
Abstract
Elastin (E) coacervation was studied close to isoelectric pH (pI = 4.7 ± 0.5), and this thermally activated self-assembly leading to phase separation involved the following three distinguishable temperatures: onset of interpolymer interaction at Tos, coacervation at Tc, and mesophase separation at Tms. Such behavior was absent in elastin solutions made at other pHs. The pH-dependent particle size histogram revealed bimodal distribution between pH 2 to 4 and 5.5 to 6.2, whereas the region pH = pI ± 0.5, a coacervate-rich domain with trimodal size distribution. The coacervation temperature TC decreased from 38 to 33 °C with increase in solution ionic strength (0–40 mM, NaCl) implying the importance of hydrophobic forces that governed the molecular self-assembly. The thermoreversibility of the turbidity profile implied that the elastin complexes did not fully dissociate into their monomeric state upon temperature reversal. Two observations were made here: (i) the hysteresis loop formed by the heating-cooling cycle had an area that decreased with ionic strength of the solution and (ii) the remnant aggregated E content increased with solution ionic strength. The present study highlights some of the key issues related to the reversibility of self-assembled soft matter systems which remains somewhat poorly explored until now.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The phenomenon of complex coacervation has been observed in a variety of complementary polyelectrolyte systems [1,2,3,4,5,6,7]. Associative interaction between oppositely charged polyions often leads to the formation of soluble interpolymer complexes which may in turn coalesce to yield macroscopic coacervate droplets [8,9,10,11,12]. In the coacervate family, it is rare to find coacervation transition in unicomplex polyampholyte systems driven by temperature. Since the coacervate material has found many applications in areas as diverse as drug encapsulation and delivery, enzyme immobilization, etc., a better understanding of all aspects of coacervation phenomenon is called for [13,14,15,16,17,18]. Ethanol-induced simple coacervation in gelatin system has been reported in the past, which belongs to the unicomplex polyampho-ion category [19,20,21]. In this system, the associative interaction was driven by intramolecular binding followed by the formation of soluble complexes of aggregated gelatin chains at room temperature. The mesophase separation in this system was characterized by signature ethanol concentrations defined as Vc, Vφ, and Vprep where the following observable changes in solution turbidity were noticed: onset of associative interactions at Vc, formation of the soluble complex at Vφ, and formation of the insoluble complex at Vprep. This is a parallel description of pH-induced complex coacervation described by Kaibara et al. [22] where the equivalent signature pHs were identified as pHc, pHφ, and pHprep.
Temperature-induced coacervation is not very common because solubility increases with temperature in most polyelectrolyte solutions. Previous studies on biopolymers such as poly (NIPAAm-co-HIPAAm) have shown thermoresponsive coacervation and lowering of LCST with salt content [23]. Wolf et al. have extensively studied the temperature-dependent behavior of aqueous poly (phosphoester) terpolymer which showed coacervation above a certain temperature. The temperature was referred to lower critical solution temperature (LCST) [24]. Swanson et al. have studied the thermoreversible phase transition in polyesters [25]. This recent literature provides a glimpse of thermally activated interpolymer coacervation.
Elastin is an important extracellular matrix protein that is responsible for extensibility and elastic recoil properties of connecting tissues, arterial walls, skin lung parenchyma, and blood vessels. Tropoelastin, which is a protein precursor of elastin, undergoes self-assembly followed by cross-linking with a lysyl oxidase to form insoluble network elastin fibrils in vivo. Elastin coacervation has drawn much attention in the past and it has been mostly recognized that the insoluble fraction of elastin, tropoelastin, is mostly responsible for causing coacervation [25]. Maeda et al. [26] have shown that the hydrophobic polymeric sequence of amino acids Val-Pro-Gly-Val-Gly and its derivatives present in the tropoelastin may cause coacervation.
Thermoresponsive polymers have numerous applications while thermoresponsive coacervates have found many biomedical applications including tissue engineering [27, 28], protein purification [29], and drug delivery [30]. A coherent picture of elastin coacervation that has emerged so far concludes the following [31]: (i) coacervation occurs primarily due to hydrophobic cooperative association of tropoelastin; (ii) the number, sequence, and contextual arrangement of hydrophobic domains are important; (iii) coacervation is an entropic driven endothermic process; and (iv) it is closely associated increased β-structures in hydrophobic domains and α-helices in cross-linking domains.
A pertinent question arises here: how thermoreversible is elastin coacervation? This is one area which has not received much attention in the past, and in the literature, there is the paucity of information on this. We provide an answer to this important question herein. Since tissue engineering involves multiple cycles of rupture and regeneration of tropoelastin self-assembly, the importance of exploration thermodynamic reversibility of elastin coacervate can be hardly stressed.
Materials and methods
Water-soluble elastin (salt free, lyophilized powder) resourced from bovine neck ligament was purchased from Sigma-Aldrich, USA, with product number E6527 and CAS no. 9007-58-3. Sodium chloride was brought from CDH, India. All chemicals were of analytic grade, and these were used without further purifications. All procedures were performed at room temperature 25 °C unless otherwise stated.
Elastin dispersion 0.1% (w/v) was prepared by dissolving biopolymer powder in deionized water at 25 °C under constant stirring for 4 h which produced a homogeneous and optically clear solution with a very light-yellow color. For salty samples, the required amount of NaCl (10–100 mM) was added to the solvent water and stirred for 5 min to which elastin powder was added and stirred for 4 h at room temperature. For pH-dependent study, the pH of each solution was adjusted as per experimental requirement using 0.1 M HCl or 0.1 M NaOH solution. All the solutions appeared optically clear and transparent after preparation. The prepared dispersions were stored in air tight borosilicate glass bottles at 4 °C for future use which in any case did not exceed 48 h. Each experiment was repeated four times to ascertain data reproducibility and the reported values are averages.
The electrophoresis measurements were done on a zeta potential instrument (ZEECOM model ZC2000, Microtec, Japan), and the UV absorption and turbidity (at 400 nm) were measured using Cary 60 spectrophotometer (Agilent, USA) for temperature-dependent study CARY single cell peltier accessory attached to the instrument were used. For each temperature measurements, 15 min of resting time was allowed for the system to reach equilibrium. PhotoCor Instruments, USA, that was operated in the multi-tau mode was used to perform dynamic light scattering (DLS) experiments. The light scattering spectrometer was mounted on a Newport vibration free optical table. Scattering angle was set at 90°, and excitation source was a 35 mW He:Ne laser (632.8 nm). Non-linear regression analysis of the measured intensity correlation functions was done to determine particle size histogram. Further details on DLS and data analysis procedures are described in ref. [32].
Results and discussion
Characterization of elastin
Since elastin is a heterogeneous biopolymer with much of its physicochemical properties dependent on the source and method of its extraction, it is necessary to first validate its physical properties. In the literature, elastin protein from various sources has been used for the coacervation studies, but there had been rarely any attempt to fully characterize the biopolymer prior to its use. We avoided this shortcoming in the present study. As the first step, we performed a pH titration of the salt-free elastin solution at room temperature and measured the solution turbidity, hydrodynamic radius (Rh), and zeta potential (ζ) of the biopolymer (Fig. 1).

Variation of solution turbidity (100-%T), mean hydrodynamic radius (Rh1), and zeta potential as a function of pH measured at room temperature 25 °C
As expected, the turbidity and particle size increased until the isoelectric pH, pI = 4.5 ± 0.3 was reached where the zeta potential recorded almost zero value. The zeta potential remained in the range of + 25 to − 25 mV corresponding to the pH region 2.5 to 9. The mean hydrodynamic radius increased from 100 to 900 nm as pI was approached, and for pH > pI, this value slowly decreased to 100 nm. If this plot is converted to a symmetry plot about the variable (pH-pI) (see Fig. 2), it depicts not a perfect symmetry of elastin charge state about its isoelectric pH. This implies that the fraction of protonated and deprotonated residues was quantitatively different at a given pH. However, this had a remarkable bearing on the average aggregate size of elastin. Below, pI the average aggregate size was much larger implying more exposure of hydrophobic domains (region I). This aspect was probed more intensely by looking at the pH-dependent particle size histogram (Fig. 3).

Symmetry plot showing a variation of zeta potential and mean size about pI of elastin (ΔpH = pH−pI). Notice the higher size of the aggregates when elastin was in its protonated state

Particle size histogram is shown as a function of pH of the solution evaluated at room temperature. Note the changing pattern of size distribution with pH change
From the size histogram data shown in Fig. 3, bimodal particle size distribution was noticed in the pH region between ≈ 2 to 4 (Fig. 3a, b) and 5.5 to 6.2 (Fig. 3e). Beyond pH = 6.2, there was only unimodal distribution (Fig. 3f). Remarkably, in the region pI ± 0.5, there were three distinct populations in the size distribution (see Fig. 3d). The dominant fraction had a mean size of Rh1 = 300 nm that shot up to 600 nm at pI, and eventually reduced to 150 nm for pH > 7. The second fraction had a mean size of Rh2 = 28 nm at pH = 2 which gradually decreased to 14 nm at pH = 6. Interestingly, a third fraction was noticed only near the pI region that had a mean size of 150 nm. For pH > 6, the particle size histogram was unimodal. Numerous studies have attributed self-assembly to the hydrophobic aggregation. Thus, for pH < pI, the observed higher aggregate size was clearly due to the abundance of intermolecular hydrophobic forces, which at pH = pI reached a maximum (Figs. 1 and 4). The coherent picture that emerged from this data revealed that the pH ≈ pI region (shaded region of Fig. 4) was the most coacervate-rich zone compared to the rest of the pH domains. Further, between pH < pI and pH > pI regions, the former had more propensity of intermolecular hydrophobic interactions and hence was more prone to coacervate.

Mean hydrodynamic radius shown as a function of solution pH. Note that close to pI region, there are three populations present in the dispersion
Coacervation
The representative temperature-dependent turbidity profiles of three salt-free elastin solutions at different pH depicted in Fig. 5. Temperature-dependent experiments were done up to 60 °C in order to avoid possible heat-induced denaturation of the protein at a higher temperature. The data clearly shows the sample prepared with pH = 4.7 (close to pI) undergoing unicomplex formation that resulted in the coacervation at TC. The other two samples did not reveal similar behavior although the pH < pI sample exhibited marginally higher turbidity compared to the pH > pI sample (Fig. 5a). The turbidity and UV-Vis absorbance profiles depicted qualitatively identical trend for the coacervating sample (Fig. 5b). From the change in the slope (\( \frac{d\left(100-\%T\right)}{\mathrm{dT}}\mathrm{vsT} \) graph, Fig. 5c) data, it was possible to identify three distinct temperatures where observable changes in the turbidity occurred. These temperatures are identified as onset of self-assembly Tos, coacervation transition TC, and mesophase separation temperature Tms.

a Temperature-dependent turbidity profiles of three samples of elastin prepared at pH close to pI, pH < pI, and pH > pI. The one at pH = 4.7 undergoes coacervation while the other two samples mostly remain stable to temperature variation. b Turbidity vs T that identifies the transition temperatures Tos, TC, and Tms, and in (C) and \( \frac{d\left(100-\%T\right)}{dT}\mathrm{vsT} \) is shown that reveals clear peaks representing aforesaid temperatures. These data are from salt-free solutions. Onset of associative self-assembly occurs at Tos, coacervation transition at TC, and mesophase separation at Tms (see text for details).
The coacervation phenomenon was probed as a function of monovalent salt sconcentration (NaCl) in the range from 10−4 to 10−1 M (data not shown) which yielded turbidity profiles similar to the one shown for the pH = 4.7 sample (Fig. 5). This data was changed to the derivative plot of \( \frac{d\left(100-\%T\right)}{\mathrm{dT}}\mathrm{vsT} \) which is depicted in Fig. 6. The peak in this plot corresponds to the coacervation temperature TC and shifts to lower temperature at higher salt concentration and similar behavior was found for the other two transition temperatures too. The dependence of TC, Tos, and Tms with different salt concentration is shown in Fig. 7 which is revealing. Between 10−4 and 4 × 10−2 M NaCl, the TC value changed by almost 5 °C making the coacervation process highly temperature tunable, since the process of coacervation is preceded by self-assembly of elastin which in turn is governed by how much of the protein is exposed and is available for self-association. This implies salt induced unfolding of this protein in the concerned salt content region, and occurrence of maximum of this at 40 mM NaCl concentration.

Plot of \( \frac{d\left(100-\%T\right)}{\mathrm{dT}}\mathrm{vsT} \) shown for samples prepared with different salt concentrations. The peaks indicate coacervation temperature TC, and it shifts to lower temperature at higher salt concentration

Dependence of onset of self-assembly, coacervation, and mesophase separation temperatures, Tos, TC, and Tms respectively shown as a function of NaCl concentration of the coacervating elastin solution. Notice the shifting of TC by as much as 5 °C making the process highly temperature tunable. No significant dependence was found for [NaCl] > 40 mM
Thermoreversibility of coacervates
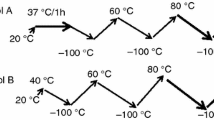
After probing the thermally activated coacervation in elastin solutions it was felt imperative to examine the thermoreversibility of the coacervates. Figure 8a depicts this behavior for salt-free solutions and Fig. 8b for salty preparations monitored through their turbidity profiles. The ABCD heating path characterizes the coacervation transition while DEFG defines the behavior on temperature reversal in Fig. 8a (cooling path). These heating-cooling profiles were identical in salty solutions too. However, two major differences were noticed: (i) the area under the loop (heating-cooling curves) decreased for higher salt concentrations (horizontal arrows in Fig. 8b) and (ii) the remnant turbidity (turbidity at room temperature, vertical arrows in Fig. 8b) was more for solutions with higher salt concentrations. What will happen if a sample is subjected to two heating-cooling cycles? Figure 8c data provides an answer to this question clearly. Some clear observations are made from this data: (i) the hysteresis loop area diminished significantly in the second cycle indicating thermoreversibility and (ii) the remnant turbidity was almost zero implying the oligomeric state of elastin at room temperature remained unaltered.

a Turbidity profile of salt-free and b for salty elastin solutions. Notice the bigger hysteresis loops in low-salt containing samples (horizontal arrow) and more remnant turbidity for higher salt containing samples (vertical arrow). c Second cycle heating-cooling diagram reveals reversibility to a large extent (I = 70 mM)
In Fig. 9, the area under the turbidity hysteresis loop (data from Fig. 8) is plotted as a function of salt concentration along with the remnant turbidity. The self-assembly of elastin is a fully hydrophobic driven process and we have already shown in Fig. 7 that at higher salt concentration coacervation occurred at a much lower temperature. Thus, more hydrophobic sites were available for interaction at higher salt containing elastin solutions. Thus, the presence of salt facilitated the unfolding of elastin which enabled exposure of hydrophobic sites that were buried inside the core of the native elastin at room temperature. For a given ionic strength of the coacervating solution, as the temperature was raised from room temperature (RT) to TC, more of elastin denatured revealing the hydrophobic sites to enable their associative interaction with their neighboring binding partners. When the temperature was reversed from TC to RT, first the coacervate got dissociated into individual elastin oligomers and further into smaller oligomers of the protein. This dissociation was more aggressive in salty solutions which enabled the elastin molecules to refold as it approached RT. However, the experimental data (Fig. 8) suggest that there was always a remnant fraction of small elastin aggregates even at RT that prevented complete refolding of elastin. This conjecture is depicted in schematic-I clearly.

The area under the heating-cooling curve loop (Fig. 8) and the remnant tropoelastin aggregates plotted as a function of the ionic strength of the coacervating solution
Phenomenology of Thermoreversibility
First, it is necessary to understand the molecular structure of elastin. Elastin (E) is constituted by linking together many small soluble precursor tropoelastin (TE) molecules of typical molecular weight of 50–70 kDa to generate the large insoluble complex. Each tropoelastin typically consists of a sequence of 36 small domains, each weighing about 2 kDa. The molecular structure consists of alternating hydrophobic and hydrophilic domains.
Tropoelastin self-assembles at physiological temperature due to the interaction between hydrophobic motifs that confer elasticity to the structure and also lead to β-turn formations. This process is thermodynamically controlled and does not require protein cleavage. The hydrophobic domains are highly stretchable from 20 to 160 nm. Baldock et al. [33] have shown that the mean persistence length of TE is 0.36 nm and it has a contour length of 166 ± 49 nm. TE monomer has a hydrodynamic radius of 5.09 nm and radius of gyration of 6.2 nm that ascribes an elongated conformation to this molecule. This molecule has Young’s modulus of about 3 kPa [33, 34].
Clarke et al. have argued the disadvantages in the use of α-elastin as a descriptor for the tropoelastin coacervation. Because α-elastin is derived from elastin partly hydrolyzed with oxalic acid, it consists mostly of hydrophobic elastin fragments that are heterogeneous and crosslinked. Tropoelastin on the other hand consists of alternate domain structures [35]. It has been further shown that unlike most proteins that show denaturation with increased temperature and become less ordered, elastin polypeptides appear to get more ordered eventually leading to the process of coacervation. This process has been described for tropoelastin, α-elastin, and synthetic polypeptides mimicking the hydrophobic domain sequences [36]. Jamieson et al. have shown that coacervation in the elastin system is not a second-order phase transition. The protein hydrolysates exist in the cold solution as monomeric α-fragments, which on warming to higher temperature form isotropic and stable aggregates preferentially at pH values near the isoelectric point [37].
In general terms, the coacervation pathway can be qualitatively visualized through a set of equilibrium steps given by
First, a fraction x of available elastin molecules [E] do undergo partial heat-induced structural change to expose the hydrophobic fragments of elastin [HE] which in turn associates with an existing oligomer [HE]n-1 to generate an elastin aggregate [E]agg. [E]N and [HE]x represent the concentration of native elastin and hydrophobic fragment of elastin respectively. In the final stage at TC these aggregates coalesce to form the coacervate mesophase [E]ms. The dissociation pathway can be described as follows
which can be approximated to a pseudo two-step process given by
When [E]RT = [E], it is fully reversible, and in the case of partial reversibility ([E]-[E]RT) will describe the remnant fraction of aggregated elastin that does not permit restoration of elastin to its native state (vertical arrows in Fig. 8b). Further, we treat [E]ms is the coacervate phase made of self-assembled elastin units. Equations (1) to (5) are a qualitative representation of kinetics of coacervation pathway. It cannot be claimed that all the [E]agg turned into [E]ms. However, in order to provide a thermodynamic description of the coacervation process, it was presumed that it involved a complete transformation of aggregates to mesophase.
However, as far as the dissociation process is concerned, the abovementioned multiple step reaction can be simply described through an equilibrium constant Keq involving an approximate two-step process where [E]ms dissociates to approach [E]RT during the cooling cycle (Eq. (5)). Thus, to first approximation
The solution turbidity depends on particle density (equivalent to concentration) among other things. Therefore, to first approximation, turbidity can be assumed to be directly proportional to particle concentration assuming that particle form factors do not change. Thus, the concentration of [E]RT and [E]ms was assumed to be proportional to the turbidity of that phase. Such simplification will facilitate quantitative estimation of the free energy of coacervation ΔG using Arrhenius description which is given by
The temperature-dependent turbidity profiles shown in Fig. 8b were fitted to Eq. (7) to determine the corresponding ΔG values and the results are plotted in Fig. 10.

a Variation of coacervation and dissociation free energy, ΔGcoa and ΔGDiss with the ionic strength of the sample. b Dependence of remnant turbidity on free energy difference ΔGchange = (ΔGcoa − |ΔGDiss|)
The heating path involved an activation free-energy ΔGcoa (coacervation free energy) while the same for the cooling path it was ΔGDiss (dissociation free energy). The data shown in Fig. 10 implies that the unicomplex formation through hydrophobic association of TE leading to coacervation was a thermally activated process. On the contrary, on cooling, these self-assembled complexes dissociated to their oligomer state spontaneously facilitated by free-energy gain. The free-energy change, ΔGchange = (ΔGcoa − |ΔGDiss|), is in fact identical to the remnant [E] oligomer content (remnant turbidity at RT). Therefore, their linear dependence with a positive slope is indicative of the fact that elastin solutions in salty environment undergo thermally activated self-assembly which on temperature reversal remain trapped in a partially unfolded state giving rise to an intrinsically disordered protein, IDP-like situation.
Secondary structure
The secondary structure of elastin in coacervate samples was monitored for heating and cooling cycle using circular dichroism (Applied Photophysics Chirascan instrument, USA) measurements in the temperature range of 30 to 60 °C (Fig. 11a). The spectra show two prominent peaks one positive band at 195 nm, and one negative band at 205 nm, and a minor negative band at 225 nm. Figure 11a clearly shows molecular ellipticity of these bands reduced with a rise in temperature indicating a decrease in helicity. In addition, the spectra clearly showed that elastin exhibited thermoreversible change as shown in the insets. There was a small hysteresis in CD spectra of elastin in heating and cooling cycle data consistent with the same noticed in turbidity. Therefore, it can be argued that there was a loss in secondary structure (helix component) of elastin with the rise in temperature and the consequent possible formation of intrinsically disordered protein-like structures cannot be ruled out [38]. This conclusion was utilized in arriving at a molecular level schematic shown in Scheme 1 and Eqs. (1–4). For a given temperature, the two profiles do overlap to a good extent (Fig. 11). This indicated that at the molecular level, the secondary structure opened up during heating and vice versa. Since the hydrophobic moieties of proteins reside inside the core of the molecule to minimize the free energy of the structures when in contact with water, it is logical to argue that at a given temperature a partially denatured structure of the elastin molecule will exist revealing only a portion of its hydrophobic moiety, which is qualitatively referred to as HE in this work. It needs to be emphasized that what we have proposed in this model is at best a conjecture based on data in hand.

Variation of circular dichroism spectra of elastin (a) heating and inset cooling cycle (b) in comparison with heating-cooling for 30 and 50 °C in salt-free solution

Representative depiction of thermoreversibility of elastin (E) coacervation. The hydrophobic motifs of elastin self-assemble (HE) to form soluble unicomplexes that coalesce to form coacervate droplets prior to mesophase separation. Legends: RT is the room temperature, E is the elastin, HE is the elastin with hydrophobic parts exposed, and Tms is the mesophase separation temperature
Conclusions
We performed an extensive study to map the pH-dependent physical properties of the elastin molecule and found that the zeta potential was not completely symmetric about its isoelectric pH. The protonated molecules were found to be in an aggregated state with a mean size that was much larger than their deprotonated counterparts. The particle size histogram revealed considerable aggregation in the pH = pI ± 0.5 region making this zone fertile for coacervation. The coacervation kinetics of elastin precursor tropoelastin was mapped as a function of solution ionic strength. This mesophase separation was found to be strongly temperature dependent and the coacervation temperature could be lowered by as much as 5 °C from its initial value of 38 °C (for salt-free sample). The samples were subjected to heating-cooling cycles that produced hysteresis loops with loop area decreasing with ionic strength which implied larger thermoreversibility of salty coacervates. However, at room temperature, the heat-cool cycled sample did reveal the presence of monomeric TE molecules only for salt-free samples, but in all other samples there was clear evidence of the existence of a remnant fraction of aggregated elastin molecules. The size of this remnant aggregates increased with the ionic strength of the samples. Estimation of free energy of the coacervation and the dissociation processes revealed that coacervate dissociation was free-energy driven while the elastin self-assembly leading to coacervation was hydrophobic and thermally activated. This study attempts to provide an understanding to the phenomenon thermoreversibity of elastin coacervates which remains poorly explored hither to.
References
De Jong HB, Kruyt HR (eds) (1949) Colloid science, 2. Elsevier, London, pp 232–238
De Jong HB, Bonner J (1935) Phosphatide auto-complex coacervates as ionic systems and their relation to the protoplasmic membrane. Protoplasma 24(1):198–218
Dubin P, Stewart RJ (2018) Complex coacervation. Soft Matter 14:329–330
Lytle TK, Sing CE (2018) Tuning chain interaction entropy in complex coacervation using polymer stiffness, architecture, and salt valency. Mol Syst Des Eng 3:183–196
Rawat K, Bohidar HB (2014) Coacervation in biopolymers. J Phys Chem Biophys 4(6):1
Cooper CL, Dubin PL, Kayitmazer AB, Turksen S (2005) Polyelectrolyte–protein complexes. Curr Opin Colloid Interface Sci 10:52–78
Pathak J, Priyadarshini E, Rawat K, Bohidar HB (2017) Complex coacervation in charge complementary biopolymers: electrostatic versus surface patch binding. Adv Colloid Interf Sci 250:40–53
Elhassan MS, Oguntoyinbo SI, Taylor J, Taylor JR (2018) Formation and properties of viscoelastic masses made from kafirin by a process of simple coacervation from solution in glacial acetic acid using water. Food Chem 239:333–342
Danielsen S, Delaney K, Fredrickson G (2018) Small ion effects on self-coacervation phenomena in block polyampholytes. Bull Am Phys Soc
Rodriguez AMB, Binks BP, Sekine T (2018) Emulsion stabilisation by complexes of oppositely charged synthetic polyelectrolytes. Soft Matter 14(2):239–254
Li Y, Dubin PL, Havel HA, Edwards SL, Dautzenberg H (1995) Complex formation between polyelectrolyte and oppositely charged mixed micelles: soluble complexes vs coacervation. Langmuir 11(7):2486–2492
Kizilay E, Kayitmazer AB, Dubin PL (2011) Complexation and coacervation of polyelectrolytes with oppositely charged colloids. Adv Colloid Interf Sci 167(1–2):24–37
Imeson AP, Watson APR, Mitchell JR, Ledward DA (1978) Protein recovery from blood plasma by precipitation with polyuronates. Int J Food Sci Technol 13(4):329–338
Tolstoguzov VB (1991) Functional properties of food proteins and role of protein-polysaccharide interaction. Food Hydrocoll 4(6):429–468
De Kruif CG, Weinbreck F, de Vries R (2004) Complex coacervation of proteins and anionic polysaccharides. Curr Opin Colloid Interface Sci 9(5):340–349
Xiao Z, Liu W, Zhu G, Zhou R, Niu Y (2014) A review of the preparation and application of flavour and essential oils microcapsules based on complex coacervation technology. J Sci Food Agric 94(8):1482–1494
Dai R, Wu G, Li W, Zhou Q, Li X, Chen H (2010) Gelatin/carboxymethylcellulose/dioctyl sulfosuccinate sodium microcapsule by complex coacervation and its application for electrophoretic display. Colloids Surf A Physicochem Eng Asp 362(1–3):84–89
Moschakis T, Biliaderis CG (2017) Biopolymer-based coacervates: structures, functionality and applications in food products. Curr Opin Colloid Interface Sci 28:96–109
Tiwari A, Bindal S, Bohidari HB (2008) Kinetics of protein-protein complex coacervation and biphasic release of salbutamol sulfate from coacervate matrix. Biomacromolecules 10(1):184–189
Mohanty B, Bohidar HB (2003) Systematic of alcohol-induced simple coacervation in aqueous gelatin solutions. Biomacromolecules 4(4):1080–1086
Gupta A, Bohidar HB (2005) Kinetics of phase separation in systems exhibiting simple coacervation. Phys Rev E 72(1):011507
Kaibara K, Okazaki T, Bohidar HB, Dubin PL (2000) pH-induced coacervation in complexes of bovine serum albumin and cationic polyelectrolytes. Biomacromolecules 1(1):100–107
Maeda T, Takenouchi M, Yamamoto K, Aoyagi T (2006) Analysis of the formation mechanism for thermoresponsive-type coacervate with functional copolymers consisting of N-isopropylacrylamide and 2-hydroxyisopropylacrylamide. Biomacromolecules 7:2230–2236
Wolf T, Rheinberger T, Wurm FR (2017) Thermoresponsive coacervate formation of random poly (phosphonate) terpolymers. Eur Polym J 95:756–765
Swanson JP, Monteleone LR, Haso F, Costanzo PJ, Liu T, Joy A (2015) A library of thermoresponsive, coacervate-forming biodegradable polyesters. Macromolecules 48:3834–3842
Maeda I, Fukumoto Y, Nose T, Shimohigashi Y, Nezu T, Terada Y, Kodama H, Kaibara K, Okamoto K (2011) Structural requirements essential for elastin coacervation: favorable spatial arrangements of valine ridges on the three-dimensional structure of elastin-derived polypeptide (VPGVG) n. J Pept Sci 17(11):735–743
Betre H, Setton LA, Meyer DE, Chilkoti A (2002) Characterization of a genetically engineered elastin-like polypeptide for cartilaginous tissue repair. Biomacromolecules 3:910–916
Meyer DE, Shin BC, Kong GA, Dewhirst MW, Chilkoti A (2001) Drug targeting using thermally responsive polymers and local hyperthermia. J Control Release 74:213–224
Meyer DE, Chilkoti A (1999) Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotechnol 17:1112–1115
Herrero-Vanrell R, Rincon AC, Alonso M, Reboto V, Molina-Martinez IT, Rodriguez-Cabello JC (2005) Self-assembled particles of an elastin-like polymer as vehicles for controlled drug release. J Control Release 102:113–122
Yeo GC, Keeley FW, Weiss AS (2011) Coacervation of tropoelastin. Adv Colloid Interf Sci 167(1–2):94–103
Bohidar HB (2002) Characterization of polyelectrolytes by dynamic light scattering Handbook of Polyelectrolytes, vol II. American Scientific Publishers, California
Baldock C, Oberhauser AF, Ma L, Lammie D, Siegler V, Mithieux SM, Tu Y, Chow JYH, Suleman F, Malfois M, Rogers S (2011) Shape of tropoelastin, the highly extensible protein that controls human tissue elasticity. Proc Natl Acad Sci 108(11):4322–4327
Graham JS, Vomund AN, Phillips CL, Grandbois M (2004) Structural changes in human type I collagen fibrils investigated by force spectroscopy. Exp Cell Res 299(2):335–342
Clarke AW, Arnspang EC, Mithieux SM, Korkmaz E, Braet F, Weiss AS (2006) Tropoelastin massively associates during coacervation to form quantized protein spheres. Biochemistry 45(33):9989–9996
Bellingham CM, Woodhouse KA, Robson P, Rothstein SJ, Keeley FW (2001) Self-aggregation characteristics of recombinantly expressed human elastin polypeptides. Biochim Biophys Acta (BBA) Protein Struct Mol Enzymol 1550(1):6–19
Jamieson AM, Downs CE, Walton AG (1973) Studies of elastin coacervation by quasi-elastic light scattering. Biochim Biophys Acta 271:34–47
Kjaergaard M, Nørholm AB, Hendus–Altenburger R, Pedersen SF, Poulsen FM, Kragelund BB (2010) Temperature-dependent structural changes in intrinsically disordered proteins: formation of α–helices or loss of polyproline II? Protein Sci 19(8):1555–1564
Acknowledgments
PK is thankful to the University Grant Commission, Government of India UGC, for a Senior Research Fellowship. KR is thankful to the Department of Science and Technology, Government of India, for the DST-Inspire Faculty Award. We are thankful to the Advanced Instrumentation Research facility (AIRF) for allowing to access their instrument.
Funding
This study was funded by the Department of Science and Technology (DST), Government of India under the DST-PURSE-II Program.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaushik, P., Rawat, K. & Bohidar, H.B. Heat-induced coacervation of elastin and its possible thermoreversibility. Colloid Polym Sci 297, 947–956 (2019). https://doi.org/10.1007/s00396-019-04518-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-019-04518-1