivity investigation of surface and interface segregation of polymer functional end groups. Macromolecules 27(19):5341–5349
Feng Y, Liu J, Wang S-Q, Ntetsikas K, Avgeropoulos A, Kostas M et al (2019) Exploring rheological responses to uniaxial stretching of various entangled polyisoprene melts. J Rheol 63(5):763–771
Foegeding EA, Luck P, Davis JP (2006) Factors determining the physical properties of protein foams. Food Hydrocoll 20(2–3):284–292
Freer EM, Wong H, Radke CJ (2005) Oscillating drop/bubble tensiometry: effect of viscous forces on the measurement of interfacial tension. J Colloid Interface Sci 282(1):128–132. https://doi.org/10.1016/j.jcis.2004.08.058
Freer EM, Yim KS, Fuller GG, Radke CJ (2004) Shear and dilatational relaxation mechanisms of globular and flexible proteins at the hexadecane/water interface. Langmuir 20(23):10159–10167
Gabriele M, Pasquino R, Grizzuti N (2011) Effects of viscosity-controlled interfacial mobility on the coalescence of immiscible polymer blends. Macromol Mater Eng 296(3–4):263–269
Grizzuti N, Buonocore G, Iorio G (2000) Viscous behavior and mixing rules for an immiscible model polymer blend. J Rheol 44(1):149–164
Jaensson NO, Anderson PD, Vermant J (2021) Computational interfacial rheology. J Nonnewton Fluid Mech 290:104507
Jalbert C, Koberstein JT, Yilgor I, Gallagher P, Krukonis V (1993) Molecular weight dependence and end-group effects on the surface tension of poly (dimethylsiloxane). Macromolecules 26(12):3069–3074
Joseph D, Arney M, Gillberg G, Hu H, Hultman D, Verdier C et al (1992) A spinning drop tensioextensometer. J Rheol 36(4):621–662
Jůza J (2019) Surface tension measurements of viscous materials by pendant drop method: time needed to establish equilibrium shape. Macromol Symp 384(1):1800150
Kleingartner JA, Lee H, Rubner MF, McKinley GH, Cohen RE (2013) Exploring the kinetics of switchable polymer surfaces with dynamic tensiometry. Soft Matter 9(26):6080–6090
Klitzing Rv, Müller H-J (2002) Film stability control. Curr Opin Colloid Interface Sci 7(1–2):42–49
Kwok D, Cheung L, Park C, Neumann A (1998) Study on the surface tensions of polymer melts using axisymmetric drop shape analysis. Polym Eng Sci 38(5):757–764
Lai L, Mei P, Wu XM, Cheng L, Ren ZH, Liu Y (2017) Interfacial dynamic properties and dilational rheology of sulfonate gemini surfactant and its mixtures with quaternary ammonium bromides at the air–water interface. J Surfactants Deterg 20(3):565–576
Langevin D (2001) Polyelectrolyte and surfactant mixed solutions. Behavior at surfaces and in thin films. Adv Coll Interface Sci 89:467–484
Laplace PS (1805) Traité de mécanique céleste: suppléments au livre X. Gauthier-Villars, Œuvres Complètes, Paris 4:771–777
LeGrand D, Gaines G Jr (1975) Immiscibility and interfacial tension between polymer liquids: dependence on molecular weight. J Colloid Interface Sci 50(2):272–279
Lei J, Gao Y, Ma Y, Zhao K, Du F (2019) Improving the emulsion stability by regulation of dilational rheology properties. Colloids Surf A 583:123906
Lucassen J, Van den Tempel M (1972) Longitudinal waves on visco-elastic surfaces. J Colloid Interface Sci 41(3):491–498
Lunkenheimer K, Kretzschmar G (1975) Neuere Ergebnisse der Untersuchung der Elastizität von löslichen Adsorptionsschichten nach der Methode der pulsierenden Blase. Z Phys Chem 256(1):593–605
Mahmoudi P, Matsen M (2017) Entropic segregation of short polymers to the surface of a polydisperse melt. Eur Phys J E 40(10):1–9
McKinley GH, Tripathi A (2000) How to extract the Newtonian viscosity from capillary breakup measurements in a filament rheometer. J Rheol 44(3):653–670
Miller E, Clasen C, Rothstein JP (2009) The effect of step-stretch parameters on capillary breakup extensional rheology (CaBER) measurements. Rheol Acta 48(6):625–639
Miller R, Loglio G, Tesei U, Schano K-H (1991) Surface relaxations as a tool for studying dynamic interfacial behaviour. Adv Coll Interface Sci 37(1–2):73–96
Miller R, Sedev R, Schano K-H, Ng C, Neumann A (1993) Relaxation of adsorption layers at solution/air interfaces using axisymmetric drop-shape analysis. Colloids Surf 69(4):209–216
Mobius D, Miller R (1998) Drops and bubbles in interfacial research. Studies in interface science. Elsevier Science & Technology 6:61–138
Molaei M, Crocker JC (2020) Interfacial microrheology and tensiometry in a miniature, 3-d printed Langmuir trough. J Colloid Interface Sci 560:407–415
Murray BS, Nelson PV (1996) A novel Langmuir trough for equilibrium and dynamic measurements on air− water and oil− water monolayers. Langmuir 12(25):5973–5976
Naillon A, de Loubens C, Chèvremont W, Rouze S, Leonetti M, Bodiguel H (2019) Dynamics of particle migration in confined viscoelastic Poiseuille flows. Phys Rev Fluids 4(5):053301
Noskov B, Bykov A (2018) Dilational rheology of monolayers of nano-and micropaticles at the liquid-fluid interfaces. Curr Opin Colloid Interface Sci 37:1–12
Oh SG, Slattery JC (1978) Disk and biconical interfacial viscometers. J Colloid Interface Sci 67(3):516–525. https://doi.org/10.1016/0021-9797(78)90242-4
Peters GW, Zdravkov AN, Meijer HE (2005) Transient interfacial tension and dilatational rheology of diffuse polymer-polymer interfaces. J Chem Phys 122(10):104901
Quintero C, Noïk C, Dalmazzone C, Grossiord J (2009) Formation kinetics and viscoelastic properties of water/crude oil interfacial films. Oil Gas Sci Technol-Revue De l’IFP 64(5):607–616
Rahman MR, Deng A, Hussak S-A, Ahmed A, Willers T, Waghmare PR (2019) On the effect of relaxation time in interfacial tension measurement. Colloids Surf A 574:239–244
Ravera F, Loglio G, Kovalchuk VI (2010) Interfacial dilational rheology by oscillating bubble/drop methods. Curr Opin Colloid Interface Sci 15(4):217–228
Renggli D, Alicke A, Ewoldt RH, Vermant J (2020) Operating windows for oscillatory interfacial shear rheology. J Rheol 64(1):141–160
Rodd LE, Scott TP, Cooper-White JJ, McKinley GH (2005) Capillary break-up rheometry of low-viscosity elastic fluids. Appl Rheol 15(1):12–27
Roland C, Santangelo P (2002) Effect of temperature on the terminal relaxation of branched polydimethysiloxane. J Non-Cryst Solids 307:835–841
Russev SC, Alexandrov N, Marinova KG, Danov KD, Denkov ND, Lyutov L, Vulchev V, Bilke-Krause C (2008) Instrument and methods for surface dilatational rheology measurements. Rev Sci Instrum 79(10):104102
Sachsenheimer D, Hochstein B, Buggisch H, Willenbacher N (2012) Determination of axial forces during the capillary breakup of liquid filaments–the tilted CaBER method. Rheol Acta 51(10):909–923
Santini E, Liggieri L, Sacca L, Clausse D, Ravera F (2007) Interfacial rheology of Span 80 adsorbed layers at paraffin oil-water interface and correlation with the corresponding emulsion properties. Colloid Surf A-Physicochem Eng Asp 309(1–3):270–279. https://doi.org/10.1016/j.colsurfa.2006.11.041
Sauer BB, Dipaolo NV (1991) Surface tension and dynamic wetting on polymers using the Wihelmy method: applications to high molecular weights and elevated temperatures. J Colloid Interface Sci 144(2):527–537
Schmidt I, Novales B, Boué F, Axelos MA (2010) Foaming properties of protein/pectin electrostatic complexes and foam structure at nanoscale. J Colloid Interface Sci 345(2):316–324
Serrien G, Geeraerts G, Ghosh L, Joos P (1992) Dynamic surface properties of adsorbed protein solutions: BSA, casein and buttermilk. Colloids Surf 68(4):219–233
Shi T, Ziegler VE, Welge IC, An L, Wolf BA (2004) Evolution of the interfacial tension between polydisperse “immiscible” polymers in the absence and in the presence of a compatibilizer. Macromolecules 37(4):1591–1599
Siahcheshm P, Goharpey F, Foudazi R (2018) Droplet retraction in the presence of nanoparticles with different surface modifications. Rheol Acta 57(11):729–743
Slattery JC, Sagis L, Oh E-S (2007) Interfacial transport phenomena. Springer Science & Business Media
Sun H-Q, Zhang L, Li Z-Q, Zhang L, Luo L, Zhao S (2011) Interfacial dilational rheology related to enhance oil recovery. Soft Matter 7(17):7601–7611
Tufano C, Peters G, Van Puyvelde P, Meijer H (2008) Transient interfacial tension and morphology evolution in partially miscible polymer blends. J Colloid Interface Sci 328(1):48–57
van Berlo F, Cardinaels R, Peters G, Anderson P (2021) Towards a universal shear correction factor in filament stretching rheometry. Rheol Acta 60(11):691–709
Vandebril S, Franck A, Fuller GG, Moldenaers P, Vermant J (2010) A double wall-ring geometry for interfacial shear rheometry. Rheol Acta 49(2):131–144
Velandia SF, Ramos D, Lebrun M, Marchal P, Lemaitre C, Sadtler V et al (2021) Exploring the link between interfacial and bulk viscoelasticity in reverse Pickering emulsions. Colloids Surf A 624:126785
Verwijlen T, Leiske D, Moldenaers P, Vermant J, Fuller G (2012) Extensional rheometry at interfaces: analysis of the Cambridge Interfacial Tensiometer. J Rheol 56(5):1225
Verwijlen T, Moldenaers P, Vermant J (2013) A fixture for interfacial dilatational rheometry using a rotational rheometer. Eur Phys J Spec Top 222(1):83–97
Vinckier I, Moldenaers P, Mewis J (1996) Relationship between rheology and morphology of model blends in steady shear flow. J Rheol 40(4):613–631
Wagner M, Wolf B (1993) Interfacial tension between polyisobutylene and poly (dimethylsiloxane): influence of chain length, temperature, and solvents. Macromolecules 26(24):6498–6502
Wagner MH, Narimissa E, Huang Q (2018) On the origin of brittle fracture of entangled polymer solutions and melts. J Rheol 62(1):221–233
Wang H, Wei X, Du Y, Wang D (2019) Experimental investigation on the dilatational interfacial rheology of dust-suppressing foam and its effect on foam performance. Process Saf Environ Prot 123:351–357
Wang S-Q (2019) Melt rupture unleashed by few chain scission events in fully stretched strands. J Rheol 63(1):105–107
Young T (1805) An assay on the cohesion of fluids. Philos Trans R Soc Lond 1805(95):65
Yousfi M, Porcar L, Lindner P, Boué F, Rharbi Y (2009) A novel method for studying the dynamics of polymers confined in spherical nanoparticles in nanoblends. Macromolecules 42(6):2190–2197
Zell A, Gier S, Rafai S, Wagner C (2010) Is there a relation between the relaxation time measured in CaBER experiments and the first normal stress coefficient? J Nonnewton Fluid Mech 165(19–20):1265–1274
Zhang H, Lamnawar K, Maazouz A (2015) Fundamental studies of interfacial rheology at multilayered model polymers for coextrusion process. AIP Conference Proceedings. AIP Publishing LLC 1664(1):100008
Ziegler VE, Wolf BA (2004) Interfacial tensions from drop retraction versus pendant drop data and polydispersity effects. Langmuir 20(20):8688–8692
Acknowledgements
The authors are thankful to Dr. Jean-Luc Bridot and Mr. Christian Boinon (TECLIS Instruments, France) for their recommendations and technical assistance regarding the interfacial rheological measurements carried out with the TRACKER instrument. The authors thank Dr. Nadia El Kissi and Mr. Vincent Verdoot from University of Grenoble Alpes for their help and their assistance regarding the capillary breakup extensional rheometry experiments. The authors also gratefully acknowledge Mr. Murat Arli from INSA Lyon for his help in the design of the high temperature interfacial cell. The authors are grateful to the engineering federation Ingelyse of Lyon University and INSA Lyon via the BQR (Bonus Qualité Recherche) for the financial support. Finally, the authors gratefully acknowledge the French Ministry of Superior Education and Research (MESRI) for the doctoral study grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
El Omari, Y., Yousfi, M., Duchet-Rumeau, J. et al. Probing the elongational rheological behaviour at interfaces of immiscible polymer melts using dilational tensiometry: effect of viscosity and temperature on the interfacial properties. Rheol Acta 61, 613–636 (2022). https://doi.org/10.1007/s00397-022-01364-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00397-022-01364-x