
Abstract
Blockade of the late Na+ current (I NaL) protects from ischemia/reperfusion damage; nevertheless, information on changes in I NaL during acute ischemia and their effect on intracellular milieu is missing. I NaL, cytosolic Na+ and Ca2+ activities (Nacyt, Cacyt) were measured in isolated rat ventricular myocytes during 7 min of simulated ischemia (ISC); in all the conditions tested, effects consistently exerted by ranolazine (RAN) and tetrodotoxin (TTX) were interpreted as due to I NaL blockade. The results indicate that I NaL was enhanced during ISC in spite of changes in action potential (AP) contour; I NaL significantly contributed to Nacyt rise, but only marginally to Cacyt rise. The impact of I NaL on Cacyt was markedly enhanced by blockade of the sarcolemmal(s) Na+/Ca2+ exchanger (NCX) and was due to the presence of (Na+-sensitive) Ca2+ efflux through mitochondrial NCX (mNCX). sNCX blockade increased Cacyt and decreased Nacyt, thus indicating that, throughout ISC, sNCX operated in the forward mode, in spite of the substantial Nacyt increment. Thus, a robust Ca2+ source, other than sNCX and including mitochondria, contributed to Cacyt during ISC. Most, but not all, of RAN effects were shared by TTX. (1) The paradigm that attributes Cacyt accumulation during acute ischemia to decrease/reversal of sNCX transport may not be of general applicability; (2) I NaL is enhanced during ISC, when the effect of Nacyt on mitochondrial Ca2+ transport may substantially contribute to I NaL impact on Cacyt; (3) RAN may act mostly, but not exclusively, through I NaL blockade during ISC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myocardial ischemia results in a characteristic pattern of metabolic and intracellular ion changes, ultimately leading to cytosolic Ca2+ (Cacyt) accumulation [5] and the resulting functional and structural derangements. Enhanced Na+ influx, exceeding the functional reserve of the Na+/K+ pump, is widely considered as the “primum movens” of this process, being coupled to Cacyt homeostasis through changes in the equilibrium potential of the sarcolemmal Na+/Ca2+ exchanger (sNCX) (coupled exchanger theory) [9, 23, 28, 31].
Several mechanisms may account for enhanced a Na+ influx during acute ischemia. While it is widely accepted that the Na+/H+ exchanger (NHE), driven by intracellular acidosis, may support large Na+ influx upon reperfusion [19, 24, 47], there is disagreement about its role during ischemia [3, 32, 47]. Several studies show that blockade of a persistent component of Na+ current (I NaL), prevents Ca2+ overload and reduces injury following reperfusion [1, 7, 16, 45]. This suggests that I NaL enhancement may contribute to increased Na+ influx during the preceding ischemia. Exposure to ischemia components (i.e., H2O2, hypoxia and ischemic metabolites) has indeed been shown to enhance I NaL in standard V-clamp experiments [26, 40, 43, 46]. On the other hand, membrane depolarization and shortening of action potential duration (APD), both correlates of acute ischemia, may reduce overall Na+ current availability and time for I NaL-mediated Na+ influx, respectively. Therefore, whether I NaL is actually enhanced during acute ischemia and contributes to cytosolic Na+/Ca2+ accumulation remains to be established. The present study aims to directly address these questions by measuring I NaL and cytosolic ionic activities (Nacyt and Cacyt) in isolated ventricular myocytes exposed to a simulated ischemia protocol.
The results obtained indicate that I NaL was enhanced during simulated ischemia, in spite of the attending action potential (AP) changes, and significantly contributed to Nacyt accumulation. However, the relationship between Nacyt and Cacyt was more complex than predicted by the coupled exchanger theory, suggesting instead a role of ischemia-induced redistribution of Ca2+ between intracellular compartments, with mitochondria contributing as a Nacyt-sensitive Ca2+ store.
Materials and methods
Cell isolation
Ventricular cardiomyocytes from male adult Sprague–Dawley rats (150–175 g) were isolated using a retrograde coronary perfusion method previously published with minor modifications [34]. Measurements were performed only in quiescent, rod-shaped, myocytes with clear striations. All experiment were approved and conducted accordingly to the guidelines stipulated by the Animal Care committee of University of Milano-Bicocca. The manuscript does not contain human data.
Simulated ischemia protocol
Cardiomyocytes were placed into a recording chamber and superfused at 36.5 °C with Tyrode’s solution containing (mM): NaCl 154, KCl 4, CaCl2 2, MgCl2 1, HEPES 5, Glucose 5.5, adjusted to pH 7.3. Cells were paced at 1 Hz, either through the patch pipette or by field stimulation, throughout the protocol.
Ischemia was simulated by superfusing myocytes with a modified Tyrode’s solution (ischemia mimic solution, ISC) containing (mM): NaCl 134, Na-lactate 20, KCl 8, CaCl2 2, MgCl2 1, HEPES 5, sucrose 37, adjusted to pH 6.8. Its composition reflects the major changes in the ischemic environment, as previously described by others [8, 10, 25, 30, 49].
ISC protocol has been performed here in normoxic condition, according to previous studies on ischemia [25] proving that the contribution of hypoxia to changes in cardiomyocyte contractility is negligible; nevertheless, its absence should be considered in the interpretation of results (see “Discussion”).
The experimental protocol included pre-ISC stabilization in normal Tyrode’s solution (about 2 min) followed by ISC superfusion for 7 min (Fig. S1). This ISC duration was selected in preliminary experiments as the maximal tolerated by the majority of cardiomyocytes; ISC wash-out (reperfusion) was almost invariably followed by contracture and death. In the following text, protocol phases are referred to as PRE (pre-ISC); 0.5ISC (0.5 min of ISC); 3ISC (3 min of ISC); 7ISC (7 min of ISC).
Cell shortening
Cardiomyocytes were field stimulated and the single-cell shortening was measured by video-edge detection system (Crescent electronics). The difference between maximal diastolic and systolic cell lengths was expressed as twitch amplitude, which was normalized within each cell to the value recorded in PRE conditions.
Electrophysiology
Myocytes were patch-clamped with borosilicate glass pipettes containing (mM): K+-aspartate 110, KCl 23, MgCl2 3, HEPES KOH 5, EGTA KOH 0.5, GTP Na+-salt 0.4, ATP Na+-salt 5, creatine phosphate Na+-salt 5, CaCl2 0.2 (calculated free-Ca2+ = 10−7 M), adjusted to pH 7.2. Series resistance was <5 MΩ and was compensated to 80% of its value.
Action potentials (AP) were recorded (I-clamp with I = 0 pA) throughout the protocol. AP waveforms recorded in PRE condition and at 7ISC, respectively, were used as templates in AP-clamp experiments.
I NaL was measured at PRE and 7ISC in AP-clamp mode as the current sensitive to 1 µM TTX [40]. To test whether ISC-induced changes in AP affected I NaL magnitude during ISC, AP-clamp was applied with two modalities: (1) the AP templates recorded at PRE and 7ISC, which included ISC-induced changes, were applied during the corresponding phases of the protocol; (2) the AP template recorded at PRE, was applied at both PRE and 7ISC, thus disregarding ISC-induced changes. Differences between I NaL recorded with the two AP-clamp modalities reflect the impact of ISC-induced membrane potential changes to I NaL.
I NaL magnitude during APs was quantified by integrating inward TTX-sensitive current from the beginning of repolarization to 90% of repolarization and dividing the result for the integration interval. This measurement, abbreviated in the following text and figures as “I NaL”, reflects mean Na+ influx rate during repolarization. Currents were normalized to cell capacitance and expressed as current density (pA/pF).
Measurement of intracellular ionic activities
Nacyt and Cacyt were measured in intact, field-stimulated (1 Hz) cardiomyocytes, loaded with Asante Natrium Green-2 (ANG-2) for Na+ and FLUO4-AM for Ca2+ measurements, respectively. Cardiomyocytes were incubated with the membrane-permeant form of the dyes for 30 min, and then washed for 15 min. ANG-2 and FLUO4-AM emissions were collected through a 535 nm band pass filter, converted to voltage, low-pass filtered (200 Hz) and digitized at 2 kHz after further low-pass digital filtering (FFT, 100 Hz) and subtraction of background luminescence [2].
For Na+ measurement, fluorescence recorded during ISC (F) was normalized to that recorded during the PRE phase (F 0) and expressed as F/F 0. Considering that, Nacyt changes were well within the range of linear dye response (Supplemental Figure S5), the uncalibrated Na+ signal was considered adequate. Because dye response is slow relative to membrane potential changes, the Na+ signal reflects an integrated value of Nacyt during the whole electrical cycle.
Ca2+ fluorescence signal was calibrated by previously described methods [34], described in the Online Resource along with the potential bias introduced by intrinsic pH sensitivity of the dye. Since dye response is fast enough, the Ca2+ signal was evaluated as diastolic Ca2+ (CaD) and Ca2+ transient amplitude (CaT, i.e., difference between systolic Ca2+ and CaD). The sarcoplasmic reticulum (SR) Ca2+ content (CaSR) was estimated at 7ISC in separate subsets of cardiomyocytes, by applying an electronically timed 10 mM caffeine pulse. The caffeine solution was Ca2+ and Na+ free, to prevent Ca2+ efflux through the sNCX. SR Ca2 fractional release (CaFR) was obtained as the ratio between CaT at 7ISC and CaSR.
Pharmacological interventions
The contribution of different mechanisms to Nacyt and Cacyt dynamics during ISC was evaluated by specific pharmacological interventions.
I NaL contribution was tested by blocking the current with either ranolazine (RAN, 10 µM) or tetrodotoxin (TTX, 1 µM). Although at this concentration TTX can be safely considered to selectively block I NaL [40], ancillary effects might be present for RAN. Therefore, whereas effects equally exerted by the two agents were considered to reflect I NaL contribution, those peculiar of RAN may possibly result from ancillary effects of the drug. RAN and TTX were applied at the beginning of the PRE phase.
Contribution of sNCX and mNCX were tested by using the selective blockers SEA0400 (SEA, 1 µM) and CGP37157 (CGP, 1 µM), respectively. Cariporide (CAR, 1 µM) and ouabain (OUAB, 1 mM) were used to inhibit the NHE and the Na+/K+ pump, respectively. RU360 (RU, 10 µM) was used to block the mitochondrial Ca2+ uniporter (MCU) [29], the main path of Ca2+ entry into mitochondria [21]. These agents were also added to the ISC solution; DMSO concentration was balanced in all the solutions.
Statistical analysis
The time courses of Nacyt and Cacyt (CaT and CaD) during the protocol, shown in figures, were obtained by averaging records from N cells and are presented as mean ± SE. Differences in twitch amplitude, Nacyt and Cacyt were statistically evaluated at 0.5ISC, 3ISC, 7ISC (Supplemental figure S1). In the case of Nacyt, peak value and the rate of rise (dNa+/dt, by linear fitting of the rising phase) were also evaluated.
Differences between means were tested by paired T test or ANOVA as appropriate (Bonferroni’s correction in post hoc comparisons). Statistical significance was defined as p < 0.05 (NS, not significant). Sample size is reported in each figure legend.
Results
Cell shortening and electrical activity
Twitch amplitude markedly decreased during early ISC (0.5ISC), to slowly recover to a stable level after 3 min (Fig. 1a). Twitch amplitude achieved a minimum at 0.5ISC (−86.9 ± 1.8% of PRE; p < 0.05), recovered at 3ISC to −12.4 ± 18.5% of PRE, without further changes at 7ISC (−10.8 ± 17.3% of PRE) (Fig. 1b).

Cell shortening and electrical activity during ISC. a Average traces ± SE of contraction amplitude (left) and statistics at discrete time points during the protocol (arrows). b Representative traces of contraction (top) and action potentials (bottom) at discrete time points (arrows in a). c Statistics for diastolic membrane potential (E diast), maximum depolarization rate (dV/dt max) and action potential duration at 90% repolarization (APD90). CTRL N = 8. °p < 0.05 vs PRE
AP were elicited throughout ISC exposure (Fig. 1b), even when mechanical activity was almost absent. ISC partially depolarized diastolic potential (E diast) and reduced dV/dt max of phase 0 (Fig. 1c). APD at 90%, repolarization (APD90) prolonged up to 0.5ISC and then shortened (Fig. 1c). RAN treatment did not measurably affect AP response to ISC (Supplemental Figure S2).
Late Na+ current
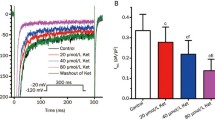
A small I NaL was present during repolarization even in PRE conditions; this component was insensitive to blockade by RAN (Fig. 2). When the 7ISC AP template was applied at 7 min of ISC, I NaL was increased by 77% (p < 0.05 vs PRE, Fig. 2b), a change completely prevented by RAN (Fig. 2b). When the PRE AP template was applied at 7ISC, I NaL increment observed was, if anything, larger than seen with the previous protocol (88%, p < 0.05 vs PRE, Supplemental Figure S3). Thus, I NaL may significantly increase during ISC in spite of the attending membrane potential changes, which, as expected, reduced overall INa availability (reduced dV/dt max, see above).

Late Na+ current (I NaL) during ISC. a Representative action potentials templates (top) and the respective TTX-sensitive currents (bottom) at PRE (black line) and 7ISC (red line) time points in CTRL and RAN groups. b Statistics for I NaL at PRE and 7ISC. N > 6 for both groups. °p < 0.05 vs PRE
Cytosolic Na+
Changes in Nacyt during ISC were assessed in intact, field-stimulated (1 Hz) cardiomyocytes in the absence (CTRL) and presence of I NaL blockade by either RAN or TTX. After an initial dip, Nacyt increased during ISC, reaching a peak at about 1–2 min, and then slowly declined (Fig. 3a). RAN and TTX significantly reduced peak Nacyt (Fig. 3a) and the rate of Nacyt increment (Fig. 3b); the effect was similar between the two agents. This suggests that I NaL enhancement significantly contributed to, but was not the only factor, in Nacyt accumulation during ISC. When both I NaL and NHE were blocked simultaneously (CAR + TTX), ISC failed to induce Nacyt accumulation (Supplemental Figure S4), thus pointing to NHE as the other Na+ influx route [35].

Effect of I NaL blockade (RAN, TTX) on cytosolic Na+ (Nacyt) during ISC. a Average traces ± SE of Nacyt during the ISC protocol in CTRL, RAN and TTX treatment groups; statistics of Nacyt changes (normalized to values at PRE) at peak Nacyt and at 7ISC time points. b Average Nacyt traces (as in a) during the early ISC phase to illustrate differences in Nacyt accumulation rate; statistics for Nacyt accumulation rate (dNacyt/dt). CTRL N = 14; RAN N = 9; TTX N = 12. *p < 0.05 vs CTRL
To test whether the Na+/K+ pump remained functional during ISC and contributed to the late Nacyt decline, cardiomyocytes were exposed to ISC in the presence of ouabain (OUAB). Under this condition, Nacyt monotonically increased throughout ISC superfusion (Supplement Figure S5), indicating that in the present settings, the Na+/K+ pump was active and contributed to limit Nacyt accumulation.
Cytosolic Ca2+
Changes in Cacyt during ISC were assessed in intact, field-stimulated (1 Hz) cardiomyocytes (Fig. 4). Both CaD and CaT increased during ISC; at variance with Nacyt, the increment was not preceded by a dip. CaD monotonically increased to achieve a more or less stable level at 3 min (Fig. 4a). CaT increment followed a sigmoidal time course, thus lagging behind CaD; it achieved a peak at about 3 min and then slowly declined (Fig. 4a). RAN slightly, but significantly, decreased CaD and visibly minimized its variability across cells, an effect not shared with TTX (Fig. 4a). The same was true for CaT even if, probably because of its larger variability, RAN effect on this parameter did not achieve significance (Fig. 4a). Both RAN and TTX tended to decrease CaSR, but when analyzed separately for each I NaL blocker their effect did not achieve statistical significance (Fig. 4b). However, when the data from RAN and TTX groups were pooled, I NaL blockade significantly reduced CaSR at 7ISC (80.7 ± 8.5 vs 60.8 ± 4.7 µM; p < 0.05, Fig. 4b). CaFR was not affected by I NaL blockade (Fig. 4b).

Effect of I NaL blockade (RAN, TTX) on cytosolic Ca2+ (Cacyt) during ISC. a Average traces ± SE of diastolic Ca2+ (CaD) and Ca2+ transient amplitude (CaT) during the ISC protocol in CTRL, RAN and TTX treatment groups; statistics of Cacyt at discrete time points (arrows) during the protocol (CTRL N = 22; RAN N = 19; TTX N = 19). b Statistics for SR Ca2+ content (CaSR) and SR Ca2+ fractional release (CaFR) at protocol end (CTRL N = 19; RAN N = 13; TTX N = 10); representative Ca2+ transient triggered by caffeine in each group. *p < 0.05 vs CTRL; # p < 0.05 vs RAN
These observations are consistent with the common notion that Cacyt increases during acute ischemia; however, neither its timing with respect to Nacyt, nor its unexpected insensitivity to I NaL blockade, were consistent with its dependency on enhanced Na+ influx. The (small) effect of RAN on CaD, not shared by TTX, might reflect an agent-specific ancillary action.
The unexpected lack of Cacyt response to reduced Na+ influx, led us to question sNCX role in mediating Cacyt accumulation during ISC. To address this point, Cacyt measurements were repeated in the presence of sNCX blockade.
Role of the sarcolemmal Na+/Ca2+ exchanger
To assess the role of sNCX during ISC, its specific inhibitor SEA [42] was also added to the ISC solution (ISC + SEA, Fig. 5).

Effect of I NaL blockade (RAN, TTX) on cytosolic Ca2+ during ISC in the presence of sNCX blockade (SEA). a Average traces ± SE of diastolic Ca2+ (CaD) and Ca2+ transient amplitude (CaT) during the ISC protocol in CTRL, RAN and TTX treatment groups; statistics of Cacyt at discrete time points (arrows) during the protocol; statistics of Cacyt at discrete time points (arrows) during the protocol (SEA N = 23; SEA + RAN N = 20; SEA + TTX N = 18). b Statistics for SR Ca2+ content (CaSR) and SR Ca2+ fractional release (CaFR) at protocol end (SEA N = 9; SEA + RAN N = 9; SEA + TTX N = 9); representative Ca2+ transient triggered by caffeine superfusion in each group. § p < 0.05 vs SEA
In the presence of SEA, ISC-induced Nacyt accumulation was reduced and Cacyt accumulation (CaD, CaT, CaSR) was markedly enhanced (Supplemental Figure S6). The direction of the reciprocal changes in Nacyt and Cacyt unequivocally indicates that, during ISC, sNCX still operated in its forward mode, thus supporting Ca2+ efflux, rather than influx. Notably, forward sNCX operation persisted in spite of the attending increase in Nacyt; moreover, the Cacyt increment induced by ISC in the presence of sNCX blockade (SEA group) was twice as large as that observed during SEA alone (Supplemental Figure S7). These findings indicate that large, sNCX-independent, Ca2+ sources contribute to Cacyt build up during ISC.
Notably, during ISC + SEA, both RAN (+RAN) and TTX (+TTX) significantly reduced Cacyt accumulation (Fig. 5a), with their effect being substantially larger than during ISC alone (Fig. 4). This suggests the contribution to Cacyt accumulation of a Na+-sensitive Ca2+ source, whose role was unveiled by sNCX blockade.
Consistent with the increase in overall cell Ca2+ content expected from sNCX blockade, CaSR at 7ISC was higher in ISC + SEA (SEA) than in ISC alone (CTRL) (116.7 ± 11.6 vs 80.7 ± 8.5 µM; p < 0.05; Supplemental Figure S6a). RAN slightly but significantly reduced CaSR even in the presence of SEA (Fig. 5b), thus suggesting its ability to modulate Cacyt independently of sNCX. This effect did not achieve significance with TTX, which, in this respect, was less efficient than RAN. CaFR was unchanged by either RAN or TTX (Fig. 5b) thus arguing against modulation of ryanodine receptors (RyRs) as a major player in the effects exerted by the two agents.
The significant effect of I NaL blockade on Cacyt in Fig. 5 suggests that, at least under sNCX inhibition, a Nacyt-sensitive intracellular compartment may contribute to its accumulation during ISC. Mitochondria are an intracellular Ca2+ compartment, potentially affected by ISC and endowed of Nacyt-sensitive Ca2+ transport. The latter is represented by mNCX, which may either uptake or release Ca2+ from mitochondria depending on the electrochemical gradient for the transport. To test this hypothesis, the experiments were repeated in the presence of mNCX blockade.
Role of the mitochondrial Na+/Ca2+ exchanger
mNCX was selectively blocked by CGP [12], which was added to the ISC solution either alone, or in the presence of SEA.
When applied alone (CGP group, Fig. 6 left), CGP did not measurably affect Cacyt accumulation during ISC (Fig. 6a); however, it significantly increased CaSR (Fig. 6b), thus suggesting a shift of Ca2+ from the mitochondrial to the SR compartment. On the other hand, when CGP was applied in the presence of SEA (+CGP group; Fig. 6 right), Cacyt accumulation and CaSR were significantly reduced. Thus, at least in the presence of the high Cacyt levels achieved under sNCX blockade, mitochondria provided a Ca2+ source, with mNCX supporting Ca2+ efflux to the cytosol [27]. CaFR was not affected by CGP (Fig. 6b), again arguing against the involvement of RyRs modulation in the observed effects.

Effect of mNCX blockade (CGP) on Cacyt and CaSR during ISC. Left effect of CGP alone; right effect of CGP in the presence of SEA. a Statistics for diastolic Ca2+ (CaD, top) and Ca2+ transient amplitude (CaT, bottom) at discrete protocol time points (CTRL N = 22; CGP N = 8; SEA N = 23; SEA + CGP N = 16); b statistics for SR Ca2+ content (CaSR) and SR fractional release (CaFR) at the end of protocol (CTRL N = 19; CGP N = 8; SEA N = 10; SEA + CGP N = 14). *p < 0.05 vs CTRL, § p < 0.05 vs SEA
In the presence of sNCX blockade, the effects of CGP, RAN and TTX on Cacyt accumulation during ISC were strikingly similar (Supplemental Figure S8). This supports the view that I NaL blockade may limit Cacyt accumulation by reducing Nacyt availability to fuel mNCX-mediated Ca2+ efflux from mitochondria.
To further test the role of mitochondria as a Ca2+ source during ISC, MCU was selectively blocked by RU [29] in the presence of sNCX blockade (+RU). RU reduced Cacyt accumulation, achieving statistical significance for CaD (Fig. 7a). In the presence of SEA + RU, CGP failed to modify Cacyt (Supplement Figure S10). These observations confirm a role of mitochondria in Cacyt increment during ISC and support the view that the effect of CGP on Cacyt (Fig. 6 right) were due to inhibition of mitochondrial Ca2+ efflux. RU also increased CaSR (Fig. 7b) likely reflecting transfer of Ca2+ from the mitochondrial compartment to the SR one.

Effect of MCU blockade (RU) on cytosolic Ca2+ during ISC (in the presence of sNCX blockade). a Average traces ± SE of diastolic Ca2+ (CaD) and Ca2+ transient amplitude (CaT) during ISC + SEA alone (SEA) and in the presence of MCU blockade (+RU); CaD and CaT statistics at discrete time points; b statistics for SR Ca2+ content (CaSR) and SR Ca2+ fractional release (CaFR) at protocol end; representative caffeine-induced Ca2+ transients. SEA N = 9; +RU N = 10. § p < 0.05 vs SEA
Discussion
The main findings of this study are that during ISC: (1) I NaL was increased in spite of AP changes; (2) I NaL blockade reduced Nacyt accumulation, but failed to affect Cacyt accumulation unless sNCX was blocked; (3) sNCX contributed to Cacyt clearance (as opposed to accumulation) throughout ISC; (4) blockade of I NaL and mNCX exerted similar effects on ISC-induced Cacyt accumulation, at least under conditions of substantial Ca2+ overload.
Relevance of ISC as a model of acute myocardial ischemia
Tissue response to acute ischemia is highly dynamic and closely dependent on a number of conditions; thus, any experimental model of acute ischemia is necessarily specific and unlikely to be of general applicability. Furthermore, an isolated myocyte, oxygenated through aqueous superfusion (low O2 solubility) and contracting without external load, cannot be strictly compared to in vivo ischemia. Nevertheless, information of general relevance on the mechanisms that can contribute to ischemic damage, can still be acquired by observing the response to conditions known to occur during it. The ischemic condition adopted in this study (ISC), although encompassing the major factors present in tissue ischemia, differs from it for the absence of hypoxia. Although hypoxia was shown to have little role in the contractile pattern during ISC application [25], it might affect the mechanisms by which such a pattern is achieved in a given time-frame. For instance, hypoxia would likely accelerate ATP decay and reactive oxide species (ROS) production, both factors known to accelerate Nacyt accumulation and facilitate reversal of sNCX transport. Therefore, failure of sNCX to switch to the reverse mode, and the modest effect of I NaL blockade, might be model-specific. However, the contribution of sNCX-independent Ca2+ sources (including mitochondria) to Cacyt accumulation in the presence of factors certainly present during real ischemia, may have general relevance. A further factor to be considered is that, whereas generated within the myocyte under true ischemia, lactic acid was applied extracellularly. This might reduce NHE contribution to Na+ loading, which was nonetheless substantial (Supplemental Figure S4).
To mimic what is reported to occur during ischemia, the ISC solution was slightly hyperosmolar [25]. The possibility that this accounted for the observed changes in the intracellular milieu was ruled out in preliminary experiments (Supplemental Figure S9).
Because of the above features, ISC reproduces conditions closer to those of a “border zone”, not directly ischemic (still energetically competent) but exposed to factors released by the neighboring ischemic area [11].
ISC-induced I NaL enhancement
A link between I NaL enhancement and ischemia/reperfusion injury has been firmly established by previous studies [1, 4, 7, 39, 50]. However, considering the opposing effect of ISC-induced membrane potential changes, I NaL enhancement by ISC was far from predictable.
Contribution of I NaL and Na+/H+ exchanger to cytosolic Na+ accumulation
About 50% of ISC-induced Nacyt accumulation was similarly prevented by RAN and TTX. Being shared by both agents, this effect is likely to result from I NaL blockade. When NHE was also blocked (Supplemental Figure S4), ISC-induced Nacyt accumulation was completely abolished; this suggests that Na+ influx via NHE accounted for the remaining 50% (this quantitative estimate does not take into account potential interactions between the two transports). Although the presence in ISC of lactic acid likely afforded relatively fast H+ equilibration across the membrane, acidosis was primarily extracellular in the present setting; this might explain the initial dip in Nacyt time course (Fig. 3). The present findings suggest that, under the present experimental conditions, NHE was still active during ISC. The monotonic increase in Nacyt during exposure to ouabain (Supplemental Figure S5) indicates that I NaL- and NHE-mediated Na+ influx were in balance with Na+ extrusion through the Na+/K+ pump, which remained active throughout the ISC period and was responsible for the late decay in Nacyt.
I NaL contribution to cytosolic Ca2+ accumulation
In spite of its remarkable effect on Nacyt, I NaL blockade unexpectedly failed to affect ISC-induced Cacyt accumulation (Fig. 4). This might simply reflect inadequacy of the I NaL-dependent Nacyt perturbation in overriding Cacyt homeostatic control; indeed, I NaL blockade tended to reduce CaSR, potentially revealing a role for SR in buffering I NaL-induced perturbation. However, the observation that the effect of I NaL blockade was unmasked by sNCX blockade implies that a Ca2+ source independent of sNCX, and at least partially sensitive to I NaL blockade (or Nacyt), must have contributed to ISC-induced Cacyt accumulation.
sNCX is often claimed to work in reverse mode during ischemia [43, 48], thereby providing a direct path for Ca2+ influx. This was clearly not the case in the present setting; however, sNCX mode may depend on the duration and extent of ischemia. Nevertheless, changes in Nacyt compatible with forward sNCX operation have been reported after sNCX knock-out in intact murine hearts subjected to no-flow ischemia (Fig. 5 in Ref. [18]).
Mitochondrial contribution to cytosolic Ca2+ accumulation
Mitochondria represent a significant Ca2+ compartment, physiologically uptaking Ca2+ through MCU [21] and extruding it to cytosol through mNCX, a Nacyt-sensitive transport [6, 27].
CGP effect in the absence of SEA suggests that, under basal conditions, mNCX blockade may promote a shift of Ca2+ from mitochondria to the SR. This implies that, during ISC, mitochondria contribute to buffer Cacyt through mNCX-mediated Ca2+ uptake. In the present setting, the impact of I NaL blockade on mitochondrial buffering was probably small enough not to affect Cacyt.
On the other hand, when sNCX was blocked, CGP reduced ISC-induced Cacyt accumulation, thus supporting the view that sizable mNCX-mediated Ca2+ efflux from mitochondria may occur during ISC in the presence of substantial Ca2+ overload [6, 37, 44]. Because mNCX flux is Nacyt-dependent, this might account for the I NaL-sensitive component of Cacyt accumulation observed under sNCX blockade.
At the conditions used in the present experiments, RU is a selective blocker of MCU, without effect on I CaL or SR Ca2+ uptake/release [29, 36]. Functional exclusion of the mitochondrial compartment by MCU blockade caused a shift of Ca2+ to the SR, reduced Cacyt accumulation and abolished the effect of mNCX blockade. Concomitance of reduced Cacyt with increased CaSR is consistent with micro-domain communication between mitochondria and SR [22].
To summarize, sNCX blockade seemingly changed the role of mitochondria during ISC from Ca2+ sink to Ca2+ source; the simplest way to explain this effect is the rather dramatic increase in overall cell Ca2+ content present in this condition, possibly reducing mitochondrial and SR Ca2+ buffering reserves. We surmise that such a Ca2+ overload might be achieved, even in the absence of sNCX blockade, during in vivo cardiac ischemia. Therefore, the specific effect of mNCX inhibition might depend on the duration and extent of ischemia; nevertheless, the contribution of mitochondria as a further Nacyt-sensitive compartment contributing to Cacyt changes may be regarded as an observation of general value.
Additional potential sources of cytosolic Ca2+ accumulation
As a Nacyt-sensitive Ca2+ compartment, mitochondria are of particular relevance to changes caused by I NaL enhancement. Nonetheless, they are unlikely to fully account for the large source of Cacyt required to support forward sNCX operation during ISC, in spite of the attending increase in Nacyt and membrane depolarization (both favoring sNCX reversal).
Because voltage-gated Ca2+ channels are potently inhibited by acidosis [20, 38] ICaL is unlikely to be enhanced during ISC; however, a H+-gated background Ca2+ conductance (TRPA1) [17] is expressed in the heart and shown to contribute to ischemia/reperfusion damage [33].
Protons compete with Ca2+ for binding to intracellular buffers, troponin C in particular [15, 41]. In the present setting, this is suggested by the virtual absence of contraction during early ISC, occurring in spite of persisting Ca2+ transients. Therefore, acidosis might support substantial release of free Ca2+ to the cytosol through a mechanism independent of transmembrane fluxes. Because sarcolemmal Na+ gradient is crucial for intracellular H+ clearance through NHE, this Ca2+ source may also be modulated, albeit indirectly, by I NaL blockade.
Discrepancy between TTX and RAN effects
RAN and TTX shared the majority of effects during ISC exposure, supporting their origin from I NaL inhibition. However, unlike TTX, RAN reduced CaD during ISC under baseline condition and limited CaSR increment during SEA exposure. This points to modulation by RAN of a Ca2+ compartment insensitive to TTX. RAN has been shown to stabilize membrane potential of mitochondria during ischemia [1, 13, 14, 51], which would enhance their ability to retain Ca2+. However, this has been attributed to limitation of Nacyt accumulation, an effect that should be shared by TTX. The possibility that RAN may affect mitochondrial performance as a Ca2+ compartment also independently of I NaL blockade may deserve further investigation.
Conclusions
Some of the observed effects of ISC may be model-specific (i.e., depend on the duration and extent of the ischemic condition) and, as such, of restricted applicability. These may include poor sensitivity of Cacyt to I NaL blockade and persistence of forward sNCX operation. Nevertheless, other observations lead to conclusions likely of more general relevance: (1) I NaL can be enhanced during acute ischemia, irrespective of membrane potential changes, and significantly contribute to Nacyt accumulation; (2) Ca2+ sources other than sNCX substantially contribute to Cacyt increment and, at least in the early phase of acute ischemia, may oppose reversal of sNCX flux; (3) under conditions of Ca2+ overload, mitochondria may act as a Nacyt-sensitive Cacyt source, thus providing a mechanism, beyond sNCX modulation, to account for I NaL-induced perturbation of intracellular milieu. A further conclusion is that most, but not all, RAN effects on intracellular milieu may result from I NaL blockade.
References
Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF (2011) Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res 64:381–392. doi:10.1016/j.phrs.2011.06.018
Alemanni M, Rocchetti M, Re D, Zaza A (2011) Role and mechanism of subcellular Ca2+ distribution in the action of two inotropic agents with different toxicity. J Mol Cell Cardiol 50:910–918. doi:10.1016/j.yjmcc.2011.02.008
Allen DG, Xiao XH (2003) Role of the cardiac Na+/H+ exchanger during ischemia and reperfusion. Cardiovasc Res 57:934–941. doi:10.1016/s0008-6363(02)00836-2
Belardinelli L, Shryock JC, Fraser H (2006) Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92(Suppl 4):iv6–iv14. doi:10.1136/hrt.2005.078790
Bernink FJ, Timmers L, Beek AM, Diamant M, Roos ST, van Rossum AC, Appelman Y (2014) Progression in attenuating myocardial reperfusion injury: an overview. Int J Cardiol 170:261–269. doi:10.1016/j.ijcard.2013.11.007
Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ (2013) NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol 59:205–213. doi:10.1016/j.yjmcc.2013.03.012
Calderon-Sanchez EM, Dominguez-Rodriguez A, Lopez-Haldon J, Jimenez-Navarro MF, Gomez AM, Smani T, Ordonez A (2016) Cardioprotective effect of ranolazine in the process of ischemia-reperfusion in adult rat cardiomyocytes. Rev Esp Cardiol (Engl Ed) 69:45–53. doi:10.1016/j.rec.2015.02.027
Caldwell JC, Burton FL, Cobbe SM, Smith GL (2012) Amplitude changes during ventricular fibrillation: a mechanistic insight. Front Physiol 3(147):1–8. doi:10.3389/fphys.2012.00147
Chen S, Li S (2012) The Na+/Ca(2)+ exchanger in cardiac ischemia/reperfusion injury. Med Sci Monit 18:RA161–RA165. doi:10.12659/MSM.883533
Cordeiro JM, Howlett SE, Ferrier GR (1994) Simulated ischaemia and reperfusion in isolated guinea pig ventricular myocytes. Cardiovasc Res 28:1794–1802. doi:10.1093/cvr/28.12.1794
Coronel R, Wilms-Schopman FJ, Fiolet JW, Opthof T, Janse MJ (1995) The relation between extracellular potassium concentration and pH in the border zone during regional ischemia in isolated porcine hearts. J Mol Cell Cardiol 27:2069–2073. doi:10.1016/0022-2828(95)90028-4
Cox DA, Conforti L, Sperelakis N, Matlib MA (1993) Selectivity of inhibition of Na(+)–Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol 21:595–599. doi:10.1097/00005344-199304000-00013
Dehina L, Descotes J, Chevalier P, Bui-Xuan B, Romestaing C, Dizerens N, Mamou Z, Timour Q (2014) Protective effects of ranolazine and propranolol, alone or combined, on the structural and functional alterations of cardiomyocyte mitochondria in a pig model of ischemia/reperfusion. Fundam Clin Pharmacol 28:257–267. doi:10.1111/fcp.12033
Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AK (2012) Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim Biophys Acta 1817:419–429. doi:10.1016/j.bbabio.2011.11.021
Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD (2013) H(+)-activated Na(+) influx in the ventricular myocyte couples Ca(2)(+)-signalling to intracellular pH. J Mol Cell Cardiol 61:51–59. doi:10.1016/j.yjmcc.2013.04.008
Hale SL, Leeka JA, Kloner RA (2006) Improved left ventricular function and reduced necrosis after myocardial ischemia/reperfusion in rabbits treated with ranolazine, an inhibitor of the late sodium channel. J Pharmacol Exp Ther 318:418–423. doi:10.1124/jpet.106.103242
Hamilton NB, Kolodziejczyk K, Kougioumtzidou E, Attwell D (2016) Proton-gated Ca(2+)-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 529:523–527. doi:10.1038/nature16519
Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E (2005) Cardiac-specific ablation of the Na+–Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res 97:916–921. doi:10.1161/01.RES.0000187456.06162.cb
Jung IS, Lee SH, Yang MK, Park JW, Yi KY, Yoo SE, Kwon SH, Chung HJ, Choi WS, Shin HS (2010) Cardioprotective effects of the novel Na+/H+ exchanger-1 inhibitor KR-32560 in a perfused rat heart model of global ischemia and reperfusion: involvement of the Akt-GSK-3beta cell survival pathway and antioxidant enzyme. Arch Pharm Res 33:1241–1251. doi:10.1007/s12272-010-0815-z
Kaibara M, Kameyama M (1988) Inhibition of the calcium channel by intracellular protons in single ventricular myocytes of the guinea-pig. J Physiol 403:621–640. doi:10.1113/jphysiol.1988.sp017268
Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427:360–364. doi:10.1038/nature02246
Kohlhaas M, Maack C (2013) Calcium release microdomains and mitochondria. Cardiovasc Res 98:259–268. doi:10.1093/cvr/cvt032
Lazdunski M, Frelin C, Vigne P (1985) The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. J Mol Cell Cardiol 17:1029–1042. doi:10.1016/S0022-2828(85)80119-X
Linz WJ, Busch AE (2003) NHE-1 inhibition: from protection during acute ischaemia/reperfusion to prevention/reversal of myocardial remodelling. Naunyn Schmiedebergs Arch Pharmacol 368:239–246. doi:10.1007/s00210-003-0808-2
Lu J, Zang WJ, Yu XJ, Chen LN, Zhang CH, Jia B (2005) Effects of ischaemia-mimetic factors on isolated rat ventricular myocytes. Exp Physiol 90:497–505. doi:10.1113/expphysiol.2004.029421
Ma J, Song Y, Shryock JC, Hu L, Wang W, Yan X, Zhang P, Belardinelli L (2014) Ranolazine attenuates hypoxia- and hydrogen peroxide-induced increases in sodium channel late openings in ventricular myocytes. J Cardiovasc Pharmacol 64:60–68. doi:10.1097/FJC.0000000000000090
Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B (2006) Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 99:172–182. doi:10.1161/01.RES.0000232546.92777.05
MacDonald AC, Howlett SE (2008) Differential effects of the sodium calcium exchange inhibitor, KB-R7943, on ischemia and reperfusion injury in isolated guinea pig ventricular myocytes. Eur J Pharmacol 580:214–223. doi:10.1016/j.ejphar.2007.10.055
Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM (1998) Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem 273:10223–10231. doi:10.1074/jbc.273.17.10223
Nakamura T, Hayashi H, Satoh H, Katoh H, Kaneko M, Terada H (1999) A single cell model of myocardial reperfusion injury: changes in intracellular Na+ and Ca2+ concentrations in guinea pig ventricular myocytes. Mol Cell Biochem 194:147–157. doi:10.1023/A:1006919929104
Namekata I, Shimada H, Kawanishi T, Tanaka H, Shigenobu K (2006) Reduction by SEA0400 of myocardial ischemia-induced cytoplasmic and mitochondrial Ca2+ overload. Eur J Pharmacol 543:108–115. doi:10.1016/j.ejphar.2006.06.012
Park CO, Xiao XH, Allen DG (1999) Changes in intracellular Na+ and pH in rat heart during ischemia: role of Na+/H+ exchanger. Am J Physiol 276:H1581–H1590
Robertson S, Thomson AL, Carter R, Stott HR, Shaw CA, Hadoke PW, Newby DE, Miller MR, Gray GA (2014) Pulmonary diesel particulate increases susceptibility to myocardial ischemia/reperfusion injury via activation of sensory TRPV1 and beta1 adrenoreceptors. Part Fibre Toxicol 11:12. doi:10.1186/1743-8977-11-12
Rocchetti M, Sala L, Rizzetto R, Staszewsky LI, Alemanni M, Zambelli V, Russo I, Barile L, Cornaghi L, Altomare C, Ronchi C, Mostacciuolo G, Lucchetti J, Gobbi M, Latini R, Zaza A (2014) Ranolazine prevents INaL enhancement and blunts myocardial remodelling in a model of pulmonary hypertension. Cardiovasc Res 104:37–48. doi:10.1093/cvr/cvu188
Salameh A, Dhein S, Beuckelmann DJ (2002) Role of the cardiac Na(+)/H(+)exchanger in [Ca(2+)](i)and [Na(+)](i)handling during intracellular acidosis. Effect of cariporide (Hoe 642). Pharmacol Res 45:35–41. doi:10.1006/phrs.2001.0908
Sanchez JA, Garcia MC, Sharma VK, Young KC, Matlib MA, Sheu SS (2001) Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J Physiol 536:387–396. doi:10.1111/j.1469-7793.2001.0387c.xd
Saotome M, Katoh H, Satoh H, Nagasaka S, Yoshihara S, Terada H, Hayashi H (2005) Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 288:H1820–H1828. doi:10.1152/ajpheart.00589.2004
Sato R, Noma A, Kurachi Y, Irisawa H (1985) Effects of intracellular acidification on membrane currents in ventricular cells of the guinea pig. Circ Res 57:553–561. doi:10.1161/01.res.57.4.553
Soliman D, Wang L, Hamming KS, Yang W, Fatehi M, Carter CC, Clanachan AS, Light PE (2012) Late sodium current inhibition alone with ranolazine is sufficient to reduce ischemia- and cardiac glycoside-induced calcium overload and contractile dysfunction mediated by reverse-mode sodium/calcium exchange. J Pharmacol Exp Ther 343:325–332. doi:10.1124/jpet.112.196949
Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L (2006) Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther 318:214–222. doi:10.1124/jpet.106.101832
Swietach P, Youm JB, Saegusa N, Leem CH, Spitzer KW, Vaughan-Jones RD (2013) Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci USA 110:E2064–E2073. doi:10.1073/pnas.1222433110
Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, Shigenobu K (2002) Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol 135:1096–1100. doi:10.1038/sj.bjp.0704574
Tang Q, Ma J, Zhang P, Wan W, Kong L, Wu L (2012) Persistent sodium current and Na+/H+ exchange contributes to the augmentation of the reverse Na+/Ca2+ exchange during hypoxia or acute ischemia in ventricular myocytes. Pflugers Arch 463:513–522. doi:10.1007/s00424-011-1070-y
Tanonaka K, Motegi K, Arino T, Marunouchi T, Takagi N, Takeo S (2012) Possible pathway of Na(+) flux into mitochondria in ischemic heart. Biol Pharm Bull 35:1661–1668. doi:10.1248/bpb.b12-00010
Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN (2006) Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol 17(Suppl 1):S169–S177. doi:10.1111/j.1540-8167.2006.00401.x
Undrovinas AI, Fleidervish IA, Makielski JC (1992) Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res 71:1231–1241. doi:10.1161/01.res.71.5.1231
van Borren MM, Baartscheer A, Wilders R, Ravesloot JH (2004) NHE-1 and NBC during pseudo-ischemia/reperfusion in rabbit ventricular myocytes. J Mol Cell Cardiol 37:567–577. doi:10.1016/j.yjmcc.2004.05.017
Wang XJ, Wang LL, Fu C, Zhang PH, Wu Y, Ma JH (2014) Ranolazine attenuates the enhanced reverse Na(+)-Ca(2)(+) exchange current via inhibiting hypoxia-increased late sodium current in ventricular myocytes. J Pharmacol Sci 124:365–373. doi:10.1254/jphs.13202FP
Wilde AA, Escande D, Schumacher CA, Thuringer D, Mestre M, Fiolet JW, Janse MJ (1990) Potassium accumulation in the globally ischemic mammalian heart. A role for the ATP-sensitive potassium channel. Circ Res 67:835–843. doi:10.1161/01.res.67.4.835
Williams IA, Xiao XH, Ju YK, Allen DG (2007) The rise of [Na(+)] (i) during ischemia and reperfusion in the rat heart-underlying mechanisms. Pflugers Arch 454:903–912. doi:10.1007/s00424-007-0241-3
Zaza A, Rocchetti M (2013) The late Na+ current—origin and pathophysiological relevance. Cardiovasc Drugs Ther 27:61–68. doi:10.1007/s10557-012-6430-0
Acknowledgements
This research was supported by grant from Gilead Inc. (Fremont, CA, US) and FAR 2015 to A. Zaza. We are grateful to Dr. Luiz Belardinelli for providing stimulating discussion throughout the execution of the study and Dr. Luca Sala for contributing to some experiments and providing insightful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experiment were approved and conducted in accordance with guidelines issued by the Animal Care Committee of the University Milano-Bicocca, in compliance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. The manuscript does not contain human data.
Conflict of interest
The study has been partially funded by Gilead, Inc. (Fremont, CA), which is the patent holder for Ranolazine. The authors declare that they have no further conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ronchi, C., Torre, E., Rizzetto, R. et al. Late sodium current and intracellular ionic homeostasis in acute ischemia. Basic Res Cardiol 112, 12 (2017). https://doi.org/10.1007/s00395-017-0602-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-017-0602-9