
Abstract
Purpose
Increasing evidence suggests the potential use of natural antioxidant compounds in the prevention/treatment of osteoporosis. This study was undertaken to investigate the effects of purified delphinidin-3-rutinoside (D3R), isolated from Solanum melongena L., on osteoblast viability and differentiation in basal conditions and its ability to protect MC3T3-E1 cells against oxidative damage induced by tert-butyl hydroperoxide (t-BHP).
Methods
MC3T3-E1 osteoblastic cells were treated with D3R (10−11–10−5 M for 24 h), followed by treatment with t-BHP (250 µM for 3 h). To test cell viability, MTT test was performed. Apoptotic cells were stained with Hoechst-33258 dye. Cytoskeleton rearrangement was stained with FICT-labelled phalloidin. Intracellular ROS production was measured using dichlorofluorescein CM-DCFA. The reduced glutathione to oxidized glutathione ratio (GSH/GSSG) contents was measured according to the OPT fluorimetric assay.
Results
D3R (10−9 M) significantly increases viability of MC3T3-E1 cells and promotes osteoblast differentiation by increasing the expression of type I collagen, alkaline phosphatase and osteocalcin. Pre-treatment with D3R (10−9 M) significantly prevented t-BHP-induced osteoblastic dysfunction and changes in the cytoskeleton organization by decreasing intracellular ROS and preventing the reduction in GSH/GSSG. D3R did not significantly modify the expression of Osteoprotegerin/RANKL system activated by t-BHP suggesting a lack of effect of D3R on osteoblast/osteoclast crosstalk. D3R protective effects against t-BHP-induced osteoblastic dysfunction were mediated by the PI3K/Akt pathway since they were completely prevented by LY294002, a PI3K/Akt specific inhibitor.
Conclusions
These findings indicate that D3R protects MC3T3-E1 cells from oxidative damage and suggest the potential utility of dietary D3R supplement to prevent osteoblast dysfunction in age-related osteoporosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Progressive free radical damage is considered a key component in the tissue degeneration associated with aging, and the skeleton is no exception [1]. The levels of reactive oxygen species (ROS) in bone increased with age and sex steroids deficiency [2], and a correlation has been reported between oxidative stress and a decrease in bone mineral density both in aged men and women [3, 4]. Furthermore, elderly osteoporotic patients displayed a marked reduction in plasma antioxidants [5]. At cellular level, oxidative stress decreases osteoblast and osteocyte lifespan and inhibits osteoblast formation suggesting a harmful role for ROS in the imbalance between bone resorption by osteoclasts and new bone deposition by osteoblasts that characterize the age-related osteoporosis. Antioxidant compounds have been proposed as attractive agents for the prevention and treatment of osteoporosis. Interestingly, parathyroid hormone (PTH), which is a strong anabolic compound, attenuates oxidative stress suggesting that antioxidant properties of PTH might contribute to its efficacy in treating age-related osteoporosis [6] A great deal of interest has been expressed towards the benefits of a diet rich in plant-based food for several chronic diseases including osteoporosis [7]. These foods are rich and exclusive sources of natural antioxidant compounds which could be responsible for their ability to attenuate age-related bone loss. Experimental studies have shown skeletal benefits of isolated nutrients such as flavonoids, lycopene, resveratrol [8, 9] and anthocyanins [10]. Furthermore, dietary antioxidant intake seems to exert a beneficial effect on bone metabolism and mineral density in postmenopausal women [11, 12].
At cellular level, ROS increase osteoclast number [13, 14] and decreased bone formation by reducing the generation and survival of osteoblasts and osteocytes, former osteoblasts that are entombed in the mineralized matrix and are responsible for sensing and adapting bone to mechanical loading [2, 15, 16]. Aubergine (Solanum melongena L.) fruit has been ranked among the top ten vegetables in terms of radical oxygen absorbance capacity [17] and has been tested for its possible health-promoting actions [18].
Among the various part of aubergine (entire fruit, pulp, skin), peeled skin possess the highest capacity in the scavenging of superoxide radicals and contains the exclusive amount of anthocyanin pigments [19] such as delphinidin-3-rutinoside (D3R), and trans- and cis-delphinine 3-(p-coumaroylrutinoside)-5-glucoside(nasunin), [20].
In our previous studies we have shown that nasunin, extracted and purified from aubergine peel of Japanese-type cultivars, exerts a strong protective effect against tert-butyl hydroperoxide (t-BHP)-induced oxidative damage in MC3T3-E1 osteoblastic cells and prevented t-BHP-induced osteoblastic dysfunction [21]. However, the structure requirements of nasunin in relation to its protective effect against oxidative damage in MC3T3-E1 cells remain to be clarified.
Previous studies on several cell types reported that the direct ability of anthocyanins to scavenge free radicals could be related to the –OH moieties on the B ring, in particular to its catechol function, to the number of OH–moieties in total [22] or to the type and extent of glycosylation and acylation [23, 24]. In the present study, we examined the effects of purified D3R, the major anthocyanin component of the aubergine skin in non-Japanese type which is a simpler delphinidin derivative than nasunin [20, 25], on MC3T3-E1 cells viability and differentiation in basal conditions. We used MC3T3-E1 osteoblast-like cell line which is the most commonly used model to study osteogenic development since the cells are characterized by distinct proliferative and differentiated stages, thus reproducing a temporal program of osteoblast differentiation as occurs during in vivo bone formation [26]. On the basis of the results obtained showing a positive effect of D3R on osteoblast differentiation, we studied the ability of D3R to counteract the cytotoxicity induced by t-BHP damage in both pre osteoblast MC3T3-E1 cells and differentiated cells and the cellular pathways involved in D3R protective effects.
It is well known that cells of the osteoblastic lineage exert a physiological control of osteoclast formation and activity [27, 28] which involves both the osteoclast stimulus Receptor Activator of NFκB Ligand (RANKL) and inhibition of this by the decoy receptor osteoprotegerin (OPG). Since, under stress conditions, RANKL levels exceed OPG production [29], we evaluated whether or not D3R might modulate the expression of OPG/RANKL mRNA in MC3T3-E1 cells.
Materials and methods
D3R purification and crystallization
Several genotypes of non-Japanese aubergine, characterized by long and dark-purple fruit containing the anthocyanin D3R in the peel, were conventionally grown in 2011 in an experimental field at CREA-Unità di Ricerca per l’Orticoltura, located in Montanaso Lombardo (Lodi, Italy). About 50 kg of fruits were harvested at the commercial ripening stage from five plants/genotypes, then carefully peeled, obtaining about 2 kg of peels containing D3R. Peels were immediately frozen in an air-blast tunnel at − 50 °C, then freeze–dried and ground. Freeze–dried powder was sieved through a 1 mm diameter sieve, and stored at − 20 °C up to analyses.
For the pigment extraction, 10 g of powder were added with 500 mL of 0.03N HCl and vigorously stirred for 1 h at room temperature. The mixture was then centrifuged at 25,000×g at 4 °C for 20 min and the supernatant was filtered on glass wool. The supernatant, representing the aubergine peel acidic aqueous extract, was divided into aliquots of 50 mL each and stored at − 80 °C until use.
For the pigment purification, the aliquots of acidic aqueous extract were treated with 50 mL of ethyl acetate to remove chlorogenic acid, the main phenolic in aubergine, and the two phases were carefully decanted into a separatory funnel and separated. The extraction was repeated three times. A total of 150 mL of ethyl acetate was sufficient to almost completely eliminate chlorogenic acid.
The red–violet pigmented fraction, mainly containing the anthocyanin, remained in the aqueous phase. The aqueous phase was evaporated in vacuo at 40 °C to eliminate residuals of ethyl acetate, and drawn to the initial volume with water.
Aqueous acidified extract from peels was further purified to obtain pure D3R-crystals following the methodology by Noda et al. [30] with some modifications.
The acidic aqueous chlorogenic-free extract was purified by elution on a RP18 silica gel column, 32-63 mesh, 60A (ICN Biomedicals; Eschwege, Germany), previously conditioned with 0.03 M HCl (3 × 20 mL), with a ratio extract vs resin 1:10, v/w, and eluted with MeOH (3 × 10 mL).
The MeOH was carefully evaporated in vacuo to a small volume and treated with 20 mL of diethyl ether (Et2O). The mixture, showing an initial precipitate, was rapidly cooled to 4 °C and centrifuged at 3000×g, to obtain a visible crystals precipitate of D3R.
The Et2O phase was then carefully removed and the precipitate was washed with cold Et2O (3 × 5 mL). After removing the solvent, D3R-crystals were dried under a stream of nitrogen and then stored in dark bottles at room temperature with silica gel as desiccant. The purity of D3R-crystals was checked by HPLC analysis and by 1H-NMR analysis, according to Braga et al. [31, see supplementary data].
Cell culture
Mouse MC3T3-E1 pre-osteoblastic cells (ATCC catalog number CRL-2593), were cultured in Dulbecco’s Modified Eagle’s Medium High Glucose (D-MEM, Euroclone, Pero, Italy), supplemented with 10% foetal bovine serum (FBS, Sigma-Aldrich, Milano, Italy), 1% L-glutamine, 100 µg/mL streptomycin and 100 U/mL penicillin at 37 °C in 5% CO2 atmosphere. This basic medium was changed every 3 days and cells were subcultured weekly. To study MC3T3-E1 differentiation, cells were cultured in medium supplemented with L-ascorbic acid 50 µg/mL and β-glycerolphosphate (10 mM, both reagents from Sigma-Aldrich). Cultures were maintained for 7 days before experiments. Cells were pre-treated with D3R for 24 h before the experiments.
Cytotoxicity assay
Cells were plated at the density of 3 × 103 cells/ in a 96-well culture plate. After treatment with D3R, cells were washed with phosphate buffered saline (PBS, Euroclone, Pero, Italy) and incubated at 37 °C with 0.5 mg/mL 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazoliumbromide (MTT, Sigma–Aldrich) for 3 h. The conversion of the tetrazolium salt MTT to a coloured formazan was used to assess cell viability. After the supernatant was removed, dimethyl sulphoxide (DMSO) was added to each well and the absorbance was recorded by a microplate spectrophotometer (WALLAC, Victor2, PerkinElmer, Waltham, MA, USA) at 550 nm. Cell phenotypic observations were made using Olympus TH4-200 inverted phase-contrast microscope, fitted with a digital camera Olympus C-4040 zoom to record any change during treatment.
Induction of oxidative stress
Oxidative stress was induced by tert-butyl hydroperoxide (t-BHP, Sigma-Aldrich, 250 µM for 3 h) and by antimycin A (AMA, Sigma-Aldrich, 70 µM for 24 h). After 24 h preincubation time, D3R was completely removed and the medium was exchanged before adding t-BHP or AMA. This avoided a direct interaction between D3R and the oxidative source in the medium. Pre-treatment with LY294002 (Sigma-Aldrich, 10 µM), an inhibitor of PI3K/Akt pathway, was performed 1 h before D3R.
Hoechst staining of apoptotic cells
Morphological changes in the nuclear chromatin of apoptotic cells were detected by Hoechst-33,258 (Sigma-Aldrich) staining. MC3T3-E1 cells (5 × 103 cells/well) were grown on 22-mm glass coverslips in 6-well plates. After treatments, the cells were fixed with 4% formaldehyde in 0.12 M sucrose, permeabilized with 0.1% TritonX100 in PBS for 5 min, and stained with 10 µg/mL Hoechst-33,258 DNA dye for 5 min. The cells then visualized using an Axioplan fluorescence microscope. The blue fluorescent Hoechst 33,258 is a cell permeable nucleic acid dye usually used to identify chromatin condensation and fragmentation by staining the condensed nuclei of apoptotic cells. Uniformly stained nuclei were scored as healthy, viable cells. Condensed or fragmented nuclei were considered as apoptotic. At least 200 cells were scored blindly without knowledge of their prior treatment.
Cytoskeleton rearrangement
MC3T3-E1 cell growth, fixation and permeabilization were carried out by the same procedure as mentioned above. The permeabilized cells were incubated with FITC-labelled phalloidin (diluted 1:100 in PBS) for 20 min at room temperature and mounted in Vectashield Mard Set Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA). The images were obtained using an Axioplan fluorescence microscope.
Alkaline phosphatase (ALP) activity and collagen content
MC3T3-E1 cells were seeded at the density of 1 × 104 in 48-well plates and treated as previously described in the “Cell culture”. After 6 days the cells were cultured with a medium containing 1% FBS and D3R (10−9 M) for 24 h followed by treatment with t-BHP (125 µΜ, for 3 h). After t-BHP was washed out with fresh medium, cells were incubated with D3R for 24 or 48 h to measure ALP or collagen content, respectively.
On harvesting, the medium was removed and cell monolayer gently washed twice with PBS. The cells were lysed with 0, 2% Triton-X-100 and centrifuged at 14,000×g for 10 min. The clear supernatant was used to measure the ALP activity and protein concentration with a commercially available ALP activity assay kit (Sigma-Aldrich) and BCA-protein assay kit (Thermo-Fisher Sientific, SpA, Rodano, Italy), respectively. ALP activity was measured as nmol/min/mg prot and results were expressed as percentage of differentiated controls.
Collagen content was quantified by Sirius Red-based colorimetric assay as previously described [21]. Cultured osteoblasts were washed with PBS, followed by fixation with Bouin’s fluid for 1 h. After fixation the culture dishes were washed by immersion in running tap water for 15 min, air dried and stained by Sirius red dye reagent (Sigma-Aldrich) for 1 h under mild shaking. The solution was removed and culture washed with 0.01 M HCl. Subsequently, the stained material was dissolved in 0.1 M NaOH and absorbance read at 550 nm. The results are expressed as percentage of differentiated control absorbance.
Intracellular ROS production
ROS production was measured using 5(6)-carboxy-2′,7-dichlorofluorescin diacetate (CM-DCFA, Sigma-Aldrich, 10 µM) as previously described [32]. The MC3T3–E1 cells were seeded in black 96-well plates and cultured for 24 h. On the day of the experiment, the cells were pre-incubated with D3R (10−9 M) for 24 h and loaded with CM-DCFA during the last 30 min of treatment. After CM-DCFA was removed, the cells were exposed to t-BHP (250 µM) for 3 h. DCF fluorescence was assessed with a spectrofluorophotometer (Victor™, PerkinElmer) at the excitation (485 nm) and emission (530 nm) wavelength.
Measurement of cell glutathione and glutathione disulphide levels
The intracellular content of glutathione (GSH) and glutathione disulphide (GSSG) was measured after t-BHP (250 µM for 3 h) according to the o-phthalaldehyde (OPT) fluorimetric assay as previously described [21]. Alteration in the GHS/GSSG ratio was used to assess the exposure of cells to oxidative stress. At pH 8, OPT conjugates with GSH and produces fluorescence that is proportional to the concentration of GSH in the sample. At pH 12 and in the presence of N-ethylmaleimide (NEM), OPT will conjugate only with GSSG and not GSH. Fluorescence was read at 420 nm with excitation set at 350 nm. The concentration was calculated from a standard curve using serial dilutions of GSH or GSSG and normalized to protein concentration measured by BCA assay.
Quantitative PCR
To investigate the effects of D3R on the expression of genes involved in osteoblasts differentiation and bone remodelling, MC3T3-E1 cells (1.5 × 105 cells/well) were plated in 6-well plates, differentiated as previously described and treated with D3R (10−9 M) or medium without the test agent for 24 h. Adherent cells were harvested and total RNA was extracted using TRIzol according to the manufacturer’s instructions (Invitrogen Life Technology, Inc., Paisley, UK). RNA pellet concentrations were assessed spectrophotometrically using microcuvette G1.0 in Eppendorf Biophotometer. The absorbance spectrum was checked for each sample in Eppendorf Biopohotometer. Specific sets of primers for ALP, collagen type 1, alpha 1 (ColA1), osteocalcin (OC), OPG and RANKL. β-catenin cDNAs were designed and synthesized (Sigma-Aldrich), according to Zhang et al. [33] and Dong et al. [34]. Quantitative PCR was performed in CFX96 Bio-Rad using 20 µL of total volume. The efficiency of each set of primers was evaluated in preliminary experiments and it was found close to 100% for target and for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Total RNA (800 ng) was retrotranscribed using the IScript Supermix kit (Bio-Rad), according to the manufacturer’s protocol. The amplification was carried out on 5 ng of total cDNA using SYBR chemistry (iTAQ Universal SYBR green supermix, Bio-Rad) according to the manufacturer’s protocol. Real-time PCR was run according to the following protocol: an initial step of 30 s at 95 °C followed by 40 cycles of 5 s at 95 °C and 30 s at 60 °C. A dissociation stage with a melt curve analysis was also performed. Four replicates were performed for each experimental point and experiments were repeated four times. Gene expression was quantified using the comparative threshold-cycle (DDCt) method considering that the targets and the reference genes have the same amplification efficiency (near to 100%).
Statistical analysis
Statistical analysis was performed with a statistic package (GraphPad Prism5, GraphPad Software San Diego, CA, USA). All data are represented as the mean ± SEM of four independent experiments. Differences between groups were assessed by one-way analysis of variance (ANOVA) followed by Bonferroni test when data were parametric. Non-parametric data were analyzed by a Kruskal–Wallis test followed by Dunn’s test. Differences with a p < 0.05 were considered to be significant.
Results
Effect of D3R on MC3T3-E1 cell viability:
We first examined the effect of increasing D3R concentrations on MC3T3-E1 cell viability in basal conditions using MTT test. As shown in Fig. 1, D3R induces a slight increase in cell viability which reached statistical significance (13% vs controls) at 10−9 M and D3R (10−9 M) was chosen for further experiments on differentiated cells.

Effects of D3R (10−11–10−5 M) on cell viability measured by MTT assay 24 h after treatment. Data are expressed as the percentage relative to control and are the mean ± SEM of four replicates within a single experiment. *p < 0.05 vs controls
Effect of D3R on differentiated MC3T3-E1 cells
D3R (10−9 M) did not significantly modify osteoblast viability in differentiated cells (data not shown), but stimulates the expression of several genes involved in osteoblast differentiation determined using real-time PCR assay. D3R treatment resulted in a significant increased expression of ALP and Col1A1, considered early stage osteogenic differentiation markers, and of OC, a late-marker of differentiation (Fig. 2a). qPCR data fit well with the stimulatory action of D3R on ALP activity and collagen content (Fig. 2b). When we examined the effects of D3R on the OPG/RANKL system, the main regulator of osteoclast differentiation and activation we found a slight, but not significant increase in the expression of OPG without any effect on RANKL. The effects of D3R on osteoblast differentiation could be due to activation of the Wnt/β-catenin signalling pathway since D3R up-regulated the mRNA expression of β-catenin (Fig. 2a) and it is well known that ALP and OC are β-catenin target genes.

Effects of D3R (10−9 M) on ALP, Col1A1, OC, β-catenin, OPG and RANKL expression (a) and ALP activity and Collagen content (b). The gene expression was measured by real-time PCR. Cells were treated with D3R for 24 h and mRNAs were collected. 100 ng of cDNA from each sample were amplified using the SYBR® Green Chemistry and a specific set of primers for ALP, Col1A1, OC, β-catenin, OPG and RANKL. Each sample was evaluated in triplicate. Data were normalized for GAPDH and expressed as mean ± SEM of the relative amounts of gene/GAPDH of each sample vs the mean control value. Four replicates were performed for each experimental point and the experiments were repeated twice. ALP activity and Collagen deposition were evaluated 24/48 h after treatment with D3R. *p < 0.05, **p < 0.01, ***p < 0.001 vs controls
Effect of D3R on t-BHP-induced cytotoxicity in MC3T3-E1 cells
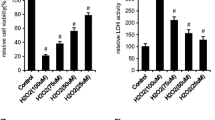
To study whether D3R would protect MC3T3-E1 cells against oxidative damage, we treated cells with increasing D3R concentrations (10−5–10−11 M) 24 h before t-BHP (250 µM for 3 h), a cytotoxic organic hydroperoxide used in our previous studies [21, 31] to induce oxidative damage. As expected, t-BHP significantly reduced MC3T3-E1 cell viability of 30%, as compared with control group as assessed by the MTT test. Pre-treatment with D3R significantly reduced the oxidative damage induced by t-BHP with a maximal protective action at 10−9 M (Fig. 3a). Similar results were obtained when we examined the ability of D3R to counteract cytotoxicity induced by AMA which generates ROS by inhibiting the mitochondrial electron transport chain complex III (Fig. 3b). When we examined the effect of D3R on t-BHP-induced apoptosis, we found that pre-treatment (24 h before) with 10−9 M D3R significantly reduced t-BHP (250 µM, for 3 h)-induced cell apoptosis (Fig. 4b). The protective action of D3R against typical nuclear changes associated with cell apoptosis induced by t-BHP is reported in Fig. 4a.

Effects of D3R (10−11–10−5 M) on t-BHP (a) or AMA-induced (b) cytotoxicity in MC3T3-E1 cells. Cells were pre-incubated with D3R 24 h before treatment with t-BHP (250 µM for 3 h) or AMA (70 µM for 24 h). Cell viability was measured by MTT assay. Data are expressed as the percentage relative to control and are the mean ± SEM of four replicates within a single experiment. ***p < 0.001 vs controls; °p < 0.05, °°p < 0.01, °°°p < 0.0001 vs t-BHP or AMA

Effects of D3R on apoptosis induced by t-BHP in MC3T3-E1 cells. Cells were pre-incubated with D3R (10−9 M) for 24 h and then treated with t-BHP (250 µM for 3 h). Apoptosis was detected by Hoechst 33,258 staining. a Panels are representative of condensed fragmented nuclei characteristic of apoptosis. Images were taken at ×20 magnification. b Quantification of apoptosis. Values are the mean ± SEM of duplicate determinations (200 cells each) of four independent experiments. ***p < 0.001 vs controls; °°°p < 0.001 vs t-BHP
Since cytoskeleton changes could be considered as a powerful tool to study cell apoptotic death in osteoblasts, we studied the effect of pre-treatment with D3R on t-BHP-induced morphological changes in MC3T3-E1 cells by examining the arrangement of the actin fibres with Phalloidin-FITC. As shown in Fig. 5, untreated MC3T3-E1 cells have a well-organized cytoskeleton with actin microfilaments placed in the direction of their main axis. D3R did not modify normal cytoskeletal morphology, but prevented the MC3T3-E1 shape modification and the rearrangement of the actin network observed in the presence of t-BHP.

Protective effects of D3R on cytoskeleton alterations induced by t-BHP in MC3T3-E1 cells. Cells were pre-incubated with D3R (10−9 M) 24 h before treatment with t-BHP (250 µM for 3 h). Representative images of cytoskeleton alterations evaluated with Axioplan fluorescence microscope (magnification ×20) detected by Phalloidin-FITC staining of the actin protein
Effect of D3R on ROS levels and detoxifying system
The significant increase in the intracellular ROS levels induced by t-BHP was prevented by D3R treatment. In D3R-treated cells, in fact, ROS levels were similar to those detected in control-treated cells (Fig. 6a). It is well known that ROS-mediated apoptotic signalling is associated with decreased cellular GSH levels and loss of cellular redox balance. When MC3T3-E1 cells were treated with t-BHP almost 60% reduction in GSH/GSSG ratio occurred (Fig. 6d) due to a decrease in GSH (Fig. 6b) and an increase in GSSG (Fig. 6c) levels. D3R treatment significantly reduced GSSG levels and prevented the reduction in GSH/GSSG ratio which remained similar to that detected in control-treated MC3T3-E1 cells.

Protective effects of D3R on intracellular ROS generation and cellular redox imbalance induced by t-BHP in MC3T3-E1 cells. Cells were pre-incubated with D3R (10− 9 M) 24 h before treatment with t-BHP (250 µM for 3 h). The intracellular ROS concentration was measured using CM-DCFA assay. Intracellular content of GSH and GSSG were measured using o-phthalaldehyde (OPT) fluorimetric assay and GSH/GSSG ratio was determined. Data are expressed as the percentage relative to control and are the mean ± SEM of six replications. *p < 0.05, **p < 0.01, ***p < 0.001 vs controls; °p < 0.05, °°p < 0.01 vs t-BHP
Inhibition of PI3K/Akt pathway block the protective effect of D3R
To study the involvement of PI3K/Akt in the D3R-mediated protection against oxidative damage, we pre-treated MC3T3-E1 cells with LY294002, a PI3K/Akt specific inhibitor. As shown in Fig. 7, LY294002 (10 µM, 1 h before D3R) worsened t-BHP cytotoxicity and the completely removed the protective action of D3R against t-BHP-induced oxidative damage.

Effects of pre-treatment with LY294002, a PI3-K antagonist, on D3R protective effects against t-BHP-induced MC3T3-E1 cytotoxicity. Cells were pre-incubated with LY294002 (10 µM) 1 h before D3R (10−9 M 24 h) and t-BHP (250 µM for 3 h) treatments. Cell viability was measured by MTT assay. Data are expressed as the percentage relative to control and are the mean + SEM of four replicates within a single experiment. ap < 0.001 vs control; bp < 0.001 vs t-BHP, cp < 0.001 vs D3R + t-BHP
Effect of D3R on t-BHP-induced cytotoxicity in differentiated MC3T3-E1 cells
To ascertain whether D3R is able to prevent MC3T3-E1 dysfunction induced by t-BHP, MC3T3-E1 cells were cultured in differentiation-inducing medium, as described in “Material and methods” section. Differentiated cells were incubated with D3R (10−9 M) 24 h before t-BHP challenge. We used t-BHP 125 µM, for 3 h, since differentiated MC3T3-E1 cells were more sensitive to damage than their undifferentiated counterparts (data not shown). t-BHP significantly reduced both collagen content (31%) and ALP activity (55%) as compared with control-treated cells. Pre-treatment with D3R significantly protects against t-BHP-induced cytotoxicity being collagen and ALP activity significantly higher than those detected in t-BHP-treated cells (Fig. 8).

D3R (10−9 M, 24 h before) prevents the t-BHP-induced decreases in ALP activity and collagen content in differentiated MC3T3-E1cells. ALP activity (a) and collagen content (b) were evaluated 24 or 48 h after treatment with t-BHP (125 µM for 3 h). Data are expressed as the percentage relative to control and are the mean ± SEM of six replications. ***p < 0.001 vs controls; °°p < 0.01, °°°p < 0.001 vs t-BHP
Effect of D3R on t-BHP-induced RANKL expression
As expected, treatment with t-BHP induced a strong reduction in the expression of OPG and a significant increase in the expression of RANKL. Pre-treatment with D3R failed to modify the action of t-BHP on mRNA levels of OPG and RANKL and did not reverse the relative ratio of OPG to RANKL, considered the critical determinant in the regulation of osteoclast biology (Fig. 9).

Effects of D3R (10−9 M) on osteoprotegerin (OPG) and RANKL expression measured by real-time PCR. Cells were pretreated with D3R for 24 h before t-BHP (250 µM for 3 h) and mRNAs were collected. 100 ng of cDNA from each sample were amplified using the SYBR® Green Chemistry and a specific set of primers for OPG and RANKL. Each sample was evaluated in triplicate. Data were normalized for GAPDH and expressed as mean ± SEM of the relative amounts (in percentage) of gene/GAPDH of each sample vs the mean control value (panel a, b). For the panel c a ratio between the OPG and RANKL expression was calculated. Four replicates were performed for each experimental point and the experiments were repeated twice. *p < 0.05, **p < 0.01, ***p < 0.001 vs controls
Discussion
In the present study we have showed that purified aubergine skin extracts of D3R, are able to induce osteoblast viability and osteogenic differentiation in basal conditions and to protect MC3T3-E1 cells against oxidative damage induced by two different compounds: t-BHP, and AMA. The organic hydroperoxide t-BHP generates reactive intermediates within cells which damage several biomolecules including lipids, thiols, DNA and proteins [35]. AMA generates ROS by inhibiting the mitochondrial electron transport chain complex III [36].
The positive action of D3R on osteoblast activity could be due to an increased expression of osteoblast differentiation markers such as ALP, Col1A1 and OC probably linked to an activation of the Wnt/β catenin signalling pathway.
Incubation of MC3T3-E1 cells with t-BHP induced a significant increase in ROS generation leading to reduced cells viability and apoptotic cell death. D3R treatment reduced intracellular ROS levels and prevented redox state alteration induced by t-BHP by increasing GSH. We also found that PI3K/Akt pathway is involved in the protective effect of D3R against t-BHP cytotoxicity since it is removed by pre-treatment with LY294002 a specific PI3K/Akt inhibitor.
When we examined the ability of D3R to modulate osteoblast/osteoclast crosstalk, we found a lack of effect of D3R on the expression of OPG/RANKL system activated by t-BHP.
Osteoblasts play a pivotal role in bone formation which involves osteoblast proliferation, differentiation and mineralization [37]. The process of osteoblast differentiation is characterized by a series of temporally and spatially coordinated events.
In the early stage, cells proliferate and the matrix matures. ALP is considered an early phenotypic marker of osteoblastogenesis [38]. ALP hydrolysed a variety of phosphate components releasing inorganic phosphate into the extracellular matrix [39]. OC a late-marker of differentiation can bind Ca2+ to regulate calcium ion homeostasis and bone mineralization [40]. Collagen type I is a necessary initiator in MC3T3-E1 cell differentiation and a required substrate for matrix mineralization [41]. D3R increased the expression of ALP, OC and Col1A1 suggesting that D3R is able to stimulate osteoblast differentiation from the early to the terminal stage, up-regulating both maturation and differentiation of MC3T3-E1 cells.
The Wnt/β-catenin signalling pathway is one of the most important regulators of osteoblast proliferation, differentiation and mineralization [42]. Targeting components of such pathway will influence bone formation and could have clinical applications including improving osteoblastogenesis in osteoporosis.
Wnt canonical signalling pathway relies on the cytosolic stabilization of β-catenin, which is a 130 amino acid protein that, in the absence of Wnt proteins, is phosphorylated by a glycogen synthase kinase (GSK-3 β) and degraded by the proteosomal machinery. Activation of the canonical Wnt signalling by Wnt proteins prevents the proteasomal degradation of β-catenin and promotes its association with TCF/Lef family of transcription factors and the expression of Wnt target genes [43]. Wnt/β-catenin signalling pathway can induce in MC3T3-E1 cells several osteoblast differentiation markers including ALP and collagen [44, 45]. We can only speculate that D3R could increase osteoblast differentiation by activating the Wnt/β-catenin signalling pathway, since we have only data regarding the D3R-induced upregulation of β-catenin mRNA and not β-catenin protein levels. This is a limitation of our study since it has been reported that the amount of β-catenin protein levels depends on the state of β-catenin destruction complex rather than the expression of β-catenin mRNA [46]. Considering the complexity of the Wnt/ β-catenin signalling pathway, further studies will be required to examine the effects of D3R on β-catenin protein levels both in the cytoplasm and in the nucleus and its ability to inactivate the destruction complex leading to GSK-3β phosphorylation and inactivation.
The potential utility of D3R as promoter of bone formation was further investigated in condition of oxidative stress. Several epidemiological evidence in humans and experimental studies in rodents indicate that oxidative stress induced by ROS, plays an important role in the development of osteoporosis [2]. ROS, in fact, have been reported to impair osteoblast activity and to contribute to the negative skeletal balance occurring during aging.
In the present study, we have shown that D3R protects MC3T3-E1 osteoblastic like cells from t-BHP-induced cytotoxicity by preventing cell death and apoptosis as shown by MTT assay and Hoechst 33,258 staining. Furthermore, D3R prevented the negative effects of t-BHP on osteoblastic function and recovered its changes in the cytoskeleton organization characterized by actin fibres destruction [32]. D3R exerts its protective effect against t-BHP-induced oxidative damage by reducing intracellular ROS and increasing intracellular antioxidant factors such as glutathione. GSH is an important intracellular antioxidant and redox potential regulator that functions by scavenging free radicals and converted itself to its oxidized form GSSG, thus protecting cells from oxidative stress [47].
t-BHP-treated cells showed an impairment of cellular glutathione redox status with a decrease in GSH levels along with the increase in its metabolite GSSG. Pre-treatment with D3R prevented this alteration and GHS redox status was maintained to a level comparable to that detected in control-treated MC3T3-E1 cells. To investigate the cellular pathways involved in D3R protection against oxidative stress we explore the possible involvement of the PI3K/Akt pathway which is implicated in the growth and survival of a range of cell types including osteoblasts [48] and participates to the prevention of oxidative stress-induced apoptosis [49].
We found that inhibition of PI3K/Akt pathway by LY294002 completely removed the protective effect of D3R against t-BHP-induced oxidative damage. Further studies will be required to clarify the Akt survival signals activated by D3R in MC3T3-E1 cells. The PI3K/Akt signalling pathway, in fact, plays a crucial role in the control of cell survival due to its direct capacity to activate several signalling molecules or to cross-talk with other signalling pathways and transcriptional networks.
PI3K/Akt signalling is involved in Nrf2-dependent transcription, and Nrf2 regulates the expression of antioxidant genes including GSH. PI3K/Akt can also regulates not only down-stream effectors such Bcl-2 family proteins which play an important anti apoptotic role [50], but also the canonical Wnt signalling pathway by inhibiting the phosphorylation of GSK-3β thus resulting in the activation of. β-catenin [51, 52]. Furthermore, Akt has been reported to increase β-catenin transcriptional capacity due to a direct ability of Akt to phosphorylate β-catenin [53]. It is possible that the protective effect of D3R against oxidative damage could involve, at least in part, the PI3K/Akt and β-catenin pathways, since preliminary data indicate that the increased expression of β-catenin mRNA induced by D3R was prevented by LY294002 (data not shown).
As previously reported [54] differentiated MC3T3-E1 osteoblast-like cells show greater sensitivity to ROS than their undifferentiated counterpart. These findings support the possibility that in normal or pathological aging, osteoblasts potentially become more vulnerable to ROS levels as they mature, which could explain the reason why older people are more likely to suffer bone fractures. The evidence that D3R significantly prevented the toxic effect of t-BHP on collagen content and ALP activity indicate that D3R could represent an interesting approach in the management of osteoblastic dysfunction associated with increased oxidative stress.
As far as the influence of anthocyanin chemical structures in relation to their cellular antioxidant activities is concerned, previous studies have shown that the radical scavenging activity of anthocyanins are correlated with the largest number of hydroxyl groups in B ring [55] and is influenced by the glycosylation pattern [24]. Previous studies have reported a higher scavenging activity of nasunin compared with D3R by using t-BHP as a free radical initiator [56] and the oxidative burst of human polymorphonuclear neutrophils [31]. In the present study on MC3T3-E1 cells, we found that the protective action of D3R against t-BHP-induced oxidative damage is comparable, in terms of potency, with that previously reported for nasunin [21]. Both purified functional food components, in fact, exert their maximum effect at 10−9 M, suggesting that their ability to protect cells against oxidative damage is independent from their glycosylation pattern constituted by simple 3-rutinose in D3R, with the addition of a glucose in five-position and of a coumaryl moiety linked to rutinose in nasunin. A possible explanation for these discrepancies could be due to the experimental protocol used in our experimental protocol showing an indirect antioxidant activity of both D3R and nasunin via increased expression/activity of defence mechanisms such GSH instead of a direct scavenging activity.
It is well known that ROS increase osteoclast number and activity and that reduced antioxidant defence in postmenopausal women is associated with bone loss [2]. Several antioxidant functional food compounds such as flavonoids, terpenoids and polyphenols have been reported to inhibit osteoclastogenesis via regulating many factors involved in the process of osteoclast differentiation and maturation including transcription factors (NFATc1, c-Fos), signalling pathways such as NF-κB, MAPKs and PI3K/Akt [57] and OPG/RANKL system [58].
RANKL and OPG synthetized by osteoblasts. RANKL plays a critical role in stimulation of osteoclast differentiation and survival through its receptor RANK. The secretion of OPG by osteoblasts prevents RANKL/RANK interaction and results in inhibition of bone resorption. Therefore, a ratio of OPG/RANKL is considered a crucial factor for bone resorption [27].
At variance from previously reported for other functional food components [58], D3R did not significantly affect the expression of OPG and RANKL in basal conditions and did not prevent the increased expression of RANKL induced by t-BHP in MC3T3-E1 cells. We cannot rule out the possibility that D3R could be involved in osteoblast/osteoclast crosstalk for several reasons. At first, we have to confirm the lack of effect of D3R on OPG/RANKL ratio by measuring the effects of D3R on OPG and RANKL protein amount. Furthermore, considering that oxidative stress could stimulate the inflammatory reaction via activation of pro-inflammatory cytokines [59] it could be interesting to study the effects of D3R on pro-inflammatory cytokines levels which, besides OPG/RANKL, regulate the cellular events involved in the bone resorption [60]. However, to address this point, we have to plan new experiments in MC3T3-E1 cells aimed to study the effects of t-BHP (250 µM, 3 h) on pro-inflammatory cytokines production and the possible effects of pre-treatment with D3R. The link between t-BHP-induced oxidative damage and production of IL 6 and TNF-α has been reported only in endothelial cells [61] using a different experimental condition (t-BHP 50 µM, 24 h). Furthermore in the experiments performed by Lee and colleagues [62] showing an inhibitory activity of berry anthocyanins on IL1b and TNF-α mRNAs levels, was used a blackcurrant fraction containing D3R (44%) and cyanidin-3 rutinoside (17%) instead of purified D3R as used in the present paper.
Previous in vivo studies have shown that commercial delphinidin intake protects against ovariectomy-induced osteopenia in mice by inhibiting osteoclast activity [10]. This discrepancy could be due to different experimental conditions, and/or to the different type of anthocyanin used. Moriwaki and colleagues [10], in fact used concentrations (0.8–66 µM) against the nM ranges used in the present experiment and examined the direct action of delphinidin on osteoclast formation and activity instead of an indirect modulatory effect of osteoblast on osteoclast through OPG/RANKL. Furthermore, it is possible that the different behaviour of D3R and delphinidin on osteoclastogenesis could be due to their very different chemical structure, since the delphinidin is the aglycon moiety of D3R, differentiated by the presence of rutinose in three-position. Further studies will be required to examine the effect of the lack or a different pattern of glycosylation on osteoclast function.
It has to be pointed out that, often, the natural extracts obtained in optimal conditions, contain the anthocyanins in glycosylated forms, increasing their stability, with only a minor part of the respective aglycons that, if present in significant amounts, could be considered as artefacts [63].
Conclusions
To our best knowledge, we have provided the first evidence that D3R promotes osteoblastic function by increasing the expression of bone specific matrix proteins (ALP, OC and Col1A1) and protects MC3T3-E1 cells against t-BHP cytotoxicity via its antioxidant capacity. The D3R protective effects against t-BHP cytotoxic depend on the activation of the PI3K/Akt signalling pathway.
These results suggest that purified D3R could be a potential preventive agent against oxidative damage in osteoblastic cells. As the present study was about the in vitro effects of D3R, further in vivo studies are needed to investigate the ability of dietary D3R supplement to regulate osteoblast function in bone debilitating diseases such as osteoporosis linked to increased oxidative damage.
References
Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (2007) Trends in oxidative aging theories. Free Radic Biol Med 43(4):477–503
Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O’Brien CA, Bellido T, Parfit AM, Weinstein RS, Jilka RL, Manolagas SC (2007) Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 282(37):27285–27297
Basu S, Michaëlsson K, Olofsson H, Johansson S, Melhus H (2001) Association between oxidative stress and bone mineral density. Biochem Biophys Res Commun 288(1):275–279
D’Amelio P, Cristofaro MA, Tamone C, Morra E, Di Bella S, Isaia G, Grimaldi A, Gennero L, Gariboldi A, Ponzetto A, Pescarmona GP, Isaia GC (2008) Role of iron metabolism and oxidative damage in postmenopausal bone loss. Bone. 43(6):1010–1015. https://doi.org/10.1016/j.bone.2008.08.107
Maggio D, Barabani M, Pierandrei M, Polidori MC, Catani M, Mecocci P, Senin U, Pacifici R, Cherubini A (2003) Marked decrease in plasma antioxidants in aged osteoporotic women: results of a cross-sectional study. J Clin Endocrinol Metab 88(4):1523–1527
Jilka RL, Almeida M, Ambrogini E, Han L, Roberson PK, Weinstein RS, Manolagas SC (2010) Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell 9(5):851–867. https://doi.org/10.1111/j.1474-9726.2010.00616.x
Slavin JL, Lloyd B (2012) Health benefits of fruits and vegetables. Adv Nutr 3(4):506–516. https://doi.org/10.3945/an.112.002154
Shen CL, von Bergen V, Chyu MC, Jenkins MR, Mo H, Chen CH, Kwun IS (2012) Fruits and dietary phytochemicals in bone protection. Nutr Res 32(12):897–910. https://doi.org/10.1016/j.nutres.2012.09.018
Hubert PA, Lee SG, Lee SK, Chun OK (2014) Dietary polyphenols, berries, and age-related bone loss: a review based on human, animal, and cell studies. Antioxidants (Basel) 3(1):144–158. https://doi.org/10.3390/antiox3010144
Moriwaki S, Suzuki K, Muramatsu M, Nomura A, Inoue F, Into T, Yoshiko Y, Niida S (2014) Delphinidin, one of the major anthocyanidins, prevents bone loss through the inhibition of excessive osteoclastogenesis in osteoporosis model mice. PLoS One 9(5):e97177. https://doi.org/10.1371/journal.pone.0097177
Hardcastle AC, Aucott L, Reid DM, Macdonald HM (2011) Associations between dietary flavonoid intakes and bone health in a Scottish population. J Bone Miner Res 26(5):941–947. https://doi.org/10.1002/jbmr.285
Sacco SM, Horcajada MN, Offord E (2013) Phytonutrients for bone health during ageing. Br J Clin Pharmacol 75(3):697–707. https://doi.org/10.1111/bcp.12033
Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR (1990) Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest 85(3):632–639
Lean JM, Jagger CJ, Kirstein B, Fuller K, Chambers TJ (2005) Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 146(2):728–735
Rodda SJ, McMahon AP (2006) Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133(16):3231–3244
Van der Horst A, Burgering BM (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 8(6):440–450
Cao GH, Sofic E, Prior RL (1996) Antioxidant capacity of tea and common vegetables. J Agric Food Chem 44(11):3426–3431
Akanitapichat P, Phraibung K, Nuchklang K, Prompitakkul S (2010) Antioxidant and hepatoprotective activities of five eggplant varieties. Food Chem Toxicol 48(10):3017–3021. https://doi.org/10.1016/j.fct.2010.07.045
Kayamori F, Igarashi K (1994) Effects of dietary nasunin on the serum-cholesterol level in rats. Biosci Biotechnol Biochem 58(3):570–571
Mennella G, Lo Scalzo R, Fibiani M, D’Alessandro A, Francese G, Toppino L, Acciarri N, de Almeida AE, Rotino GL (2012) Chemical and bioactive quality traits during fruit ripening in eggplant (S. melongena L.) and allied species. J Agric Food Chem 60(47):11821–11831
Casati L, Pagani F, Braga PC, Lo Scalzo R, Sibilia V (2016) Nasunin, a new player in the field of osteoblast protection against oxidative stress. J Funct Foods 23:474–484
Yi L, Chen CY, Jin X, Zhang T, Zhou Y, Zhang QY, Zhu JD, Mi MT (2012) Differential suppression of intracellular reactive oxygen species-mediated signaling pathway in vascular endothelial cells by several subclasses of flavonoids. Biochimie 94(9):2035–2044. https://doi.org/10.1016/j.biochi.2012
He J, Giusti MM (2010) Anthocyanins: natural colorants with health-promoting properties. Annu Rev Food Sci Technol 1:163 – 87 doi. https://doi.org/10.1146/annurev.food.080708.100754
Jing P, Qian B, Zhao S, Qi X, Ye L, Mónica Giusti M, Wang X (2015) Effect of glycosylation patterns of Chinese eggplant anthocyanins and other derivatives on antioxidant effectiveness in human colon cell lines. Food Chem 172:183–189. https://doi.org/10.1016/j.foodchem.2014.08.100
Azuma K, Ohyama A, Ippoushi K, Ichiyanagi T, Takeuchi A, Saito T, Fukuoka H (2008) Structures and antioxidant activity of anthocyanins in many accessions of eggplant and its related species. J Agric Food Chem 56(21):10154–10159. https://doi.org/10.1021/jf801322m
Quarles LD, Yohay DA, Lever LW, Caton R, Wenstrup RJ (1992) Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: an in vitro model of osteoblast development. J Bone Miner Res 7(6):683–692
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423(6937):337–342
Mrak E, Casati L, Pagani F, Rubinacci A, Zarattini G, Sibilia V (2015) Ghrelin increases beta-catenin level through protein kinase A activation and regulates OPG expression in rat primary osteoblasts. Int J Endocrinol 2015:547473. https://doi.org/10.1155/2015/547473
Bai XC, Lu D, Liu AL, Zhang ZM, Li XM, Zou ZP, Zeng WS, Cheng BL, Luo SQ (2005) Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J Biol Chem 280(17):17497–17506
Noda Y, Kneyuki T, Igarashi K, Mori A, Packerm L (2000) Antioxidant activity of nasunin, an anthocyanin in eggplant peels. Toxicology 148(2–3):119–123
Braga PC, Lo Scalzo R, Dal Sasso M, Lattuada N, Greco V, Fibiani M (2016) Characterization and antioxidant activity of semi-purified extracts and pure delphinidin-glycosides from eggplant peel (Solanum melongena L.). J Funct Foods 20:411–421
Dieci E, Casati L, Pagani F, Celotti F, Sibilia V (2014) Acylated and unacylated ghrelin protect MC3T3-E1 cells against tert-butyl hydroperoxide-induced oxidative injury: pharmacological characterization of ghrelin receptor and possible epigenetic involvement. Amino Acids 46(7):1715–1725
Zhang JK, Yang L, Meng GL, Fan J, Chen JZ, He QZ, Chen S, Fan JZ, Luo ZJ, Liu J (2012) Protective effect of tetrahydroxystilbene glucoside against hydrogen peroxide-induced dysfunction and oxidative stress in osteoblastic MC3T3-E1 cells. Eur J Pharmacol 689(1–3):31–37. https://doi.org/10.1016/j.ejphar.2012.05.045
Dong CL, Liu HZ, Zhang ZC, Zhao HL, Zhao H, Huang Y, Yao JH, Sun TS (2015) The influence of microRNA-150 in osteoblast matrix mineralization. J Cell Biochem 116(12):2970–2979. https://doi.org/10.1002/jcb.25245
Brambilla L, Cantoni O (1998) Mitochondrial formation of hydrogen peroxide is causally linked to the antimycin A-mediated prevention of tert-butylhydroperoxide-induced U937 cell death. FEBS Lett 431(2):245–249
Aon MA, Cortassa S, Marbán E, O’Rourke B (2003) Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278(45):44735–44744. https://doi.org/10.1074/jbc.M302673200
Titorencu I, Pruna V, Jinga VV, Simionescu M (2014) Osteoblast ontogeny and implications for bone pathology: an overview. Cell Tissue Res 355(1):23–33. https://doi.org/10.1007/s00441-013-1750-3
Stein GS, Lian JB, Owen TA (1990) Relationship of cell growth to the regulation of tissue-specific gene expression during osteoblast differentiation. FASEB J 4(13):3111–3123
Farley JR, Hall SL, Tanner MA, Wergedal JE (1994) Specific activity of skeletal alkaline phosphatase in human osteoblast-line cells regulated by phosphate, phosphate esters, and phosphate analogs and release of alkaline phosphatase activity inversely regulated by calcium. J Bone Miner Res 9(4):497–508
Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, Bradley A, Karsenty G (1996) Increased bone formation in osteocalcin-deficient mice. Nature 382(6590):448–452
Rodan GA, Noda M (1991) Gene expression in osteoblastic cells. Crit Rev Eukaryot Gene Expr 1(2):85–98
Bodine PV, Komm BS (2006) Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord 7:33–39
Novak A, Dedhar S (1999) Signaling through beta-catenin and Lef/Tcf. Cell Mol Life Sci 56(5–6):523–537
Westendorf JJ, Kahler RA, Schroeder TM (2004) Wnt signaling in osteoblasts and bone diseases. Gene 341:19–39
Tian Y, Xu Y, Fu Q, He M (2011) Parathyroid hormone regulates osteoblast differentiation in a Wnt/β-catenin-dependent manner. Mol Cell Biochem 355(1–2):211–216. https://doi.org/10.1007/s11010-011-0856-8
Mei G, Zou Z, Fu S, Xia L, Zhou J, Zhang Y, Tuo Y, Wang Z, Jin D (2014) Substance P activates the Wnt signal transduction pathway and enhances the differentiation of mouse preosteoblastic MC3T3-E1 cells. Int J Mol Sci 15(4):6224–6240. https://doi.org/10.3390/ijms15046224
Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N, Fujii J, Myers A, Biswal S (2009) Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med 46(4):443–453
McGonnell IM, Grigoriadis AE, Lam EW, Price JS, Sunters A (2012) A specific role for phosphoinositide 3-kinase and AKT in osteoblasts? Front Endocrinol 3:88. https://doi.org/10.3389/fendo.2012.00088
Wang B, Shravah J, Luo H, Raedschelders K, Chen DD, Ansley DM (2009) Propofol protects against hydrogen peroxide-induced injury in cardiac H9c2 cells via Akt activation and Bcl-2 up-regulation. Biochem Biophys Res Commun 389(1):105–111. https://doi.org/10.1016/j.bbrc.2009.08.097
Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE (2000) Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem 275(15):10761–10766
Smith E, Frenkel B (2005) Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3beta-dependent and -independent manner. J Biol Chem 280(3):2388–2394
Sunters A, Armstrong VJ, Zaman G, Kypta RM, Kawano Y, Lanyon LE, Price JS (2010) Mechano-transduction in osteoblastic cells involves strain-regulated estrogen receptor alpha-mediated control of insulin-like growth factor (IGF) I receptor sensitivity to Ambient IGF, leading to phosphatidylinositol 3-kinase/AKT-dependent Wnt/LRP5 receptor-independent activation of beta-catenin signaling. J Biol Chem 285(12):8743–8758. https://doi.org/10.1074/jbc.M109.027086
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282(15):11221–11229
Fatokun AA, Stone TW, Smith RA (2008) Responses of differentiated MC3T3-E1 osteoblast-like cells to reactive oxygen species. Eur J Pharmacol 587(1–3):35–41. https://doi.org/10.1016/j.ejphar.2008.03.024
Yi L, Chen CY, Jin X, Mi MT, Yu B, Chang H, Ling WH, Zhang T (2010) Structural requirements of anthocyanins in relation to inhibition of endothelial injury induced by oxidized low-density lipoprotein and correlation with radical scavenging activity. FEBS Lett 584(3):583–90. https://doi.org/10.1016/j.febslet.2009.12.006.
Yoshiki Y, Okubo K, Igarashi K (1995) Chemiluminescence of anthocyanins in the presence of acetaldehyde and tert-butyl hydroperoxide. J Biolumin Chemilumin 10(6):335–338. https://doi.org/10.1002/bio.1170100605
An J, Yang H, Zhang Q, Liu C, Zhao J, Zhang L, Chen B (2016) Natural products for treatment of osteoporosis: the effects and mechanisms on promoting osteoblast-mediated bone formation. Life Sci 147:46–58. https://doi.org/10.1016/j.lfs.2016.01.024
An J, Hao D, Zhang Q, Chen B, Zhang R, Wang Y, Yang H (2016) Natural products for treatment of bone erosive diseases: the effects and mechanisms on inhibiting osteoclastogenesis and bone resorption. Int Immunopharmacol 36:118–131. https://doi.org/10.1016/j.intimp.2016.04.024
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol Med 49(11):1603–1616. https://doi.org/10.1016/j.freeradbiomed.2010.09.006
Knowles HJ, Athanasou NA (2009) Canonical and non-canonical pathways of osteoclast formation. Histol Histopathol 24(3):337–346. https://doi.org/10.14670/HH-24.337
Liu H, Mao P, Wang J, Wang T, Xie CH2 (2016) Azilsartan, an angiotensin II type 1 receptor blocker, attenuates tert-butyl hydroperoxide-induced endothelial cell injury through inhibition of mitochondrial dysfunction and anti-inflammatory activity. Neurochem Int 94:48–56. https://doi.org/10.1016/j.neuint.2016.02.005
Lee SG, Kim B, Yang Y, Pham TX, Park YK, Manatou J, Koo SI, Chun OK, Lee JY (2014) Berry anthocyanins suppress the expression and secretion of proinflammatory mediators in macrophages by inhibiting nuclear translocation of NF-κB independent of NRF2-mediated mechanism. J Nutr Biochem 25(4):404–411. https://doi.org/10.1016/j.jnutbio.2013.12.001
Bakker J, Timberlake CF (1985) The distribution of anthocyanins in grape skin extracts of port wine cultivars as determined by high performance liquid chromatography. J Sci Food Agric 36(12):1315–1324
Acknowledgements
This work was supported by funds from PROGETTO CARIPLO GIOVANI 2015−0834 to Lavinia Casati. The authors thank the expertise and technical support of Dr. Giuseppe L. Rotino (CREA-ORL, Montanaso Lombardo) for providing the aubergine fruits and Prof. Giovanna Speranza (Dipartimento di Chimica, Università degli Studi di Milano) for the analysis by 1H-NMR of D3R crystals purity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Casati, L., Pagani, F., Fibiani, M. et al. Potential of delphinidin-3-rutinoside extracted from Solanum melongena L. as promoter of osteoblastic MC3T3-E1 function and antagonist of oxidative damage. Eur J Nutr 58, 1019–1032 (2019). https://doi.org/10.1007/s00394-018-1618-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-018-1618-0