
Abstract
Purpose
Several clinical studies suggested that light-to-moderate alcohol intake could alleviate nonalcoholic fatty liver disease (NAFLD), but the underlying mechanism is still poorly understood.
Methods
Mice fed a high-fat diet (HFD) were submitted or not to moderate ethanol intake for 3 months (ca. 10 g/kg/day) via drinking water. Biochemical, analytical and transcriptomic analyses were performed in serum and liver.
Results
Serum ethanol concentrations in ethanol-treated HFD mice comprised between 0.5 and 0.7 g/l throughout the experiment. NAFLD improvement was observed in ethanol-treated HFD mice as assessed by reduced serum transaminase activity. This was associated with less microvesicular and more macrovacuolar steatosis, the absence of apoptotic hepatocytes and a trend towards less fibrosis. Liver lipid analysis showed increased amounts of fatty acids incorporated in triglycerides and phospholipids, reduced proportion of palmitic acid in total lipids and higher desaturation index, thus suggesting enhanced stearoyl-coenzyme A desaturase activity. mRNA expression of several glycolytic and lipogenic enzymes was upregulated. Genome-wide expression profiling and gene set enrichment analysis revealed an overall downregulation of the expression of genes involved in collagen fibril organization and leukocyte chemotaxis and an overall upregulation of the expression of genes involved in oxidative phosphorylation and mitochondrial respiratory chain complex assembly. In addition, mRNA expression of several proteasome subunits was upregulated in ethanol-treated HFD mice.
Conclusions
Moderate chronic ethanol consumption may alleviate NAFLD by several mechanisms including the generation of non-toxic lipid species, reduced expression of profibrotic and proinflammatory genes, restoration of mitochondrial function and possible stimulation of proteasome activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol abuse is associated with a large array of ailments including liver diseases, psychiatric disorders, cardiomyopathy and pancreatitis [1, 2]. Regarding the liver, sustained high alcohol consumption (> 20 and 30 g/day of alcohol for women and men, respectively) almost always leads to steatosis, which can then progress in some patients to steatohepatitis, cirrhosis and hepatocellular carcinoma [2, 3]. Importantly, a recent systematic analysis of data providing from 195 different countries and territories indicated that alcohol drinking, regardless of amount, actually increases the overall risk to health across population [4]. However, the latter study and others found some protective effects of light-to-moderate alcohol consumption (i.e. < 20 and 30 g/day of alcohol for women and men, respectively) on ischemic heart disease and diabetes [4,5,6]. Moreover, several clinical studies suggested that light-to-moderate alcohol intake could have beneficial effects on nonalcoholic fatty liver disease (NAFLD) [7,8,9,10,11,12], which is commonly associated with obesity. For instance, light-to-moderate alcohol consumption has been reported to reduce the degree of fatty liver [10], the levels of serum transaminases [11, 12] and the risk of developing nonalcoholic steatohepatitis (NASH) [7, 8, 11, 12], which is characterized by the presence of necroinflammation and fibrosis, in addition to fatty liver. However, as pointed out by some authors, clinical data reporting these beneficial effects on NAFLD should be interpreted with caution because of the existence of several potential sources of bias, confounding factors and errors [7, 11]. Moreover, other studies reported no beneficial effects of moderate alcohol drinking on NAFLD [12,13,14]. For example, a 47-month longitudinal cohort study reported that modest alcohol use was associated with less improvement in steatosis and blood level of aspartate aminotransferase (AST), compared with no alcohol consumption [13], while a Mendelian randomization study showed that moderate alcohol consumption had no beneficial effect on hepatic steatosis and necroinflammatory activity [14].
Only 4 experimental studies carried out in rodents previously reported the beneficial effects of light-to-moderate intake of alcohol on NAFLD [15,16,17,18]. In 2009, we first reported that moderate ethanol consumption for 6 months reduced the gain of body weight, liver triglycerides and diabetes in leptin-deficient obese and diabetic ob/ob mice [15]. However, these beneficial effects were not associated with reduced levels of plasma transaminases. A recent study also carried out in ob/ob mice reported that moderate alcohol intake for 6 weeks decreased plasma levels of alanine aminotransferase (ALT) and AST, liver triglycerides and hepatic inflammation [16]. Investigations performed in rats fed a high-fat diet (HFD) reported that low ethanol consumption for 12 weeks lessened serum levels of ALT and lactate dehydrogenase (LDH), whereas liver triglycerides were unchanged [17]. Finally, investigations in rats fed a high-fat high-cholesterol diet showed that a moderate ethanol consumption for 8 weeks alleviated liver fibrosis and reduced the mRNA expression of several profibrotic genes [18]. Notably, the beneficial effects of moderate ethanol exposure in rodents are not restricted to the liver and can also be observed in other organs and tissues such as heart, colon, white adipose tissue and blood vessels [19,20,21]. Lower oxidative stress and inflammation, as well as improvement of lipid metabolism could be involved in these favorable effects [19,20,21].
The mechanisms whereby light-to-moderate alcohol consumption could be beneficial on NAFLD are poorly understood. Hence, the aim of our study was to improve our knowledge regarding these mechanisms by carrying out investigations in HFD obese mice submitted or not to moderate ethanol intake (ca. 10 g/kg/day) for 3 months via drinking water. In particular, we performed a thorough histological examination of livers associated for the first time with hepatic fatty acid analysis and unsupervised genome-wide expression profiling.
Materials and methods
Animals and ethanol administration
Thirty 7-week-old male C57BL/6J-+/+ mice, weighing 20–23 g, were purchased from Janvier (Le-Genest-St-Isle, France). After arrival, the mice were acclimatized in the animal care facility of the University of Rennes 1 (ARCHE), accredited by the French veterinary authorities (agreement n° A35 23840) and fed ad libitum on a standard diet (SD) bringing 3 kcal/g of food (Teklad global 16% protein rodent diet, ENVIGO, Huntingdon, United Kingdom). After 1 week of acclimatization, mice were split into three groups of 10 animals. While one group was kept on the SD, the two others were fed a 60% high-fat diet (HFD) bringing 5.5 kcal/g (260HF V19, Safe Diet, Augy, France). In this diet, 60% of the energy came from lipids, mainly anhydrous milk fat with 233 g/kg of saturated fatty acids (SFAs). The main nutrient composition of these diets is given in Supplementary Table 1. Details regarding the protocol of ethanol exposure (ca. 10 g/kg/day corresponding to 15.9% of calories from alcohol) and blood withdrawal are provided in Supplementary methods 1.
Serum analyses
Immediately after collection, blood was centrifuged for 10 min at 1000g and serum was immediately used for most biochemical analyses, or stored at − 80 °C until use for insulin and adiponectin measurement. Activity of ALT and AST, total cholesterol, triglycerides and glucose levels were measured on an automatic analyzer (Cobas 8000, Roche Diagnostics, Mannheim, Germany) using the appropriate reagents purchased from Roche Diagnostics. Total antioxidant capacity was assessed using the Biovision TAC Colorimetric Assay Kit (K274-100, Milpitas, CA). Ethanol levels were measured using the Ethanol assay kit purchased from Abcam (Paris, France). Insulin and adiponectin were respectively determined using the Ultra-Sensitive Insulin Mouse ELISA Kit and the Adiponectin Mouse ELISA kit purchased from Crystal Chem (Downers Grove, IL).
Liver histology and fibrosis quantification
To evaluate steatosis, necroinflammation, apoptosis and fibrosis, liver fragments were fixed in 4% neutral formalin and embedded in paraffin. Then, 4 µm thick sections were cut and stained with hematoxylin–eosin–saffron (HES) or Sirius red. The sections were then digitally scanned with the NanoZoomer 2.0 RS (Hamamatsu Corp. Japan) and thoroughly examined by an experienced pathologist (V.T.-S.). The degree of hepatic steatosis was evaluated in ten different randomly chosen fields (200×) as the percentage of hepatocytes containing one or several lipid vacuoles. The classification of steatosis into three different categories (i.e. microvesicular, mediovesicular and macrovacuolar) was determined as previously described [22, 23]. Necroinflammation, portal fibrosis and perisinusoidal fibrosis were evaluated on the entire slide and scored 0 (absent), 1 (mild) or 2 (moderate), as previously reported [23]. Finally, apoptosis was scored 0 (absent) or 1 (present) depending on the presence of apoptotic bodies in the hepatic parenchyma.
To have more information regarding fibrosis, deposition of fibrillary collagen was assessed using second-harmonic generation (SHG) microscopy available at the light microscopy-imaging center (MRic facility of Biosit, University of Rennes 1), as previously described [24, 25]. More details on the SHG methodology used in our study are provided in Supplementary methods 2.
Activity of respiratory chain complexes I and II in liver
For protein extraction, liver samples were rinsed in phosphate-buffered saline (PBS) and then lysed using a Dounce homogenizer with the extraction buffer supplied by Abcam, as recommended by the manufacturer. Mitochondrial respiratory chain (MRC) complex I activity was measured with 5 µg of liver proteins using the Complex I Enzyme Activity Dipstick Assay kit from Abcam (Paris, France), as recently described [26, 27]. Activity of the MRC complex II, also referred to as succinate dehydrogenase (SDH), was measured with 100 µg of liver proteins using the Complex II Enzyme Activity Microplate Assay kit from Abcam, as recently reported [26, 27].
Hepatic glutathione, carbonylated proteins, lipid peroxidation and CYP2E1 protein expression
Reduced (GSH) and oxidized (GSSG) glutathione were measured in liver tissue extracts using the Glutathione Assay Kit purchased from Cayman Chemicals (Montigny-le-Bretonneux, France). Hepatic carbonylated proteins were also assessed as valuable markers of oxidative stress [28]. Protein carbonyl quantification was carried out using the Protein Carbonyl Elisa Kit purchased from Enzo Life Sciences (Villeurbanne, France). Lipid peroxidation was assessed using the Lipid Peroxidation (MDA) Colorimetric/Fluorometric Assay Kit (K739) from BioVision. Protein expression of hepatic cytochrome P450 2E1 (CYP2E1) and heat shock cognate 70 (HSC70) (used as loading control) was assessed by western blot analysis as previously described [15], using respectively the antibodies from Oxford Biomedical Research (Oxford, MI) and Santa Cruz Biotechnology (Dallas, TX). The dilution used for the antibodies was 1:1000 for CYP2E1 and 1:500 for HSC70.
RNA extraction and transcriptomic analysis
For our transcriptomic analysis, 5 HFD mice and 5 ethanol-treated HFD mice were randomly chosen in each group of mice. Total RNA was extracted from 10 to 15 mg of mouse liver with the RNeasy Mini Kit (Qiagen, Courtaboeuf, France). Genome-wide expression profiling was then performed using the low-input QuickAmp labeling kit and SurePrint G3 Mouse GE v2 8 × 60K microarrays purchased from Agilent Technologies (Santa Clara, CA), as previously described [29]. Further information on transcriptomic analysis and gene set enrichment analysis (GSEA) is provided in Supplementary methods 3.
Hepatic lipid extraction and fatty acid analysis
For hepatic fatty acid analysis, all HFD and ethanol-treated HFD mice (n = 10 per group) were investigated. Hepatic lipid extraction and fatty acid analysis were performed as previously described [30,31,32]. Further information regarding the experimental procedures is provided in Supplementary methods 4.
Statistical analysis
All results are expressed as mean ± SEM (standard error of mean). Comparisons between groups were performed using one-way analysis of variance (ANOVA) with a post hoc Newman–Keuls test, two-way ANOVA with a post hoc Bonferroni test, Student t test or nonparametric Mann–Whitney U test, as appropriate. All statistical analyses and graphics were performed using GraphPad Prism5 software (GraphPad Software, San Diego, CA, USA).
Results
Body weight, food consumption and energy intake
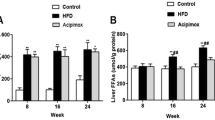
As expected, mice fed the HFD (henceforth referred to as HFD mice) gained more body weight compared with mice fed the SD (SD mice) and the body weight gain was significant from the second week of HFD (Fig. 1a). However, HFD mice consuming ethanol gained significantly less body weight during the last month compared with naive HFD mice (Fig. 1a). This difference was apparently not related to lower food consumption in ethanol-treated obese mice in this period of time (Fig. 1b). HFD induced a transient increase in food consumption (Fig. 1b), as already observed in different rodent models of diet-induced obesity (DIO) [33, 34]. Caloric intake was lower in SD mice compared with HFD mice and within the latter group, ethanol-treated HFD mice consumed overall more energy than HFD mice (Fig. 1c), due to the calories provided by ethanol (7 kcal/g).

Body weight, body weight gain, food consumption and energy intake in mice fed the standard diet (SD), mice fed the high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). a Body weight (mean ± SEM for 9–10 mice per group) was measured every week or 2 weeks (left y axis) and the body weight gain (right y axis) was subsequently calculated. $Significantly different between HFD and SD mice, *significantly different between HFD + E and naive HFD mice, p < 0.01 with a two-way ANOVA test. b Food consumption (mean ± SEM for 9–10 mice per group) was measured every week. $Significantly different between HFD and SD mice, p < 0.05 with a two-way ANOVA test. c Energy intake (kcal/mouse/day) corresponding to food and ethanol (if any) consumption in the different groups of animals (mean ± SEM for 9–10 mice per group). $Significantly different between HFD and SD mice, ¶Significantly different between HFD and SD mice, †Significantly different between HFD + E and SD mice, p < 0.05 with a two-way ANOVA test
Serum parameters and liver weight
Different serum parameters were serially measured during the 4-month experiment (Fig. 2a). Serum ethanol concentrations in ethanol-treated HFD mice were comprised between 0.5 and 0.7 g/l (ca. 11 and 15 mM) throughout the experiment. However, it should be underlined that ethanol concentrations were measured in the morning after an overnight fast. Hence, it cannot be excluded that these concentrations might have been higher if measured in the fed state and during the dark cycle when the highest ethanol consumption usually occurs in mice [35, 36]. Serum glucose levels were increased in naive and ethanol-treated HFD mice during the course of the experiment although hyperglycemia was reduced in ethanol-treated HFD mice after 3 months but not afterwards. Serum total cholesterol was increased in naive and ethanol-treated HFD mice throughout the experiment but hypercholesterolemia was lower in ethanol-treated HFD mice after 4 months. Serum triglyceride levels were enhanced in ethanol-treated HFD mice during the first 3 months but returned to normal levels after 4 months. Although serum levels of triglycerides were enhanced in ethanol-treated HFD mice compared with HFD mice at months 2 and 3, it seemed unlikely that this unfavorable metabolic profile might be due to the higher energy intake brought only by ethanol during the overnight fast. Indeed, this higher caloric intake would also have induced higher serum triglycerides at month 4 but also unfavorable profiles on serum glucose and total cholesterol throughout the experiment. Serum ALT activity was higher in naive and ethanol-treated HFD mice during the experiment but a significant reduction of this activity was observed in ethanol-treated HFD mice after 3 and 4 months. At these periods of time, AST activity was significantly increased in naive HFD mice, but not in ethanol-treated HFD mice, compared with SD mice. There was no modification of the serum total antioxidant capacity whatever the groups of mice and the timing of measurement (Supplementary Fig. 2). At the end of the 4-month experiment, serum insulin levels and liver weight were enhanced in HFD mice compared with the SD mice (Fig. 2b, c). However, there was no difference between naive and ethanol-treated HFD mice (Fig. 2b, c). Interestingly, a trend (p = 0.055) toward higher serum adiponectin levels was found in HFD mice treated with ethanol compared to the other groups of mice (Fig. 2b).

a Serum parameters serially measured during the 4-month experiment in mice fed the standard diet (SD), mice fed the high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). Serum ethanol levels were measured only in the 10 ethanol-treated HFD mice. Values are mean ± SEM for 8–10 mice per group for glucose, triglycerides and cholesterol and 7–10 mice per group for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities. $Significantly different from SD mice, *significantly different from naive HFD mice, p < 0.05 with a one-way ANOVA test. b Serum insulin and adiponectin (mean ± SEM for 7–10 mice per group) measured at the end of the 4-month experiment. $Significantly different from SD mice, p < 0.001 with a one-way ANOVA test. c Liver weight (mean ± SEM for 9–10 mice per group) measured at the end of the 4-month experiment. $Significantly different from SD mice, p < 0.001 with a one-way ANOVA test
Liver steatosis, necroinflammation and apoptosis
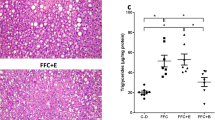
A thorough evaluation of steatosis, necroinflammation, apoptosis and fibrosis was performed at the end of the experiment in 9 SD mice, 10 naive HFD mice and 10 ethanol-treated HFD mice. As expected, steatosis was present in a vast majority of hepatocytes in DIO mice (Fig. 3a). However, the percentage of hepatocytes with steatosis was slightly but significantly reduced in ethanol-treated HFD mice (87%) compared with naive HFD mice (94%) (Fig. 3a). More strikingly, the pattern of steatosis was modified in ethanol-treated HFD mice, which presented significantly less microvesicular steatosis compared to naive HFD mice (Fig. 3b, c). Indeed, the percentage of steatotic hepatocytes presenting microvesicular steatosis was respectively 49 and 23% in naive and ethanol-treated HFD mice (Fig. 3b). On the other hand, ethanol-treated HFD mice presented significantly more mediovesicular and macrovacuolar steatosis compared to naive HFD mice (Fig. 3b, c). Regarding necroinflammation, there was however no difference between naive and ethanol-treated HFD mice. Indeed, in each group, there were 9 mice scored 1 and 1 mouse scored 2 for this liver lesion. The inflammatory infiltrates, which were observed in the portal tracts and the lobules, consisted mainly of lymphocytes and Kupffer cells, with few neutrophils. In contrast, ethanol drinking reduced the apoptosis score. Indeed, there were 4 naive HFD mice presenting apoptotic bodies in the hepatic parenchyma (score 1), whereas this pathological feature was not observed in ethanol-treated HFD mice. In keeping with these results, our microarray data showed a significant (p < 0.05) downregulation of different genes involved in apoptosis in ethanol-treated HFD mice compared with naive HFD mice. This was for instance the case for Card11 (− 43%), Bcl2l14 (− 36%), Aen (− 26%), Tnfsf12 (− 23%), Acin1 (− 22%) and Bid (− 11%). In contrast, there was a significant upregulation of some anti-apoptotic genes such as Faim2 (+ 35%), Aven (+ 19%), Tmbim4 (+ 13%) and Dad1 (+ 11%). However, GSEA did not reveal an overall change in the expression of genes involved in cell death.

Features of hepatic steatosis in mice fed the standard diet (SD), mice fed the high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). a Percentage of hepatocytes presenting steatosis. Values are mean ± SEM for 9–10 mice per group. *Significantly different from naive HFD mice, p < 0.01 with a one-way ANOVA test. b Pattern of steatosis in HFD and HFD + E mice. Percentage of steatotic hepatocytes presenting microvesicular, mediovesicular and macrovacuolar steatosis, respectively. Values are mean ± SEM for 10 mice per group. *Significantly different from naive HFD mice, p < 0.01 with a two-way ANOVA test. c Representative pictures of a naive SD mouse liver with no steatosis, a naive HFD mouse liver with predominant microvesicular steatosis and an ethanol-treated HFD mouse liver with predominant mediovesicular and macrovacuolar steatosis. Sections were stained with HES and pictures were taken at ×100 and ×200 magnification, as indicated. Scale bars (250 μm) are indicated on each picture
Assessment of liver fibrosis by histology and SHG microscopy
Portal and perisinusoidal fibrosis was either absent (score 0) or mild (score 1) in all DIO mice (data not shown). Whereas the mean score of perisinusoidal fibrosis was similar between naive and ethanol-treated HFD mice, that of portal fibrosis tended to be lower in the last group (Fig. 4a). To have a quantitative assessment of fibrosis, liver fibrillar collagen deposition was assessed using SHG microscopy and the percentage of liver area with fibrosis was calculated for each mouse, as previously reported [24, 25]. A trend towards reduced area of fibrosis was observed in ethanol-treated HFD mice compared to naive HFD mice (Fig. 4b, c) but the difference did not reach statistical difference (p = 0.10).

Hepatic fibrosis in mice fed the high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). a Score of perisinusoidal and portal fibrosis. Values are mean ± SEM for 10 mice per group. b Percentage of area of fibrosis assessed by second-harmonic generation (SHG) microscopy. Values are mean ± SEM for 10 mice per group, p = 0.10 with a t test. c Representative pictures of two naive HFD mouse livers and two HFD + E mouse livers with less portal fibrosis. Scale bars (250 μm) are indicated on each picture
Investigations on liver lipids and fatty acids
Liver fatty acid analysis was subsequently carried out to better understand the altered pattern of fatty liver in ethanol-treated HFD mice. Whereas the mass of fatty acids from total lipids was similar between ethanol-treated HFD mice and naive HFD mice, more fatty acids were found in triglycerides and phospholipids (Fig. 5a). The five most abundant hepatic fatty acids in HFD mice were, by decreasing order of magnitude, oleic acid (C18:1n-9; 44.1%), palmitic acid (C16:0; 27.1%), cis-vaccenic acid (C18:1n-7; 6.7%), palmitoleic acid (C16:1n-7; 6.0%) and linoleic acid (C18:2n-6; 4.8%) (Fig. 5b and data not shown). The proportion of the remaining fatty acids was near or below 2% (data not shown). There was a significant decrease in the proportion of palmitic acid and an increase in that of oleic acid in ethanol-treated HFD mice compared with naive HFD mice (Fig. 5b). Overall, the proportion of saturated fatty acids (SFAs) was significantly reduced in ethanol-treated HFD mice, whereas that of monounsaturated fatty acids (MUFAs) was enhanced (Fig. 5b). Moreover, the Δ9-desaturation index, calculated as the ratio (C16:1 n-7 + C18:1n-9)/(C16:0 + C18:0), was significantly enhanced in ethanol-treated HFD mice compared with naive HFD mice (Fig. 5c), suggesting higher hepatic stearoyl-CoA desaturase (SCD1) activity by ethanol intake. However, Scd1 mRNA expression was not increased in ethanol-treated HFD mice (data not shown). Interestingly, our genome-wide expression profiling showed in ethanol-treated HFD mice an upregulation of genes encoding 4 enzymes involved in glycolysis and conversion of pyruvate to acetyl-coenzyme A (Gpi1, Aldob, Pklr, Pdhb), 4 enzymes implicated in fatty acid synthesis and elongation (Acly, Acaca, Me1, Elovl6) and one enzyme participating in triglyceride formation (Lpin1) (Fig. 5d). In addition, we found an increased mRNA expression of thyroid hormone responsive protein (Thrsp, also referred to as Spot14), cell death-inducing DFFA-like effector a (Cidea) and adipogenin (Adig), three proteins involved in lipid synthesis and storage (Fig. 5d).

Analysis of hepatic fatty acids in high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). a Mass of fatty acids from total lipids and in the triglyceride and phospholipid fractions. Values are mean ± SEM for 10 mice per group. *Significantly different from naive HFD mice, p < 0.05 with a t test. b Proportion of palmitic acid (C16:0), oleic acid (C18:1n-9), saturated fatty acids (SFAs) and monounsaturated fatty acids (MUFAs) in total lipids. Values are mean ± SEM for 10 mice per group. *Significantly different from naive HFD mice, p < 0.05 with a t test. c Δ9-Desaturation index calculated as the ratio (C16:1n-7 + C18:1n-9)/(C16:0 + C18:0). Values are mean ± SEM for 10 mice per group. *Significantly different from naive HFD mice, p < 0.05 with a t test. d Hepatic mRNA levels of genes involved in glycolysis and lipogenesis extracted from the transcriptomic analysis (GSE116417). Values are mean ± SEM for 5 mice per group. *Significantly different from naive HFD mice, p < 0.05 with a Mann–Whitney test
Microarray analysis, GSEA and MRC complex activity
Our microarray analysis complemented by GSEA revealed in naive HFD mice a significant enrichment of the GO gene sets corresponding to “collagen fibril organization” and “extracellular structure organization”, in comparison with ethanol-treated HFD mice (Fig. 6a). Accordingly, a marked reduction of Col1a1, Col1a2, Col2a1 and Col3a1 mRNA levels was for instance observed in ethanol-treated HFD mice (Fig. 6a). GSEA also disclosed in naive HFD mice a significant enrichment of the GO gene sets related to “cell chemotaxis” and “leukocyte chemotaxis”, in comparison with ethanol-treated HFD mice with reduced Cx3cl1, Cxcl1, Ccr2 and Cx3cr1 mRNA expression in the latter group of mice (Fig. 6b). In contrast, GSEA showed in ethanol-treated HFD a significant enrichment of the GO gene sets corresponding to “oxidative phosphorylation” (OXPHOS) and “MRC complex assembly”, compared with naive HFD mice (Fig. 7a). For example, increased mRNA levels of Nd1, Nd3, Nd5, Cox5a, Cox7a2, Atp5d, Atp8 and Ndufs4 were observed in ethanol-treated HFD mice (Fig. 7a). Regarding MRC complexes activity, no difference was found between the groups for complex I activity, whereas HFD-induced reduction of complex II activity was significantly restored in ethanol-treated HFD mice (Fig. 7b).

mRNA expression of hepatic genes involved in fibrosis and cell chemotaxis in high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). Genome-wide expression profiling performed in naive (n = 5) and ethanol-treated (n = 5) HFD mice (GSE116417) was used for subsequent gene set enrichment analysis (GSEA). a GSEA revealing in naive HFD mice a significant enrichment of the gene ontology (GO) gene sets corresponding to “collagen fibril organization” and “extracellular structure organization”, in comparison with ethanol-treated HFD mice. The hepatic mRNA expression of four representative genes involved in fibrosis, extracted from the transcriptomic analysis (GSE116417), is shown below the GSEA plots. *Significantly different from naive HFD mice, p < 0.05 with a Mann–Whitney test. b GSEA revealing in naive HFD mice a significant enrichment of the GO gene sets related to “cell chemotaxis” and “leukocyte chemotaxis”, in comparison with ethanol-treated HFD mice. The hepatic mRNA expression of four representative genes involved in cell chemotaxis, extracted from the transcriptomic analysis (GSE116417), is shown below the GSEA plots. *Significantly different from naive HFD mice, p < 0.05 with a Mann–Whitney test

mRNA expression of hepatic genes involved in mitochondrial function and mitochondrial respiratory chain (MRC) complex I and II activities in high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). a Genome-wide expression profiling performed in naive (n = 5) and ethanol-treated (n = 5) HFD mice (GSE116417) was used for subsequent gene set enrichment analysis (GSEA). GSEA showing in ethanol-treated HFD a significant enrichment of the gene ontology (GO) gene sets corresponding to “oxidative phosphorylation” (OXPHOS) and “MRC complex assembly”, compared with naive HFD mice. The hepatic mRNA expression of eight representative genes involved in oxidative phosphorylation and MRC complex assembly, extracted from the transcriptomic analysis (GSE116417), is shown below the GSEA plots. *Significantly different from naive HFD mice, p < 0.05 with a Mann–Whitney test. b MRC complex I and II activities. Values are mean ± SEM for 9–10 mice per group. $Significantly different from SD mice, *significantly different from naive HFD mice, p < 0.05 with a one-way ANOVA test
Investigations on oxidative stress and proteasome gene expression
Investigations were also carried out on oxidative stress and cellular defense systems in liver. Whereas there was no difference between the groups for total liver GSH, the GSSG/GSH ratio was significantly decreased in ethanol-treated HFD mice compared with SD mice (Fig. 8a). Liver carbonylated proteins were enhanced in DIO mice compared with SD mice but there was no difference between naive and ethanol-treated HFD mice (Fig. 8a). Hepatic levels of malondialdehyde (MDA), reflecting lipid peroxidation, tended to be higher in HFD mice compared to SD mice but there was no statistical difference between the different groups of animals. The protein expression of CYP2E1 was slightly (18%) but significantly reduced in ethanol-treated HFD mice compared with naive HFD mice (Fig. 8b), while cyp2e1 mRNA expression was not different between these groups (1.00 ± 0.06 and 0.98 ± 0.07 in naive HFD mice and ethanol-treated HFD mice, respectively). Our microarray analysis complemented by GSEA also revealed in naive HFD mice a moderate but significant enrichment (p = 0.016) of the GO gene set corresponding to “response to oxidative stress” in comparison with ethanol-treated HFD mice (Supplementary Fig. 3a). A reduction of Gpx3, Gpx7, Gpx8, Hmox1, Ogg1 and Sod3 mRNA expression was for example observed in the latter group of mice (Supplementary Fig. 3b). Finally, GSEA showed in ethanol-treated HFD a significant enrichment of the GO gene set corresponding to the “proteasome complex”, compared with naive HFD mice (Fig. 8c). For instance, increased mRNA levels of Psmb4, Psmb7, Psmc1, Psmc4, Psmd9 and Psmd13 were observed in ethanol-treated HFD mice (Fig. 8c).

Markers of hepatic oxidative stress and cellular defense systems in mice fed the standard diet (SD), mice fed the high-fat diet (HFD) and ethanol-treated HFD mice (HFD + E). a Total glutathione, namely reduced glutathione (GSH) plus oxidized glutathione (GSSG), GSSG/GSH ratio and carbonylated proteins and levels of malondialdehyde (MDA). Values are mean ± SEM for 9–10 mice per group for GSH and carbonylated proteins and 7–10 mice per group for MDA levels. $Significantly different from SD mice, p < 0.05 with a one-way ANOVA test. b Protein expression of cytochrome P450 2E1 (CYP2E1) and heat shock cognate 70 (HSC70), used as loading control. Representative western blot for 5 mice per group. CYP2E1/HSC70 ratios showed in the graph are mean ± SEM for 10 mice per group. *Significantly different from naive HFD mice, p < 0.05 with a t test. c Gene set enrichment analysis (GSEA) showing in ethanol-treated HFD a significant enrichment of the gene ontology (GO) gene set corresponding to the “proteasome complex”, compared with naive HFD mice. The hepatic mRNA expression of 6 representative genes involved in the proteasome complex, extracted from the transcriptomic analysis (GSE116417), is shown below the GSEA plots. Values are mean ± SEM for 5 mice per group. *Significantly different from naive HFD mice, p < 0.05 with a Mann–Whitney test
Discussion
Light-to-moderate chronic ethanol consumption could have beneficial effects on NAFLD in patients [7, 8, 10,11,12], although some studies did not confirm these effects [12,13,14]. In this study, we used a mouse model of DIO in an attempt to better understand how moderate alcohol consumption could be beneficial on NAFLD. Thanks to different biochemical, analytical and transcriptomic analyses performed in mouse liver, our investigations suggested that moderate chronic ethanol consumption may alleviate NAFLD by several mechanisms including the generation of non-toxic lipid species such as triglycerides enriched in MUFAs, reduced expression of profibrotic and proinflammatory genes and restoration of mitochondrial function.
Murine models of NAFLD and moderate ethanol consumption
Our DIO model fulfilled the different recommended items required for a relevant mouse model of NASH, in particular, regarding overweight, hyperinsulinemia, hyperglycemia and raised serum transaminase activity [37]. However, it is noteworthy that our naive HFD mice presented borderline NASH rather than definite NASH, as discussed afterwards.
Regarding ethanol exposure (ca. 10 g/kg/day corresponding to 15.9% of calories from alcohol), it might be considered as high when compared to humans. However, previous investigations indicated that our protocol of alcohol administration can be considered as a bona fide model of moderate chronic ethanol consumption. First, although data are scarce, it has been shown that mice metabolize ethanol at a greater rate than do rats and humans [38]. Second, serum ethanol concentrations in ethanol-treated HFD mice were between 0.5 and 0.7 g/l, which are below the toxic range in humans (starting from 0.8 to 1 g/l) [39, 40]. Third, exposure of the livers to high ethanol concentrations would have led to increased CYP2E1 expression at the mRNA and protein levels [41,42,43], which was not observed in ethanol-treated HFD mice. Finally, ethanol exposure in our murine model was similar (or even lower) to previous studies in rodents investigating the effects of moderate alcohol consumption on different tissues and parameters [19, 44,45,46].
A caveat of our investigations was that ethanol-treated HFD mice ingested ethanol before blood and liver collection since these animals were allowed to drink during the different episodes of overnight fast scheduled in the study. Thus, some results observed in ethanol-treated HFD mice might have been directly induced by the presence of ethanol (and possibly the corresponding calories) during the few hours preceding the biological sample collection, in addition to the long-term effects of the moderate consumption of ethanol.
Moderate chronic ethanol consumption alleviates NAFLD in obese mice
Moderate chronic ethanol consumption in HFD mice significantly decreased serum ALT and AST activity, thus indicating NAFLD alleviation, although the mean necroinflammation score was similar between ethanol-treated and naive HFD mice. However, no apoptotic bodies was observed in ethanol-treated HFD mice, in contrast to naive HFD mice. Because hepatic apoptosis can be associated with increased serum transaminase activity [47, 48], our data suggested that lower serum ALT and AST activity in ethanol-treated HFD mice could be linked to reduced hepatic apoptosis. In keeping with this hypothesis, these mice presented a downregulation of several genes involved in apoptosis and an upregulation of some anti-apoptotic genes. Finally, ethanol consumption tended to reduce hepatic fibrosis, in particular, in portal areas. Interestingly, a recent study in a rat model of NAFLD reported that a moderate chronic ethanol consumption improved liver fibrosis and decreased the mRNA expression of several profibrotic genes such as TGF-β, α-SMA and collagen [18].
NAFLD alleviation is associated with increased serum adiponectin and higher hepatic levels of triglycerides and MUFAs
The mechanisms whereby light-to-moderate alcohol consumption could be beneficial on NAFLD are poorly understood, possibly because only a few experimental investigations have tackled this issue [15,16,17,18]. Some clinical and experimental investigations suggested a role of insulin resistance improvement, possibly via enhanced circulating adiponectin levels [15, 49]. Interestingly, adiponectin is able not only to alleviate fatty liver but also hepatic necroinflammation and fibrosis [50, 51]. In this study, serum adiponectin levels tended to be higher in ethanol-treated HFD mice compared to naive HFD mice but this was not associated with lower serum insulin concentrations.
Our fatty acid analysis revealed that moderate chronic ethanol consumption significantly increased the amount of both hepatic triglycerides and phospholipids (as measured by their fatty acid amounts), consistent with larger lipid droplets. This was accompanied with a significant decrease in the proportion of palmitic acid and an increase in that of oleic acid in ethanol-treated HFD mice compared with naive HFD mice as well as a greater Δ9-desaturation index, suggesting higher SCD1 activity. Thus, ethanol-induced NAFLD improvement might be related to the lower hepatic levels of palmitic acid and higher levels of triglycerides since these lipid species are respectively harmful and beneficial for the hepatocytes, as discussed afterwards. Interestingly, higher SCD1 activity in ethanol-treated HFD mice might also have explained the significant increase in serum triglyceride levels in these mice. Indeed, high SCD1 activity favors triglyceride secretion from hepatocytes [52, 53]. However, further investigations will be required to explain why hypertriglyceridemia was transient in ethanol-treated HFD mice and no longer observed after 4 months. Nonetheless, reduced body weight and possibly fat mass might have played a role.
NAFLD improvement may occur through lower levels of SFAs including palmitic acid
Numerous investigations previously reported that excess of palmitic acid is deleterious for the liver, in particular, by inducing hepatocyte oxidative stress, endoplasmic reticulum (ER) stress and eventually apoptosis [54,55,56]. Moreover, lipotoxicity-induced mitochondrial dysfunction could play a major role in the pathogenesis of NASH [55,56,57,58]. By inducing cell death and favoring the release of extracellular vesicles, palmitic acid could also play a key role in the pathogenesis of NASH by promoting inflammation and fibrosis [55, 56, 59]. It is noteworthy that a lipidomic analysis in NAFLD patients showed a trend toward higher hepatic levels of palmitic acid and lower levels of oleic acid in NASH compared with simple fatty liver [60]. On the other hand, increased conversion of palmitate to MUFAs via higher SCD1 activity favors accretion of neutral lipids (triglycerides or cholesterol ester) and protects against palmitate-induced cell death [61,62,63]. Interestingly, SCD1 knockout mice fed a methionine–choline deficient (MCD) diet had less steatosis but increased hepatocellular apoptosis, liver injury, and fibrosis compared with wild-type mice [64]. Similarly, diacylglycerol acyltransferase 2 (DGAT2) antisense oligonucleotide treatment in MCD diet-fed mice decreased steatosis but enhanced hepatic free fatty acids, oxidative stress, lobular necroinflammation and fibrosis [65]. Hence, these investigations and others [54,55,56] support a protective role of triglyceride accumulation against SFA-induced lipotoxicity.
NAFLD alleviation is associated with larger lipid droplets and less microvesicular steatosis suggesting restoration of mitochondrial function
NAFLD alleviation in ethanol-treated HFD mice was associated with larger lipid droplets. Lipid droplet growth is deemed to be due to the relocalization of several triglyceride synthesis enzymes (e.g. GPAT4, AGPAT3, DGAT2) from the ER to lipid droplets [66, 67]. Further investigations will be needed to determine whether this mechanism occurred in ethanol-treated HFD mice. Nevertheless, these mice presented higher expression of several enzymes involved in lipogenesis such as ATP citrate lyase (Acly), acetyl-CoA carboxylase 1 (Acaca), malic enzyme (Me1) and Lipin 1 (Lpin1). Thus, besides enzyme relocalization, enhanced expression of lipogenic enzymes might have favored lipid droplet expansion in ethanol-treated HFD mice. In keeping with this assumption, our previous investigations in ob/ob mice showed that moderate chronic ethanol consumption led to an increase in the hepatic mRNA levels of Acaca, fatty acid synthase (Fas) and Scd1, which was associated with an enlargement of the lipid droplet size [15]. Lipid droplet growth is also dependent on the expression of the CIDE proteins, which promote the formation of large lipid droplets in different tissues including liver [68, 69]. Interestingly, Cidea mRNA expression was increased by 2.5-fold in ethanol-treated HFD mice compared with naive HFD mice.
Whatever its etiology, microvesicular steatosis is thought to be deleterious for the liver, in particular, as the consequence of mitochondrial dysfunction and energy shortage [70,71,72]. Interestingly, a previous study in NAFLD patients showed that the presence of microvesicular steatosis was significantly associated with megamitochondria, ballooning cell injury, more advanced fibrosis and diagnosis of NASH [73]. In the present study, the occurrence of less microvesicular steatosis in ethanol-treated HFD mice might be linked to an improvement of mitochondrial function via transcriptional mechanisms. Indeed, GSEA revealed in these mice a significant enrichment of the expression of genes involved in OXPHOS and MRC complex assembly. Moreover, this transcriptional effect might have explained why MRC complex II activity was no longer decreased in ethanol-treated HFD mice compared with naive HFD mice. However, it cannot be excluded that restoration of mitochondrial function in ethanol-treated HFD mice might also be the mere consequence of reduced levels of SFAs including palmitic acid.
Other potential mechanisms involved in NAFLD alleviation
GSEA also disclosed that ethanol drinking lessened the hepatic expression of a wide range of genes involved in fibrosis and leucocyte chemotaxis. Although moderate chronic ethanol consumption might have directly lessened the expression of these genes, a reduction of apoptosis might have been indirectly involved. Indeed, hepatocyte apoptosis can favor both inflammation and fibrosis [74, 75]. Nevertheless, the downregulation of proinflammatory and profibrotic genes in ethanol-treated HFD mice was not associated with a significant improvement of the necroinflammation and fibrosis scores, although there was a trend toward less fibrosis in these mice. However, it is noteworthy that both necroinflammation and fibrosis was mild in our DIO mouse model, indicating borderline NASH. Hence, any improvement of these lesions might be difficult to appraise. Accordingly, more prolonged exposure to HFD and ethanol might have been needed to significantly alleviate these lesions.
Liver GSSG/GSH was reduced in ethanol-treated HFD mice compared to naive HFD mice, thus suggesting an alleviation of some components of oxidative stress such as reduced CYP2E1 protein expression. This assumption was also supported by a global reduction in ethanol-treated HFD mice of the mRNA expression of different genes involved in the response to oxidative stress. Nevertheless, ethanol consumption significantly enhanced the mRNA levels of several genes encoding proteasome subunits, which may reflect an adaptive response to get rid of some damaged proteins [76, 77]. Since the proteasome plays a major role in cell protection, our results suggest that NAFLD alleviation by moderate chronic ethanol consumption might be, at least in part, linked to proteasome stimulation. Previous investigations reported that low ethanol exposure or moderate oxidative stress could activate the proteasome [78, 79]. In our study, however, ethanol-treated HFD mice presented evidence of lower oxidative stress and increased proteasome activity at the gene-expression level. Because the transcription factor nuclear factor erythroid 2-like 2 (NFE2L2, also referred to as Nrf2) is able to activate the expression of genes involved in both biological processes [76, 77], our data suggest a lack of robust Nrf2 activation in ethanol-treated HFD mice. Accordingly, a supervised GSEA with a dataset of 481 Nrf2 target genes (data set NFE2L2.V2) [80] did not reveal in these mice a significant enrichment of genes related to the Nrf2 pathway. Further investigations will be needed to determine why oxidative stress and proteasome activity seemed to be disconnected in ethanol-treated HFD mice. Nevertheless, our previous investigations in ob/ob mice showed that fatty liver improvement induced by moderate ethanol consumption over 6 months was associated with some signs of mild oxidative stress such as reduced aconitase activity and higher MnSOD protein expression [15]. Hence, additional investigations will be required to determine whether further prolongation of the moderate alcohol drinking protocol might lead to stronger hepatic oxidative stress and loss of the beneficial effects on liver afforded by ethanol. Moreover, it should be underlined that hepatic fat accumulation can favor in the long-term the occurrence of unfavorable extra-hepatic effects such as dyslipidemia (including hypertriglyceridemia and hypercholesterolemia), systemic inflammation and cardiovascular diseases [81, 82].
Conclusion
Our study performed in DIO mice suggests that moderate chronic ethanol consumption may alleviate NAFLD progression, in particular, as regards hepatic cytolysis and fibrosis. Several mechanisms could be involved including a reduction in the levels of deleterious SFAs (including palmitic acid) and a concomitant generation of non-toxic lipid species such as MUFAs. NAFLD alleviation in ethanol-treated HFD mice was associated with larger lipid droplets and less microvesicular steatosis, in keeping with a possible improvement of mitochondria function. Genome-wide expression profiling and GSEA revealed an overall upregulation of genes involved in the proteasome complex, thus suggesting possible stimulation of proteasome activity. Because there was also evidence of lower oxidative stress in ethanol-treated HFD mice, further investigations will be required to determine why moderate chronic ethanol intake could induce an opposite effect on proteasome activity and oxidative stress.
References
Rehm J (2011) The risks associated with alcohol use and alcoholism. Alcohol Res Health 34:135–143
Seitz HK, Bataller R, Cortez-Pinto H et al (2018) Publisher correction: Alcoholic liver disease. Nat Rev Dis Primers 4:18. https://doi.org/10.1038/s41572-018-0021-8
Louvet A, Mathurin P (2015) Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 12:231–242. https://doi.org/10.1038/nrgastro.2015.35
GBD 2016 Alcohol Collaborators (2018) Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 392:1015–1035. https://doi.org/10.1016/S0140-6736(18)31310-2
Bonnet F, Disse E, Laville M et al (2012) Moderate alcohol consumption is associated with improved insulin sensitivity, reduced basal insulin secretion rate and lower fasting glucagon concentration in healthy women. Diabetologia 55:3228–3237. https://doi.org/10.1007/s00125-012-2701-3
Poli A, Marangoni F, Avogaro A et al (2013) Moderate alcohol use and health: a consensus document. Nutr Metab Cardiovasc Dis 23:487–504. https://doi.org/10.1016/j.numecd.2013.02.007
Ajmera VH, Terrault NA, Harrison SA (2017) Is moderate alcohol use in nonalcoholic fatty liver disease good or bad? A critical review. Hepatology 65:2090–2099. https://doi.org/10.1002/hep.29055
Dunn W, Sanyal AJ, Brunt EM et al (2012) Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). J Hepatol 57:384–391. https://doi.org/10.1016/j.jhep.2012.03.024
Kwon HK, Greenson JK, Conjeevaram HS (2014) Effect of lifetime alcohol consumption on the histological severity of non-alcoholic fatty liver disease. Liver Int 34:129–135. https://doi.org/10.1111/liv.12230
Moriya A, Iwasaki Y, Ohguchi S et al (2015) Roles of alcohol consumption in fatty liver: a longitudinal study. J Hepatol 62:921–927. https://doi.org/10.1016/j.jhep.2014.11.025
Seitz HK, Mueller S, Hellerbrand C, Liangpunsakul S (2015) Effect of chronic alcohol consumption on the development and progression of non-alcoholic fatty liver disease (NAFLD). Hepatobiliary Surg Nutr 4:147–151. https://doi.org/10.3978/j.issn.2304-3881.2014.12.01
Kwon I, Jun DW, Moon J-H (2018) Effects of moderate alcohol drinking in patients with nonalcoholic fatty liver disease. Gut Liver. https://doi.org/10.5009/gnl18175
Ajmera V, Belt P, Wilson LA et al (2018) Among patients with nonalcoholic fatty liver disease, modest alcohol use is associated with less improvement in histologic steatosis and steatohepatitis. Clin Gastroenterol Hepatol 16:1511.e5–1520.e5. https://doi.org/10.1016/j.cgh.2018.01.026
Sookoian S, Flichman D, Castaño GO, Pirola CJ (2016) Mendelian randomisation suggests no beneficial effect of moderate alcohol consumption on the severity of nonalcoholic fatty liver disease. Aliment Pharmacol Ther 44:1224–1234. https://doi.org/10.1111/apt.13828
Fromenty B, Vadrot N, Massart J et al (2009) Chronic ethanol consumption lessens the gain of body weight, liver triglycerides, and diabetes in obese ob/ob mice. J Pharmacol Exp Ther 331:23–34. https://doi.org/10.1124/jpet.109.155168
Kanuri G, Landmann M, Priebs J et al (2016) Moderate alcohol consumption diminishes the development of non-alcoholic fatty liver disease (NAFLD) in ob/ob mice. Eur J Nutr 55:1153–1164. https://doi.org/10.1007/s00394-015-0929-7
Osaki A, Okazaki Y, Kimoto A et al (2014) Beneficial effect of a low dose of ethanol on liver function and serum urate in rats fed a high-fat diet. J Nutr Sci Vitaminol 60:408–412. https://doi.org/10.3177/jnsv.60.408
Sun F, Zhuang Z, Zhang D et al (2018) Chronic moderate alcohol consumption relieves high-fat high-cholesterol diet-induced liver fibrosis in a rat model. Clin Exp Pharmacol Physiol 45:1046–1055. https://doi.org/10.1111/1440-1681.12976
Godfrey J, Jeanguenin L, Castro N et al (2015) Chronic voluntary ethanol consumption induces favorable ceramide profiles in selectively bred alcohol-preferring (P) rats. PLoS One 10:e0139012. https://doi.org/10.1371/journal.pone.0139012
Klarich DS, Penprase J, Cintora P et al (2017) Effects of moderate alcohol consumption on gene expression related to colonic inflammation and antioxidant enzymes in rats. Alcohol 61:25–31. https://doi.org/10.1016/j.alcohol.2017.02.179
Justice M, Ferrugia A, Beidler J et al (2019) Effects of moderate ethanol consumption on lipid metabolism and inflammation through regulation of gene expression in rats. Alcohol Alcohol 54:5–12. https://doi.org/10.1093/alcalc/agy079
Buron N, Porceddu M, Roussel C et al (2017) Chronic and low exposure to a pharmaceutical cocktail induces mitochondrial dysfunction in liver and hyperglycemia: differential responses between lean and obese mice. Environ Toxicol 32:1375–1389. https://doi.org/10.1002/tox.22331
Trak-Smayra V, Paradis V, Massart J et al (2011) Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp Pathol 92:413–421. https://doi.org/10.1111/j.1365-2613.2011.00793.x
Gailhouste L, Le Grand Y, Odin C et al (2010) Fibrillar collagen scoring by second harmonic microscopy: a new tool in the assessment of liver fibrosis. J Hepatol 52:398–406. https://doi.org/10.1016/j.jhep.2009.12.009
Tabet E, Genet V, Tiaho F et al (2016) Chlordecone potentiates hepatic fibrosis in chronic liver injury induced by carbon tetrachloride in mice. Toxicol Lett 255:1–10. https://doi.org/10.1016/j.toxlet.2016.02.005
Bucher S, Le Guillou D, Allard J et al (2018) Possible involvement of mitochondrial dysfunction and oxidative stress in a cellular model of NAFLD progression induced by benzo[a]pyrene/ethanol CoExposure. Oxid Med Cell Longev 2018:4396403. https://doi.org/10.1155/2018/4396403
Le Guillou D, Bucher S, Begriche K et al (2018) Drug-induced alterations of mitochondrial DNA homeostasis in steatotic and nonsteatotic HepaRG cells. J Pharmacol Exp Ther 365:711–726. https://doi.org/10.1124/jpet.117.246751
Baraibar MA, Ladouce R, Friguet B (2013) Proteomic quantification and identification of carbonylated proteins upon oxidative stress and during cellular aging. J Proteom 92:63–70. https://doi.org/10.1016/j.jprot.2013.05.008
Bucher S, Tête A, Podechard N et al (2018) Co-exposure to benzo[a]pyrene and ethanol induces a pathological progression of liver steatosis in vitro and in vivo. Sci Rep 8:5963. https://doi.org/10.1038/s41598-018-24403-1
Rioux V, Catheline D, Bouriel M, Legrand P (2005) Dietary myristic acid at physiologically relevant levels increases the tissue content of C20:5n-3 and C20:3n-6 in the rat. Reprod Nutr Dev 45:599–612. https://doi.org/10.1051/rnd:2005048
Rioux V, Lemarchal P, Legrand P (2000) Myristic acid, unlike palmitic acid, is rapidly metabolized in cultured rat hepatocytes. J Nutr Biochem 11:198–207. https://doi.org/10.1016/S0955-2863(00)00065-6
Rioux V, Pédrono F, Blanchard H et al (2013) Trans-vaccenate is Δ13-desaturated by FADS3 in rodents. J Lipid Res 54:3438–3452. https://doi.org/10.1194/jlr.M042572
Ravinet Trillou C, Delgorge C, Menet C et al (2004) CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord 28:640–648. https://doi.org/10.1038/sj.ijo.0802583
Guerville M, Leroy A, Sinquin A et al (2017) Western-diet consumption induces alteration of barrier function mechanisms in the ileum that correlates with metabolic endotoxemia in rats. Am J Physiol Endocrinol Metab 313:E107–E120. https://doi.org/10.1152/ajpendo.00372.2016
Goldstein DB, Kakihana R (1977) Circadian rhythms of ethanol consumption by mice: a simple computer analysis for chronopharmacology. Psychopharmacology 52:41–45. https://doi.org/10.1007/BF00426598
Jelic P, Shih MF, Taberner PV (1998) Diurnal variation in plasma ethanol levels of TO and CBA mice on chronic ethanol drinking or ethanol liquid diet schedules. Psychopharmacology 138:143–150. https://doi.org/10.1007/s002130050656
Haczeyni F, Yeh MM, Ioannou GN et al (2018) Mouse models of non-alcoholic steatohepatitis: a reflection on recent literature. J Gastroenterol Hepatol 33:1312–1320. https://doi.org/10.1111/jgh.14122
Livy DJ, Parnell SE, West JR (2003) Blood ethanol concentration profiles: a comparison between rats and mice. Alcohol 29:165–171. https://doi.org/10.1016/S0741-8329(03)00025-9
Stead AH, Moffat AC (1983) A collection of therapeutic, toxic and fatal blood drug concentrations in man. Hum Toxicol 2:437–464. https://doi.org/10.1177/096032718300200301
Schulz M, Iwersen-Bergmann S, Andresen H, Schmoldt A (2012) Therapeutic and toxic blood concentrations of nearly 1,000 drugs and other xenobiotics. Crit Care 16:R136. https://doi.org/10.1186/cc11441
Ronis MJ, Huang J, Crouch J et al (1993) Cytochrome P450 CYP 2E1 induction during chronic alcohol exposure occurs by a two-step mechanism associated with blood alcohol concentrations in rats. J Pharmacol Exp Ther 264:944–950
Badger TM, Huang J, Ronis M, Lumpkin CK (1993) Induction of cytochrome P450 2E1 during chronic ethanol exposure occurs via transcription of the CYP 2E1 gene when blood alcohol concentrations are high. Biochem Biophys Res Commun 190:780–785. https://doi.org/10.1006/bbrc.1993.1117
Roberts BJ, Song BJ, Soh Y et al (1995) Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. J Biol Chem 270:29632–29635. https://doi.org/10.1074/jbc.270.50.29632
Wang Y, Seitz HK, Wang X-D (2010) Moderate alcohol consumption aggravates high-fat diet induced steatohepatitis in rats. Alcohol Clin Exp Res 34:567–573. https://doi.org/10.1111/j.1530-0277.2009.01122.x
Sanchez Vega MC, Chong S, Burne THJ (2013) Early gestational exposure to moderate concentrations of ethanol alters adult behaviour in C57BL/6J mice. Behav Brain Res 252:326–333. https://doi.org/10.1016/j.bbr.2013.06.003
Stragier E, Martin V, Davenas E et al (2015) Brain plasticity and cognitive functions after ethanol consumption in C57BL/6 J mice. Transl Psychiatry 5:e696. https://doi.org/10.1038/tp.2015.183
Haouzi D, Lekehal M, Tinel M et al (2001) Prolonged, but not acute, glutathione depletion promotes Fas-mediated mitochondrial permeability transition and apoptosis in mice. Hepatology 33:1181–1188. https://doi.org/10.1053/jhep.2001.24235
Lacronique V, Mignon A, Fabre M et al (1996) Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat Med 2:80–86. https://doi.org/10.1038/nm0196-80
Nova E, Baccan GC, Veses A et al (2012) Potential health benefits of moderate alcohol consumption: current perspectives in research. Proc Nutr Soc 71:307–315. https://doi.org/10.1017/S0029665112000171
Fang H, Judd RL (2018) Adiponectin regulation and function. Compr Physiol 8:1031–1063. https://doi.org/10.1002/cphy.c170046
Polyzos SA, Kountouras J, Zavos C, Tsiaousi E (2010) The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab 12:365–383. https://doi.org/10.1111/j.1463-1326.2009.01176.x
Legrand P, Catheline D, Fichot MC, Lemarchal P (1997) Inhibiting delta9-desaturase activity impairs triacylglycerol secretion in cultured chicken hepatocytes. J Nutr 127:249–256. https://doi.org/10.1093/jn/127.2.249
Attie AD, Krauss RM, Gray-Keller MP et al (2002) Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J Lipid Res 43:1899–1907. https://doi.org/10.1194/jlr.M200189-JLR200
Cazanave SC, Gores GJ (2010) Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin Lipidol 5:71–85. https://doi.org/10.2217/clp.09.85
Hirsova P, Ibrahim SH, Gores GJ, Malhi H (2016) Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res 57:1758–1770. https://doi.org/10.1194/jlr.R066357
Marra F, Svegliati-Baroni G (2018) Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 68:280–295. https://doi.org/10.1016/j.jhep.2017.11.014
Begriche K, Massart J, Robin M-A et al (2013) Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58:1497–1507. https://doi.org/10.1002/hep.26226
Simões ICM, Fontes A, Pinton P et al (2018) Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol 95:93–99. https://doi.org/10.1016/j.biocel.2017.12.019
Hirsova P, Ibrahim SH, Krishnan A et al (2016) Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 150:956–967. https://doi.org/10.1053/j.gastro.2015.12.037
Puri P, Baillie RA, Wiest MM et al (2007) A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46:1081–1090. https://doi.org/10.1002/hep.21763
Listenberger LL, Han X, Lewis SE et al (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA 100:3077–3082. https://doi.org/10.1073/pnas.0630588100
Busch AK, Gurisik E, Cordery DV et al (2005) Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapoptosis. Diabetes 54:2917–2924. https://doi.org/10.2337/diabetes.54.10.2917
Alkhouri N, Dixon LJ, Feldstein AE (2009) Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol 3:445–451. https://doi.org/10.1586/egh.09.32
Li ZZ, Berk M, McIntyre TM, Feldstein AE (2009) Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem 284:5637–5644. https://doi.org/10.1074/jbc.M807616200
Yamaguchi K, Yang L, McCall S et al (2007) Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45:1366–1374. https://doi.org/10.1002/hep.21655
Thiam AR, Farese RV, Walther TC (2013) The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 14:775–786. https://doi.org/10.1038/nrm3699
Wilfling F, Wang H, Haas JT et al (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24:384–399. https://doi.org/10.1016/j.devcel.2013.01.013
Yang H, Galea A, Sytnyk V, Crossley M (2012) Controlling the size of lipid droplets: lipid and protein factors. Curr Opin Cell Biol 24:509–516. https://doi.org/10.1016/j.ceb.2012.05.012
Gao G, Chen F-J, Zhou L et al (2017) Control of lipid droplet fusion and growth by CIDE family proteins. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1197–1204. https://doi.org/10.1016/j.bbalip.2017.06.009
Fromenty B, Pessayre D (1995) Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther 67:101–154. https://doi.org/10.1016/0163-7258(95)00012-6
Begriche K, Massart J, Robin M-A et al (2011) Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol 54:773–794. https://doi.org/10.1016/j.jhep.2010.11.006
Hegarty R, Deheragoda M, Fitzpatrick E, Dhawan A (2018) Paediatric fatty liver disease (PeFLD): all is not NAFLD—pathophysiological insights and approach to management. J Hepatol 68:1286–1299. https://doi.org/10.1016/j.jhep.2018.02.006
Tandra S, Yeh MM, Brunt EM et al (2011) Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol 55:654–659. https://doi.org/10.1016/j.jhep.2010.11.021
Canbay A, Friedman S, Gores GJ (2004) Apoptosis: the nexus of liver injury and fibrosis. Hepatology 39:273–278. https://doi.org/10.1002/hep.20051
Zhan S-S, Jiang JX, Wu J et al (2006) Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 43:435–443. https://doi.org/10.1002/hep.21093
Chapple SJ, Siow RCM, Mann GE (2012) Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol 44:1315–1320. https://doi.org/10.1016/j.biocel.2012.04.021
Jung T, Höhn A, Grune T (2014) The proteasome and the degradation of oxidized proteins: part II—protein oxidation and proteasomal degradation. Redox Biol 2:99–104. https://doi.org/10.1016/j.redox.2013.12.008
Osna NA, Haorah J, Krutik VM, Donohue TM (2004) Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology 40:574–582. https://doi.org/10.1002/hep.20352
Donohue TM, Thomes PG (2014) Ethanol-induced oxidant stress modulates hepatic autophagy and proteasome activity. Redox Biol 3:29–39. https://doi.org/10.1016/j.redox.2014.10.006
Malhotra D, Portales-Casamar E, Singh A et al (2010) Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 38:5718–5734. https://doi.org/10.1093/nar/gkq212
Byrne CD, Targher G (2015) NAFLD: a multisystem disease. J Hepatol 62:S47–S64. https://doi.org/10.1016/j.jhep.2014.12.012
Than NN, Newsome PN (2015) A concise review of non-alcoholic fatty liver disease. Atherosclerosis 239:192–202. https://doi.org/10.1016/j.atherosclerosis.2015.01.001
Acknowledgements
This work was supported by a grant from the Agence Nationale de la Recherche (ANR STEATOX project ANR-13-CESA-0009). Simon Bucher was a recipient of a joint fellowship from the Région Bretagne (ARED) and ANR. We are grateful to Alain Fautrel and Marine Seffals (histopathology platform H2P2), Laurence Bernard-Touami (animal care facility ARCHE), Stéphanie Dutertre (microscopy Rennes imaging center platform MRIC), all from the SFR Biosit UMS CNRS 3480-INSERM 018 SFR, for their excellent technical support. We also wish to thank Nicolas Collet (Laboratoire de Biochimie-Toxicologie, CHU de Rennes) for his skilled technical assistance for the serum analyses. We are grateful to INSERM (Institut National de la Recherche et de la Santé Médicale) for its constant financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bucher, S., Begriche, K., Catheline, D. et al. Moderate chronic ethanol consumption exerts beneficial effects on nonalcoholic fatty liver in mice fed a high-fat diet: possible role of higher formation of triglycerides enriched in monounsaturated fatty acids. Eur J Nutr 59, 1619–1632 (2020). https://doi.org/10.1007/s00394-019-02017-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-019-02017-1