
Abstract
Increasing evidence suggests a role for oxidative stress in age-related decrease in osteoblast number and function leading to the development of osteoporosis. This study was undertaken to investigate whether ghrelin, previously reported to stimulate osteoblast proliferation, counteracts tert-butyl hydroperoxide (t-BHP)-induced oxidative damage in MC3T3-E1 osteoblastic cells as well as to characterize the ghrelin receptor (GHS-R) involved in such activity. Pretreatment with ghrelin (10−7–10−11 M) significantly increased viability and reduced apoptosis of MC3T3-E1 cells cultured with t-BHP (250 μM) for three hours at the low concentration of 10−9 M as shown by MTT assay and Hoechst-33258 staining. Furthermore, ghrelin prevented t-BHP-induced osteoblastic dysfunction and changes in the cytoskeleton organization evidenced by the staining of the actin fibers with Phalloidin-FITC by reducing reactive oxygen species generation. The GHS-R type 1a agonist, EP1572 (10−7–10−11 M), had no effect against t-BHP-induced cytotoxicity and pretreatment with the selective GHS-R1a antagonist, d-Lys3-GHRP-6 (10−7 M), failed to remove ghrelin (10−9 M)-protective effects against oxidative injury, indicating that GHS-R1a is not involved in such ghrelin activity. Accordingly, unacylated ghrelin (DAG), not binding GHS-R1a, displays the same protective actions of ghrelin against t-BHP-induced cytotoxicity. Preliminary observations indicate that ghrelin increased the trimethylation of lys4 on histones H3, a known epigenetic mark activator, which may regulate the expression of some genes limiting oxidative damage. In conclusion, our data demonstrate that ghrelin and DAG promote survival of MC3T3-E1 cell exposed to t-BHP-induced oxidative damage. Such effect is independent of GHS-R1a and is likely mediated by a common ghrelin/DAG binding site.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aging is believed to be a major factor involved in the reduction of bone mass and quality leading to osteoporosis. As aging progresses, there is an increase in osteoblast and osteocyte apoptosis as well as a decrease in osteoblast number and bone formation (Manolagas 2010; Manolagas and Parfitt 2010). Thus, the amount of bone resorbed by osteoclasts is not fully restored with new bone deposited by osteoblasts, this imbalance causing bone loss (Seeman 2003).
Although the pathophysiological mechanisms underlying the adverse effects of aging on bone mass and strength are not well understood, previous studies have shown that oxidative stress resulting from excessive levels of reactive oxygen species (ROS) or dysfunction of antioxidant defense is involved in this disease (Almeida 2012; Manolagas 2010).
In mice, it has been reported that increased cellular oxidative stress induces low-turnover osteopenia (Nojiri et al. 2011) and that bone mass progressively declines in association with decreased antioxidant enzyme levels (Almeida et al. 2007). In the femora of ovariectomized rats, increased lipid peroxidation and decreased levels of enzymatic antioxidants were found (Muthusami et al. 2005). These experimental data fit well with clinical studies showing that increased oxidative stress is negatively associated with bone mineral density in aged men and women (Basu et al. 2001). Furthermore, elderly osteoporotic women showed high levels of lipid peroxidation (Ozgocmen et al. 2007) and a marked decrease in plasma levels of antioxidants as compared to age-matched controls (Maggio et al. 2003).
Formation of ROS occurs primarily in the mitochondria from the escape of electrons passing through the electron transport chain during normal metabolisms. ROS are also generated during fatty acid oxidation or in response to external stimuli such as inflammation. Increased mitochondrial damage with age may results in excessive ROS production that damages proteins, lipids and DNA, leading to cell death (Muller et al. 2007; Almeida et al. 2007).
Several studies have shown that free radicals and ROS can affect osteoblast growth and function (Bai et al. 2004; Manolagas 2010), suggesting the potential use of antioxidants in the prevention and/or treatment of osteoporosis.
Ghrelin, identified in 1999 as an endogenous ligand of the growth hormone secretagogue receptor (GHS-R), derives from a 117 amino acid pre-prohormone which is cleaved into a 94 amino acid proghrelin peptide. This proghrelin peptide is further cleaved and gives rise to two major forms of 28 amino acid ghrelin (Hosoda et al. 2000). The ghrelin peptide acylated at serine-3 by ghrelin-O-acyltransferase (GOAT) (Yang et al. 2008), usually referred as ghrelin in the literature, and a non-acylated form (DAG) which circulates at tenfold higher levels than ghrelin (Holmes et al. 2009; Patterson et al. 2005). The octanoylation is critical for the binding to the GHS-R 1a, in inducing GH secretion, food intake (Kojima and Kangawa 2005) and inhibition of gastric acid secretion (Sibilia et al. 2006). DAG, in fact, seems to be devoid of any endocrine and gastrointestinal activities.
Indeed, DAG was reported to exert some non-endocrine action, sharing with ghrelin a protective effect on cardiomyocytes and endothelial cells (Baldanzi et al. 2002) and an inhibitory action on inflammatory pain (Sibilia et al. 2012). Therefore, the existence of a ghrelin receptor subtype different from GHS-R 1a, yet to be identified (Seim et al. 2011), that could mediate some of the effects of both peptides has been suggested.
Since ghrelin inhibits oxidative damage in various peripheral tissues (El Eter et al. 2007; Kawczynska-Drozdz et al. 2006; Zhang et al. 2011; Tong et al. 2012) and in the brain (Obay et al. 2008), it has been proposed as an antioxidant agent.
The role of ghrelin, if any, in the protection against oxidative stress in osteoblasts has never been investigated previously. This is surprising, since ghrelin and synthetic GH secretagogues play a role in bone formation. Ghrelin and its receptor GHS-R 1a are expressed in several osteoblasts and osteoblastic cell lines, and ghrelin directly promotes both osteoblast proliferation and differentiation (Kim et al. 2005; Maccarinelli et al. 2005; Fukushima et al. 2005; Costa et al. 2011). Furthermore, long-term treatment with ghrelin (Fukushima et al. 2005) or synthetic GH secretagogues increases BMD (Svensson et al. 2000) and prevents gonadectomy-induced bone loss in the rat (Sibilia et al. 1999, 2002).
In the present study, we investigated the effects of ghrelin on the survival of osteoblastic MC3T3-E1 cells exposed to tert-butyl hydroperoxide (t-BHP). Tert-butyl hydroperoxide is an organic hydroperoxide widely used as a pro-oxidant to induce oxidative stress in a variety of cells (Altman et al. 1994; Sestili et al. 1998; Haidara et al. 2008).
In order to characterize the ghrelin receptor involved in ghrelin cytoprotection, we examined the effects of DAG, which does not bind the classic GHS-R1a (Bednarek et al. 2000; Ghigo et al. 2005) and the GHS-R 1a agonist, EP1572 (Broglio et al. 2002; Sibilia et al. 2006, 2012) on t-BHP-induced cytotoxicity. The ability of the specific GHS-R 1a antagonist, d-Lys3-GHRP-6 (Carreira et al. 2006; Muccioli et al. 2007; Sibilia et al. 2006, 2012), to reverse the ghrelin protective effects against oxidative stress was also examined. The possible effect of ghrelin on the intracellular levels of H3K4me3, an important epigenetic mark, has been also evaluated in a pilot experiment.
Materials and methods
Drugs
Ghrelin, DAG and EP1572 (Aib-DTrp-DgTrp-CHO) were synthesized by conventional solid-phase synthesis and purified to at least 98 % purity by HPLC by Neosystem (Strasburg, France). d-Lys3- GHRP-6 was purchased from Bachem AG (Budendorf, Switzerland). Tert-butyl hydroperoxide (t-BHP) and Trolox were purchased from Sigma-Aldrich Chemical, Italy. Trolox was dissolved in ethanol (70 %) with a final ethanol concentration in the medium less than 0.5 %.
Cell culture
Mouse MC3T3-E1 osteoblastic cells (ATCC catalog number CRL-2593) were cultured in Dulbecco’s modified Eagle’s medium high glucose (D-MEM, Euroclone, Italy), and supplemented with 10 % fetal bovine serum (FBS, Sigma-Aldrich Chemical, Italy), 1 % l-glutamine, 100 µg/ml streptomycin and 100 U/ml penicillin at 37 °C in 5 % CO2 atmosphere. This basic medium was changed every 3 days, and cells were subcultured weekly.
Cell viability assay
Cell was plated at the density of 3 × 103 cells/well in a 96-well culture plate. After treatment, cells were washed with phosphate-buffered saline (PBS, Euroclone, Italy) and incubated at 37 °C with 0.5 mg/ml 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich Chemical, Italy) for 3 h. The conversion of the tetrazolium salt MTT to a colored formazan was used to assess cell viability. After the supernatant was removed, dimethyl sulfoxide was added to each well and the absorbance was recorded by a microplate spectrophotometer (WALLAC) at 550 nm.
Cell phenotypic observations were made using Olympus TH4-200 inverted phase contrast microscope, fitted with a digital camera Olympus C-4040 zoom to record any change during treatment.
Hoechst staining of apoptotic cells
Morphological changes in the nuclear chromatin of apoptotic cells were detected by Hoechst-33258 (Sigma-Aldrich Chemical., Italy) staining. MC3T3-E1 cells (5 × 103 cells/well) were grown on 22-mm glass coverslips in 6-well plates. After treatment, the cells were fixed with 4 % formaldehyde in 0.12 M sucrose, permeabilized with 0.1 % TritonX100 in PBS for 5 min, and stained with 10 μg/ml Hoechst-33258 DNA dye for 5 min. The cells then visualized using an Axioplan fluorescence microscope. The blue fluorescent Hoechst 33258 is a cell permeable nucleic acid dye usually used to identify chromatin condensation and fragmentation by staining the condensed nuclei of apoptotic cells. Uniformly stained nuclei were scored as healthy, viable cells. Condensed or fragmented nuclei were considered as apoptotic. At least 200 cells were scored blindly without knowledge of their prior treatment.
Cytoskeleton rearrangement
MC3T3-E1 cell growth, fixation and permeabilization were carried out by the same procedure as mentioned above. The permeabilized cells were incubated with FICT-labeled phalloidin (diluted 1:100 in PBS) for 20 min at room temperature and mounted in Vectashield HardSet Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA). The images were obtained using an Axioplan fluorescence microscope.
Intracellular ROS production
ROS production was measured using 5(6)-carboxy-2′,7-dichlorofluorescin diacetate (CM-DCFA, Sigma-Aldrich Chemical, Italy 10 μM) according to the method described by Wang and Joseph (1999). The MC3T3–E1 cells were seeded in black 96-well plates and cultured for 24 h. On the day of the experiment, the cells were preincubated with ghrelin or DAG (10−9 M) for 2 h and loaded with CM-DCFA during the last 30 min of treatment. After CM-DCFA was removed, the cells were exposed to t-BHP (250 μM) for 3 h. DCF fluorescence was assessed with a spectrofluorophotometer (Victor™, PerkinElmer, Waltham, MA, USA) at the excitation (485 nm) and emission (530 nm) wavelengths.
Epigenetic studies
Samples for Western blotting of histones were prepared as follow: Cells were collected in PBS-EDTA, and cell pellets were homogenized in Laemmli buffer 1X (62,5 mM Tris–HCl pH 6.8, 7.5 % glycerol, 2 % SDS, 0.125 M dithiothreitol), subjected to ultrasonic bath for 10 min prior to heat samples at 95 °C for 5 min. The resulting whole cell extracts were then centrifuged at 13,200 rpm for 10 min, and supernatants were saved and used directly for immunoblotting. Protein extracts were resolved by SDS/15 %PAGE. Protein gels were electrotransferred to 0.2 μm pore nitrocellulose membranes by standard wet procedures. After transfer, membranes were stained with Ponceau S to verify the correct loading and transfer, and then blocked and probed overnight at 4 °C with the primary specific antibodies: anti-H3K4me3 (ref. Ab8580, Abcam) and anti-H4 (ref. 10158, Abcam). Membranes were processed with WesternDot™ 625 Western Blot Kits (Life Technologies) as described by the manufacturer. Fluorescent signals were quantified using ChemiDoc system (Bio-Rad).
Statistical analysis
Statistical analysis was performed with a statistic package (GraphPad Prism5, GraphPad Software San Diego, CA, USA). All data are represented as the mean ± SEM of four independent experiments. Differences between groups were assessed by one-way analysis of variance (ANOVA) followed by Bonferroni test when data were parametric. Nonparametric data were analyzed by a Kruskal–Wallis test followed by Dunn’s test. A probability of p < 0.05 was considered to be significant.
Results
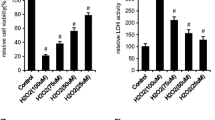
Figure 1 shows dose- and time-dependent effect of t-BHP on MC3T3-E1 cell viability as assessed by the MTT test. The concentration of 250 μM t-BHP for 3 h reduces cell viability of 59.5 ± 2.4 %, as compared with control group, and this effect remains at the same extent when the incubation was carried out for 6 h. The concentration of 250 μM t-BHP for 3 h was used in subsequent experiments. In our previous experiments, we have shown that ghrelin significantly increases cells viability starting from 6 h of incubation (data not shown). To avoid the confounding stimulatory action of ghrelin on cell viability, we pretreated MC3T3-E1 cells with increasing concentrations of the peptide 2 h before treatment with t-BHP. As shown in Fig. 2a, ghrelin exerts a maximal protective effect against t-BHP cytotoxicity when cells were incubated with 10−9 and 10−11 M. Greater ghrelin concentrations did not further increase its protective effect but, on the contrary, were less effective. We chose the concentration of 10−9 M ghrelin for further experiments. The effectiveness of our experimental conditions has been validated by the use of Trolox, a well-known antioxidant compound (Cos et al. 2003). Interestingly, we found that a concentration of 10−5 M Trolox is required to exert a protective action against t-BHP cytotoxicity comparable to that of 10−11 M ghrelin (Fig. 2b).

Time-and dose-dependent effects of t-BHP on MC3T3-E1 cell viability. Cells were treated with increasing concentrations of t-BHP (125–500 µM) and incubated up to 180 min. Data are expressed as the percentage relative to control and are the mean ± SEM of four replicates within a single experiment that was repeated at least three times. ***p < 0.001 vs. control; && p < 0.01 vs. t-BHP 250 µM

Effects of ghrelin (a) and Trolox (b) on t-BHP-induced cytotoxicity on MC3T3-E1 cells. Cells were preincubated with ghrelin (10−5–10−11 M) or Trolox (10−5–10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). Cell viability was measured by MTT assay. Data are expressed as the percentage relative to control and are the mean ± SEM of four replicates within a single experiment that was repeated at least three times. *p < 0.05, **p < 0.01, ***p < 0.001 vs. control; ••p < 0.01, •••p < 0.001 vs. t-BHP
We then investigated the effect of ghrelin on t-BHP-induced apoptosis. As shown in Fig. 3b, treatment with t-BHP (250 μM) for 3 h caused a significant induction of cell apoptosis (28.0 ± 3.3 %) as compared with control (5.9 ± 1.0 % apoptotic cells) that was significantly reduced by pretreatment (2 h before) with 10−9 M ghrelin (10.7 ± 1.3 %). The protective action of ghrelin against typical nuclear changes associated with cell apoptosis induced by t-BHP is reported in Fig. 3a.

Ghrelin inhibits apoptosis induced by t-BHP in MC3T3-E1 cells. Cells were preincubated with ghrelin (10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). Apoptosis was detected by Hoechst 33258 staining. a Panels are representative of condensed or fragmented nuclei characteristic of apoptosis. Images were taken at 20× magnification. b Quantification of apoptosis. Values are the mean ± SEM of duplicate determinations (200 cells each) of three independent experiments. ***p < 0.001 vs. control; •••p < 0.001 vs. t-BHP
In the studies performed to investigate the effects of t-BHP on the morphology of MC3T3-E1 cells, we have shown that MC3T3-E1 control monolayers are arranged in a star-like manner and present round nuclei. Many processes connect each cell with their neighboring. Treatment with t-BHP induced cytoplasm condensation and loss of cell–cell contact. In addition, many cells died and detached from the monolayer. Pretreatment with ghrelin decreased t-BHP-induced morphological changes (Fig. 4a).

Protective effects of ghrelin on morphological and cytoskeleton alterations induced by treatment with t-BHP in MC3T3-E1 cells. Cells were preincubated with ghrelin (10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). Representative images of a morphological changes evaluated with inverted phase contrast microscope (magnification 20×), b cytoskeleton alterations evaluated with Axioplan fluorescence microscope (magnification 20×) detected by Phalloidin-FITC staining of the actin protein
To test the possibility that t-BHP-induced morphological changes could be due to the cytoskeleton protein modifications, we examined the cytoskeleton network by staining the actin fibers with Phalloidin-FITC.
As shown in Fig. 4b, under normal conditions, MC3T3-E1 cells have actin microfilaments placed in the direction of their main axis. The MC3T3-E1 shape modification and the rearrangement of the actin network observed in the presence of t-BHP were prevented by ghrelin treatment. Ghrelin-treated MC3T3-E1 cells, in fact, displayed an actin filament pattern similar to that observed in control cells.
To examine whether the protective effect of ghrelin against t-BHP-induced cytotoxicity depends on an interaction with GHS-R1a, MC3T3-E1 cells were treated with increasing concentrations of the GHS-R1a agonist, EP1572. EP1572 had no effect on cell viability when administered alone (data not shown) and did not modify the reduction in cell viability induced by t-BHP (Fig. 5a). Furthermore, the selective GHS-R1a antagonist, d-Lys3-GHRP-6 (10−7 M), applied for 30 min before ghrelin (10−9 M) failed to remove the protective effects of ghrelin against oxidative injury (Fig. 5b).

Lack of involvement of the GHS-R 1a in the protective effect of ghrelin against t-BHP-induced cytotoxicity in MC3T3-E1 cells. a The GHS-R1a agonist, EP1572 (10−5–10−11 M) was administered 2 h before treatment with t-BHP (250 µM for 3 h). b MC3T3-E1 cells were pretreated with the GHS-R1a antagonist, d-Lys3-GHRP-6 (10−7 M), 30 min before ghrelin (10−9 M) and subsequently incubated for 2 h before t-BHP (250 µM, for 3 h). Cell viability was measured by MTT assay. Data are expressed as the percentage relative to control and are the mean ± SEM of four replicates within a single experiment that was repeated at least three times. ***p < 0.001 vs. control; •••p < 0.001 vs. t-BHP
The possibility that the cytoprotective effect of ghrelin is linked to an interaction with a receptor different from GHS-R1a was confirmed by the results obtained with DAG which did not bind GHS-R1a. Pretreatment with increasing DAG concentrations (10−5–10−11 M) significantly increased MC3T3-E1 viability as compared with t-BHP-treated cells (Fig. 6).

DAG protects MC3T3-E1 cells from t-BHP-induced cytotoxicity. The cells were preincubated with DAG (10−5–10−11 M) for 2 h before treatment with t-BHP (250 µM for 3 h). Cell viability was measured by MTT assay. Data are expressed as the percentage relative to control and are the mean ± SEM of four replicates within a single experiment that was repeated at least three times. *p 0.05, ***p 0.001 vs. control; •••p < 0.001 vs. t-BHP
DAG 10−9 M significantly reduced the proportion of apoptotic cells (13.1 ± 1.1 %) as compared with t-BHP-treated cells (26.0 ± 3.0 %, Fig. 7) and prevented both the typical morphological changes (Fig. 8a) and the disassembling of actin filaments (Fig. 8b) induced by the cytotoxic effect of t-BHP.

DAG inhibits apoptosis induced by t-BHP in MC3T3-E1 cells. Cells were preincubated with DAG (10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). Apoptosis was detected by Hoechst 33258 staining. a Panels are representative of condensed or fragmented nuclei characteristic of apoptosis. Images were taken at 20× magnification. b Quantification of apoptosis. Values are the mean ± SEM of duplicate determinations (200 cells each) of three independent experiments. ***p < 0.001 vs. control; •••p < 0.001 vs. t-BHP

Protective effects of DAG on morphological and cytoskeleton alterations induced by treatment with t-BHP in MC3T3-E1 cells. Cells were preincubated with DAG (10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). Representative images of a morphological changes evaluated with inverted phase contrast microscope (magnification 20×), b cytoskeleton alterations evaluated with Axioplan fluorescence microscope (magnification 20×) detected by Phalloidin-FITC staining of the actin protein
Intracellular ROS production plays an important role in t-BHP-induced oxidative injury. To study whether ghrelin and DAG exert their protective action against t-BHP by reducing ROS production, the intracellular ROS concentration was measured. CM-DCFA assay showed that treatment with t-BHP significantly increased intracellular ROS production in MC3T3-E1 cells. In the presence of ghrelin or DAG, ROS production was similar to the one detected in untreated control cells (Fig. 9).

Ghrelin and DAG reduce the generation of intracellular ROS induced by t-BHP in MC3T3-E1 cells. Cells were preincubated with ghrelin (10−9 M) or DAG (10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). The intracellular ROS concentration was measured using CM-DCFA assay. Data are expressed as the percentage relative to control and are the mean ± SEM of six replications. ***p < 0.001 vs. control; ••p < 0.01 vs. t-BHP
In the last series of experiments, we studied the possible effect of ghrelin on the cellular content of H3K4me3 in MC3T3-E1 cells treated with t-BHP. As shown in Fig. 10, ghrelin significantly increased the amount of trimethylation of lys4 on histones H3, an important epigenetic mark of gene activation.

Western blot analysis of H3K4me3 levels in MC3T3-E1 cells. Cells were preincubated with ghrelin (10−9 M) 2 h before treatment with t-BHP (250 µM for 3 h). The relative amount of H3K4me3 was normalized by the signal obtained for total H4 with an anti-H4 antibody. Data are expressed as the percentage relative to control and are the mean ± SEM. *p < 0.05 vs. control
Discussion
This study was aimed to evaluate the effects of ghrelin on t-BHP-induced cytotoxicity and to determine whether GHS-R1a mediates its protective effects against oxidative injury.
For this purpose, we used ghrelin, synthetic analogs acting as GHS-R1a agonist (EP1572) or antagonist, (d-Lys3-GHRP-6), and the natural ghrelin isoform DAG which does not bind GHS-R1a.
We have shown that ghrelin, in either the acylated or unacylated form, protects MC3T3-E1 cells from t-BHP-induced toxic activity raising the possibility that its protective effect could be independent of the presently known GHS-R1a. Accordingly, EP1572 has no effect on t-BHP-induced oxidative injury and the selective GHS-R1a antagonist, d-Lys3-GHRP-6, did not prevent the protective effect elicited by ghrelin treatment.
In recent years, the beneficial effects of ghrelin on bone tissue have received increasing attention (Cocchi et al. 2005; van der Velde et al. 2008; Nikolopoulos et al. 2010; Delhanty et al. 2013).
It has been reported that ghrelin stimulates proliferation and differentiation (Fukushima et al. 2005; Kim et al. 2005; Maccarinelli et al. 2005) and inhibits apoptosis of MC3T3-E1 cells (Kim et al. 2005; Liang et al. 2013). These results together with previous studies, showing that ghrelin decreases oxidative injury in various peripheral tissues (El Eter et al. 2007; Kawczynska-Drozdz et al. 2006; Iseri et al. 2008; Zhang et al. 2011) and in the brain (Obay et al. 2008), lead us to examine the possibility that ghrelin could protect osteoblast against t-BHP-induced oxidative damage.
Oxidative stress plays an important role in the pathogenesis of age-related bone loss not only in rodents (Banfi et al. 2008) but also in humans (Basu et al. 2001; Maggio et al. 2003) leading to increased osteoblast and osteocyte apoptosis and reduced rate of bone formation (Manolagas and Parfitt 2010). In line with these reports, t-BHP reduced osteoblastic viability and increased their apoptosis. In this study, we showed that ghrelin protects MC3T3-E1 cells against t-BHP-induced oxidative stress. Interestingly, ghrelin increased viability of t-BHP-treated cells in a way that was not directly dose-dependent. In fact, the maximal protective activity of ghrelin was observed at low concentrations. This pattern, previously shown in other reports (Maccarinelli et al. 2005; Kim et al. 2005; Costa et al. 2011), could be due to the existence of multiple ghrelin receptor subtypes with differential affinities for the peptide. By increasing ghrelin concentrations, the peptide could recognize receptor subtypes having different or no activity on cell viability thus tempering the protective effect prevailing at low concentrations. Previous studies have shown that ghrelin protects MC3T3-E1 cells from apoptosis induced by TNF-α (Kim et al. 2005) and serum deprivation (Liang et al. 2013). In this study, we provided the first direct evidence that ghrelin reduced t-BHP-induced nuclear morphological changes and chromatin condensation associated with cell apoptosis as shown by Hoechst 33258 staining. Further studies will be required to examine the cell death pathway involved in the t-BHP cytotoxicity and the endocellular mechanisms involved in the protective effect of ghrelin against oxidative stress in MC3T3-E1 cells.
t-BHP-induced cytotoxicity is accompanied by osteoblast dysfunction and changes in the cytoskeleton organization with destruction of actin fibers.
Ghrelin treatment reduced the osteoblastic dysfunction induced by t-BHP and partially prevented the disruption of the cytoskeleton. Further studies will be required to examine whether longer ghrelin incubation time (24 and 48 h) could completely prevent t-BHP-induced cytoskeletal disruption.
Ghrelin and GHS-R1a are expressed in MC3T3-E1 cells (Fukushima et al. 2005; Maccarinelli et al. 2005), and many biological actions of ghrelin are linked to the binding of ghrelin to GHS-R1a. Therefore, the association of GHS-R1a with ghrelin’s protective action against t-BHP-induced cytotoxicity was examined. Treatment with EP1572, acting as GHS-R1a agonist, did not prevented the reduction in cell viability induced by t-BHP. Furthermore, in the presence of the GHS-R1a antagonist, d-Lys3-GHRP-6, ghrelin was still able to exert its protective effect against oxidative injury.
The lack of the involvement of GHS-R1a in mediating the protective effects of ghrelin is in line with the results obtained with DAG which does not bind GHS-R1a (Muccioli et al. 2007).
It is unlikely that DAG could exert its protective effect in MC3T3-E1 cells through an interaction with specific binding sites not recognized by ghrelin, as previously reported in cardiomyocytes (Lear et al. 2010). Our data, in fact, showed that both ghrelin and DAG were equally protective against oxidative damage, suggesting the existence of an unknown ghrelin receptor, different from GHS-R1a, that DAG might share with ghrelin.
Evidence for an alternative ghrelin receptor has been previously reported. Ghrelin and DAG have been shown to stimulate cell signaling in cells that do not express GHS-R1a and in GHS-R-knockout animal models (Baldanzi et al. 2002; Delhanty et al. 2006; Granata et al. 2007). Furthermore, DAG shares with ghrelin an inhibitory action on inflammatory pain (Sibilia et al. 2012) and stimulatory effect on adipogenesis (Thompson et al. 2004).
Furthermore, Costa et al. (2011) have shown that ghrelin promotes rat osteoblast proliferation through a GHS-R1a-independent pathway, confirming the data previously reported by Delhanty et al. (2006) in bone cells from humans.
Several protective effects have been proposed to explain the anti-oxidant activity of ghrelin in various peripheral tissues and in the brain (Obay et al. 2008; Tong et al. 2012). Our data showing that both ghrelin and DAG significantly reduce ROS levels in t-BHP-treated cells indicate that the peptides could enhance the endogenous antioxidant defense mechanisms by up-regulating intracellular antioxidant enzymes.
Further studies will be designed to clarify the signaling pathways involved in the protective effect of both peptides against t-BHP.
Ghrelin binding to GHS-R1a has been shown to activate different intracellular pathways including PI3 K, PKA, MAPK and AMPK (Baldanzi et al. 2002; Malagón et al. 2003; Carreira et al. 2004). Interestingly, Granata et al. (2007) demonstrated that activation of cAMP/PKA and PI3K/Akt pathway signaling is involved in the cytoprotective effects of both ghrelin and DAG on pancreatic β-cells via a specific ghrelin/DAG binding site, that is not GHS-R1a.
Our preliminary results showing that the levels of H3 K4me3 are significantly increased by ghrelin, suggest a possible involvement of epigenetic modifications in the protective effect of ghrelin against oxidative injury. It is noteworthy that Huang et al. (2012) showed a significant correlation between this epigenetic mark and the high expression of genes involved in the peroxisome proliferator-activated receptors α (PPARα) and PPARs, which regulate ROS production (Aleshin and Reiser 2013, in press).
In conclusion, we have shown for the first time, that ghrelin may protect MC3T3-E1 cells from oxidative stress induced by t-BHP by reducing the intracellular ROS levels. This effect is independent from GHS-R1a, and it is likely mediated by a common ghrelin/DAG binding site. Even if the mechanism involved in ghrelin antioxidant activity remains to be clarified, this study provides a theoretical basis for exogenous application of ghrelin to maintain osteoblast activity during aging.
The increase in ROS levels, in fact, may represent a key pathogenic event in the dramatic decrease in bone formation observed in age-related bone loss.
References
Aleshin S, Reiser G (2013) Role of the peroxisome proliferator-activated receptors (PPAR)-α, β/δ and γ triad in regulation of reactive oxygen species signaling in brain. Biol Chem. doi:10.1515/hsz-2013-0215 (Epub ahead of print)
Almeida M (2012) Aging mechanisms in bone. Bonekey Rep1
Almeida M, Han L, Martin-Millan M et al (2007) Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 282:27285–27297
Altman SA, Zastawny TH, Randers L et al (1994) tert-Butyl hydroperoxide-mediated DNA base damage in cultured mammalian cells. Mutat Res 306:35–44
Bai XC, Lu D, Bai J et al (2004) Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem Biophys Res Commun 314:197–207
Baldanzi G, Filigheddu N, Cutrupi S et al (2002) Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol 159:1029–1037
Banfi G, Iorio EL, Corsi MM (2008) Oxidative stress, free radicals and bone remodeling. Clin Chem Lab Med 46:1550–1555
Basu S, Michaëlsson K, Olofsson H et al (2001) Association between oxidative stress and bone mineral density. Biochem Biophys Res Commun 288:275–279
Bednarek MA, Feighner SD, Pong SS et al (2000) Structure-function studies on the new growth hormone-releasing peptide, ghrelin: minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J Med Chem 43:4370–4376
Broglio F, Boutignon F, Benso A et al (2002) EP1572: a novel peptido-mimetic GH secretagogue with potent and selective GH-releasing activity in man. J Endocrinol Invest 25:26–28
Carreira MC, Camiña JP, Smith RG, Casanueva FF (2004) Agonist-specific coupling of growth hormone secretagogue receptor type 1a to different intracellular signaling systems. Role of adenosine. Neuroendocrinology 79:13–25
Carreira MC, Camiña JP, Díaz-Rodríguez E et al (2006) Adenosine does not bind to the growth hormone secretagogue receptor type-1a (GHS-R1a). J Endocrinol 191:147–157
Cocchi D, Maccarinelli G, Sibilia V et al (2005) GH-releasing peptides and bone. J Endocrinol Invest 28(8 Suppl):11–14
Cos P, Hermans N, Calomme M et al (2003) Comparative study of eight well-known polyphenolic antioxidants. J Pharm Pharmacol 55:1291–1297
Costa JL, Naot D, Lin JM et al (2011) Ghrelin is an osteoblast mitogen and increases osteoclastic bone resorption in vitro. Int J Pept 2011:605193. doi:10.1155/2011/605193
Delhanty PJ, van der Eerden BC, van der Velde M et al (2006) Ghrelin and unacylated ghrelin stimulate human osteoblast growth via mitogen-activated protein kinase (MAPK)/phosphoinositide 3-kinase (PI3K) pathways in the absence of GHS-R1a. J Endocrinol 188:37–47
Delhanty PJ, van der Eerden BC, van Leeuwen JP (2013) Ghrelin and bone. Biofactors. doi:10.1002/biof.1120 (Epub ahead of print)
El Eter E, Al Tuwaijiri A, Hagar H, Arafa M (2007) In vivo and in vitro antioxidant activity of ghrelin: attenuation of gastric ischemic injury in the rat. J Gastroenterol Hepatol 22:1791–1799
Fukushima N, Hanada R, Teranishi H et al (2005) Ghrelin directly regulates bone formation. J Bone Miner Res 20:790–798
Ghigo E, Broglio F, Arvat E et al (2005) Ghrelin: more than a natural GH secretagogue and/or an orexigenic factor. Clin Endocrinol 62:1–17
Granata R, Settanni F, Biancone L et al (2007) Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-kinase/Akt signaling. Endocrinology 148:512–529
Haidara K, Marion M, Gascon-Barré M et al (2008) Implication of caspases and subcellular compartments in tert-butyl hydroperoxide induced apoptosis. Toxicol Appl Pharmacol 229:65–76
Holmes E, Davies I, Lowe G, Ranganath LR (2009) Circulating ghrelin exists in both lipoprotein bound and free forms. Ann Clin Biochem 46:514–516
Hosoda H, Kojima M, Matsuo H, Kangawa K (2000) Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res Commun 279:909–991
Huang Y, Min S, Lui Y et al (2012) Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Gene Immun 13:311–320. doi:10.1038/gene.87
Işeri SO, Sener G, Saglam B et al (2008) Ghrelin alleviates biliary obstruction-induced chronic hepatic injury in rats. Regul Pept 146:73–79
Kawczynska-Drozdz A, Olszanecki R, Jawien J et al (2006) Ghrelin inhibits vascular superoxide production in spontaneously hypertensive rats. Am J Hypertens 19(7):764–767
Kim SW, Her SJ, Park SJ et al (2005) Ghrelin stimulates proliferation and differentiation and inhibits apoptosis in osteoblastic MC3T3-E1 cells. Bone 37:359–369
Kojima M, Kangawa K (2005) Ghrelin: structure and function. Physiol Rev 85:495–522
Lear PV, Iglesias MJ, Feijóo-Bandín S et al (2010) Des-acyl ghrelin has specific binding sites and different metabolic effects from ghrelin in cardiomyocytes. Endocrinology 151:3286–3298
Liang QH, Liu Y, Wu SS et al (2013) Ghrelin inhibits the apoptosis of MC3T3-E1 cells through ERK and AKT signaling pathway. Toxicol Appl Pharmacol 272:591–597
Maccarinelli G, Sibilia V, Torsello A et al (2005) Ghrelin regulates proliferation and differentiation of osteoblastic cells. J Endocrinol 184:249–256
Maggio D, Barabani M, Pierandrei M et al (2003) Marked decrease in plasma antioxidants in aged osteoporotic women: results of a cross-sectional study. J Clin Endocrinol Metab 88:1523–1527
Malagón MM, Luque RM, Ruiz-Guerrero E et al (2003) Intracellular signaling mechanisms mediating ghrelin-stimulated growth hormone release in somatotropes. Endocrinology 144:5372–5380
Manolagas SC (2010) From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31:266–300
Manolagas SC, Parfitt AM (2010) What old means to bone. Trends Endocrinol Metab 6:369–374
Muccioli G, Baragli A, Granata R et al (2007) Heterogeneity of ghrelin/growth hormone secretagogue receptors. Toward the understanding of the molecular identity of novel ghrelin/GHS receptors. Neuroendocrinology 86:147–164
Muller FL, Lustgarten MS, Jang Y et al (2007) Trends in oxidative aging theories. Free Radic Biol Med 43:477–503
Muthusami S, Ramachandran I, Muthusamy B et al (2005) Ovariectomy induces oxidative stress and impairs bone antioxidant system in adult rats. Clin Chim Acta 360:81–86
Nikolopoulos D, Theocharis S, Kouraklis G (2010) Ghrelin, another factor affecting bone metabolism. Med Sci Monit 16:147–162
Nojiri H, Saita Y, Morikawa D et al (2011) Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res 26:2682–2694
Obay BD, Taşdemir E, Tümer C et al (2008) Dose dependent effects of ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptides 29:448–455
Ozgocmen S, Kaya H, Fadillioglu E et al (2007) Role of antioxidant systems, lipid peroxidation, and nitric oxide in postmenopausal osteoporosis. Mol Cell Biochem 295:45–52
Patterson M, Murphy KG, le Roux CW et al (2005) Characterization of ghrelin-like immunoreactivity in human plasma. J Clin Endocrinol Metab 90:2205–2211
Seeman E (2003) Invited Review: pathogenesis of osteoporosis. J Appl Physiol 95:2142–2151
Seim I, Josh P, Cunningham P et al (2011) Ghrelin axis genes, peptides and receptors: recent findings and future challenges. Mol Cell Endocrinol 340:3–9
Sestili P, Guidarelli A, Dachà M, Cantoni O (1998) Quercetin prevents DNA single strand breakage and cytotoxicity caused by tert-butylhydroperoxide: free radical scavenging versus iron chelating mechanism. Free Radic Biol Med 25:196–200
Sibilia V, Cocchi D, Pagani F et al (1999) Hexarelin, a growth hormone-releasing peptide, counteracts bone loss in gonadectomized male rats. Growth Horm IGF Res 9:219–227
Sibilia V, Cocchi D, Villa I et al (2002) Bone effects of hexarelin, a GH-releasing peptide, in female rats: influence of estrogen milieu. Eur J Endocrinol 146:855–862
Sibilia V, Muccioli G, Deghenghi R et al (2006) Evidence for a role of the GHS-R1a receptors in ghrelin inhibition of gastric acid secretion in the rat. J Neuroendocrinol 18:122–128
Sibilia V, Pagani F, Mrak E et al (2012) Pharmacological characterization of the ghrelin receptor mediating its inhibitory action on inflammatory pain in rats. Amino Acids 43:1751–1759
Svensson J, Lall S, Dickson SL et al (2000) The GH secretagogues ipamorelin and GH-releasing peptide-6 increase bone mineral content in adult female rats. J Endocrinol 165:569–577
Thompson NM, Gill DA, Davies R et al (2004) Ghrelin and des-octanoyl ghrelin promote adipogenesis directly in vivo by a mechanism independent of the type 1a growth hormone secretagogue receptor. Endocrinology 145:234–242
Tong XX, Wu D, Wang X et al (2012) Ghrelin protects against cobalt chloride-induced hypoxic injury in cardiac H9c2 cells by inhibiting oxidative stress and inducing autophagy. Peptides 38:217–227
van der Velde M, Delhanty P, van der Eerden B et al (2008) Ghrelin and bone. Vitam Horm 77:239–258
Wang H, Joseph JA (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical Biol Med 27:612–616
Yang J, Brown MS, Liang G et al (2008) Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132:387–396
Zhang Q, Huang WD, Lv XY, Yang YM (2011) Ghrelin protects H9c2 cells from hydrogen peroxide-induced apoptosis through NF-κB and mitochondria-mediated signaling. Eur J Pharmacol 654:142–149
Acknowledgments
This work was supported by funds from FIRST 2009 to VS and from Project ASI DC-DTE-2011-033 for biomedical and biotechnological studies to FC.
Conflict of interest
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dieci, E., Casati, L., Pagani, F. et al. Acylated and unacylated ghrelin protect MC3T3-E1 cells against tert-butyl hydroperoxide-induced oxidative injury: pharmacological characterization of ghrelin receptor and possible epigenetic involvement. Amino Acids 46, 1715–1725 (2014). https://doi.org/10.1007/s00726-014-1734-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1734-y