
Abstract
IgG4-related disease (IgG4-RD) is capable of causing great morbidity and even mortality if the condition remains undiagnosed or poorly treated, yet is typically a treatment-responsive disorder. Glucocorticoids have not been studied rigorously and practices with regard to dosing and duration of treatment remain largely empiric. In addition, IgG4-RD patients are often particularly susceptible to and intolerant of the deleterious effects of glucocorticoid therapy. B cell depletion with anti-CD20 monoclonal antibodies appears to be a rapid, effective means of obtaining disease control and limiting patients’ glucocorticoid exposure, but this option is frequently not available. Other therapies targeting the B cell lineage may also be efficacious, and one is under study. The means by which depletion or inhibition of B cells and their progeny ameliorate IgG4-RD is coming into focus now through careful mechanistic studies of samples from treated patients. The mechanistic understanding of IgG4-RD will bring an array of specific targets for therapeutic intervention. Plasmablast-directed therapy with a CD19 monoclonal antibody is currently in clinical trials. CD4 + cytotoxic T lymphocytes and fibrosis, both observed nearly universally in the tissue of IgG4-RD patients, present two unexploited vulnerabilities in controlling and even reversing the effects of the disease. Further development of such therapies is a major goal of the next few years.
Zusammenfassung
Eine IgG4-bedingte Erkrankung (IgG4-RD) kann eine hohe Morbidität und sogar Mortalität zur Folge haben, wenn sie nicht diagnostiziert oder falsch behandelt wird. In der Regel handelt es sich jedoch um eine Störung, die auf eine Therapie anspricht. Glukokortikoide wurden noch nicht eingehend untersucht, und Vorgehensweisen bezüglich Dosierung und Behandlungsdauer bleiben weitgehend empirisch. Zudem sind IgG4-RD-Patienten häufig besonders anfällig für die schädlichen Auswirkungen der Glukokortikoidtherapie und tolerieren diese nicht. Die B‑Zellen-Depletion mit monoklonalen Anti-CD20-Antikörpern scheint eine schnelle, effektive Maßnahme zu sein, um die Krankheit zu kontrollieren und die Exposition der Patienten gegenüber Glukokortikoiden zu begrenzen, aber diese Behandlungsmöglichkeit ist meist nicht verfügbar. Andere Therapien, welche auf die B‑Zell-Linien zielen, könnten ebenfalls wirksam sein; eine davon wird derzeit untersucht. Die Maßnahmen, durch welche die Depletion oder Inhibition von B‑Zellen und ihrer Abkömmlinge eine IgG4-RD verbessert, rückt nun durch genaue machanistische Studien von Proben behandelter Patienten in den Fokus. Das mechanistische Verständnis von IgG4-RD wird eine Vielzahl an spezifischen Zielen für therapeutische Interventionen bringen. Die Plasmablasten-gesteuerte Therapie mit einem monoklonalen CD19-Antikörper wird derzeit in klinischen Studien untersucht. Cd4 + zytotoxische T‑Zellen und Fibrose, die im Gewebe von nahezu allen IgG4-Patienten gefunden wurden, stellen bei der Kontrolle und sogar bei der Reversibilität der Auswirkungen dieser Erkrankung zwei nichtuntersuchte Schwachstellen dar. Die Weiterentwicklung solcher Therapien ist ein wesentliches Ziel der nächsten Jahre.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IgG4-related disease (IgG4-RD) is an immune-mediated, fibroinflammatory condition that can affect nearly any organ system [21]. Therapy for this disease has focused to date on glucocorticoids, which appear to be highly effective in inducing remission yet have not been studied thoroughly in rigorous, prospective studies and frequently cannot be employed without causing substantial morbidity. The paucity of data regarding the use of disease-modifying anti-rheumatic drugs (DMARDs) provide little support for the use of these medications in clinical practice, albeit they are often used in the hope of reducing glucocorticoid dependency. More recently, B cell depletion has shown evidence of efficacy in glucocorticoid sparing and has even been used as monotherapy, with considerable success. As more features of IgG4-RD pathophysiology become unveiled, newer therapeutic targets are emerging.
Disease pathophysiology
IgG4-RD was once regarded as a Th2-mediated disease, but there is now considerable doubt about that notion. Circulating CD4 + GATA3 + T lymphocytes, analyzed among a cohort of 39 IgG4-RD patients with biopsy-proven disease, were found to be productive of stereotypic Th2 cytokines (IL-4, IL-5, IL-13) in only the subset of patients with a history of atopic disease [18]. In addition, the Th2 cells of patients with IgG4-RD have been found to be polyclonally expanded, suggesting only responses to a lifetime of environmental allergen exposures rather than response to a specific triggering antigen [18]. In short, concomitant atopic disease probably constituted a crucial confounder in earlier studies supporting a predominant role for Th2 pathways in the pathophysiology of IgG4-RD.
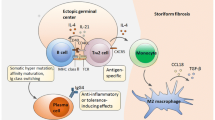
A current theoretical model of IgG4-RD pathophysiology is shown in Fig. 1.

Current model of the immunologic mechanism of IgG4-RD: 1. Antigen presentation, activation of self-antigen detecting naïve T cell and differentiation into follicular helper T cells and CD4 cytotoxic T lymphocytes (CTLs). 2. T–B collaboration as follicular helper T cells drive class switching, somatic hypermutation and plasmablast differentiation of activated, self-antigen detecting B cells. 3. CD4 CTLs and plasmablasts circulate to the site of inflammation. 4. T×B collaboration in which activated plasmablasts present antigen to CD4 CTLs and drive further clonal expansion. 5. Activated CD4 CTLs secrete profibrotic cytokines (IL-1β, TGF-β1, IFN-γ) stimulating myofibroblasts to generate storiform fibrosis and cytotoxic molecules (granzyme, perforin) causing tissue damage
Plasmablasts, defined by cell surface expression of CD19, CD27, and CD38 but negative for CD20, are elevated to high concentrations in the peripheral blood of patients with IgG4-RD [16, 23]. These plasmablasts are oligoclonally expanded (as demonstrated by next-generation sequencing), express IgG4, and show intense somatic hypermutation [16]. Moreover, they swiftly respond to B cell depletion and demonstrate clonal divergence upon repopulation after B cell repletion. In the setting of such somatic hypermutation and strong evidence for class switching, an important role for follicular helper T (Tfh) cells is anticipated. Indeed, experimental evidence indicates that a subset of Tfh cells drives the class switch within germinal centers, through the production of IL-4 (submitted).
Oligoclonal proliferation of a novel CD4 + effector memory T cell is also present in both the peripheral blood and affected tissues of patients with IgG4-RD [17]. Through both gene profiling and flow cytometry of these effector memory T cells, a fusion signature of modified Th1, cytolytic and myeloid cells was observed [17]. These cytotoxic T lymphocyte (CD4 CTL) cells demonstrate striking oligoclonal expansion by next-generation sequencing [17], implying that this disease is triggered in a given patient by an antigen (or perhaps a collection of antigens, which differ from patient to patient) not yet identified. These CD4 + CTLs elaborate fibrosis-mediating cytokines (TGF-β, IFN-γ, IL-1β) and in theory could therefore serve as the primary driver of the storiform fibrosis, generally present in abundance even in IgG4-RD patients with early disease.
Role of IgG4 is likely to be an attempt to quell inflammatory response
Finally, the thinking with regard to the importance of the IgG4 molecule in this condition has swung 180° in the past few years. IgG4, rather than being positioned squarely in the center of pathophysiological models of disease, is believed to subserve a role of attempting to dampen and downregulate the inflammatory response. Such a role is consistent with the current orthodoxies concerning the nature of IgG4, namely, that it differs from other IgG subclasses by its relative inability to fix complement—at least via the classical pathway of complement activation—or bind Fc receptors. Ironically, the principal role of IgG4 in this disease that bears its name likely lies not in the mediation of inflammation but rather in an attempt—usually ineffectual—to quell it.
In summary, current thought about the disease pathophysiology of IgG4-RD involves an intricate dance between various cells of the B and T cell lineages, including B cells, plasmablasts, plasma cells, CD4 + CTLs, and Tfh cells—as well as interactions with cells such as myofibroblasts, macrophages, and eosinophils. The sum of these pathways leads to the histopathological findings characteristic of IgG4-RD in essentially every organ system: a lymphoplasmacytic infiltrate, frequent mild-to-moderate tissue (as well as peripheral) eosinophilia, obliterative phlebitis (and occasionally arteritis), and storiform fibrosis. This model of pathophysiology not only explains in large measure the known efficacy of certain medications (e. g., glucocorticoids and rituximab), it also provides the rationale for other therapeutic targets, some of which are currently under study. We now discuss current and future treatment options for patients with IgG4-RD, beginning with glucocorticoids.
Glucocorticoids
Induction therapy
Glucocorticoids are regarded as highly-effective agents for the exertion of disease control in IgG4-RD, and for the induction of remission in many. In a retrospective, multicenter study of 25 IgG4-RD patients in France, prednisone at a starting dose of approximately 47 mg/day (0.67 mg/kg for a 70-kg patient) was effective in controlling the disease in 90 % of patients [6]. In that study, treatment response was defined by the presence of at least two of the following features: improved clinical status, reduction in serum IgG4 concentration, and improved radiologic findings. Even higher response rates have been reported in autoimmune pancreatitis [3, 10, 20]. Another retrospective study examined the effect of prednisone in 30 patients with IgG4-related sclerosing cholangitis and found that 97 % of patients experienced either improvement or resolution of strictures and liver function tests on treatment [7].
Glucocorticoids are generally a cornerstone of remission induction efforts
Thus, glucocorticoids are generally a cornerstone of remission induction efforts [15], and a typical starting prednisone dose is 40 mg/day. To quantify response to therapy in an objective manner, we routinely use the IgG4-Related Disease Responder Index (IgG4-RD RI), which was modeled after the Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG) [1]. An international validation of this tool is currently being conducted. Despite their considerable efficacy, a number of important caveats to the use of glucocorticoids exist, which are discussed below.
Maintenance therapy
Most experts agree upon continuing the initial glucocorticoid dose for 2–4 weeks followed by a gradual taper [15]. Many of the therapeutic studies cited above employed a taper of 5 mg per week until off [6, 7, 20]. A similar strategy includes tapering by 10 mg every 2 weeks until the achievement of a daily dose of 20 mg, continuing 20 mg/day for 2 weeks, and then continuing to taper by 5 mg every 2 weeks to discontinuation [15]. Although practice in the US is to taper the glucocorticoid completely off within 2–3 months, Japanese clinicians often continue prednisone at a low to moderate dose (2.5–10.0 mg daily) for up to several years [11]. There has been no study of IgG4-RD patients begun on a dose of prednisone considered sufficient to control the disease and then followed prospectively through a prescribed prednisone taper to discontinuation of the medicine.
Two major issues with regard to glucocorticoid therapy require elaboration. First, IgG4-RD patients often have baseline comorbidities and frailties that make them poor glucocorticoid candidates. Because IgG4-RD often affects older individuals, patients often already suffer from obesity, glucose intolerance, hypertension, osteoporosis, and other relative contraindications to prolonged glucocorticoid courses. In addition, type 1 (IgG4-related) autoimmune pancreatitis, one of the more common disease manifestations, often devolves into endocrine and exocrine insufficiency, further complicating glucocorticoid treatment.
A large percentage of patients relapse during or after glucocorticoid taper
In a prospective trial of glucocorticoids from Japan, 28 % of patients developed either new diabetes or exacerbations of previously known diabetes. In the cohort of patients from France described above, 67 % of patients experienced side effects from glucocorticoid therapy [6]. Thus, the comorbid conditions of each patient and the potential for glucocorticoid intolerance must be considered on an individual basis when deciding on the suitability of treatment, as well as the initial dose and duration of glucocorticoid therapy.
Second, although only a minority of patients fail to respond to glucocorticoid treatment, a large percentage relapse during or after the glucocorticoid taper. Without ongoing maintenance therapy, between 30 and 60 % of patients relapse within 3 months of discontinuing glucocorticoid monotherapy [10, 20]. Even with low-dose maintenance glucocorticoids studied retrospectively in patients with autoimmune pancreatitis, 23 % relapsed while on treatment [10]. These glucocorticoid issues—the substantial risk of adverse effects and their lack of efficacy in maintaining disease control in many patients—are major factors in the selection of a therapeutic regimen.
Conventional disease-modifying antirheumatic drugs
The 2015 Consensus Guidance Statement on the Management and Treatment of IgG4-RD [15] states: “There are few data overall to support the notion that conventional steroid-sparing agents are effective in IgG4-RD.” In fact, in the largest series available, no benefit in relapse-free survival was observed by adding a DMARD to glucocorticoids [8].
Adding a DMARD to glucocorticoids is not associated with improved relapse-free survival
Small, retrospective case series of azathioprine, methotrexate, and mycophenolate mofetil comprise essentially the only evidence base for the use of these medications. We typically reserve them for patients with major contraindications to glucocorticoids who are not able to obtain authorization for use of rituximab or enrollment in an ongoing trial of Xmab5871, described below.
B cell-targeted therapy
The discovery of oligoclonally expanded plasmablasts in patients with IgG4-RD [16] and their correlation with disease activity [23] elucidate further how targeting cells of the B cell lineage might work in IgG4-RD (and other diseases). Plasmablasts are circulating plasma cells that arise from activated CD20 + B cells. Plasmablasts are antibody-secreting cells and typically develop in short order into plasma cells. Anti-CD20 monoclonal antibodies function via antibody-dependent cell-mediated cytotoxicity and cause direct depletion of circulating and tissue resident B cells, thereby eliminating the progenitor to the plasmablast. Despite the fact that they lack CD20, plasmablasts fall quickly following B cell depletion. The decline of these cells—and perhaps more importantly their recurrent rise over time in some patients—correlates better with disease activity than does the serum IgG4 concentration. Use of the total plasmablast concentration as a means of monitoring disease activity means that plasmablasts comprise the first cell in any rheumatologic condition to serve as a useful biomarker [16].
Rituximab is potentially useful in treating IgG4-RD
The potential utility of rituximab in IgG4-RD was demonstrated initially in case series [13, 14] and then in a prospective, open-label trial involving 30 patients [2]. Of the patients enrolled, 77 % achieved the primary outcome in the trial, defined as a decline in the IgG4-RD Responder Index (IgG4-RD RI) of ≥2 points, no disease flares before 6 months, and no glucocorticoid use between months 2 and 6. Twenty-six of the 30 patients enrolled were treated without glucocorticoids, yet 97 % of patients demonstrated at least some evidence of a therapeutic response. Moreover, 47 % achieved a complete remission at 6 months, defined by an IgG4-RD RI of 0 and no additional glucocorticoid treatment [2].
In addition to interfering with antigen presentation by plasmablasts, B cell depletion may also achieve its effect through the reduction of immune complex formation. The potential importance of immune complex formation as a possible disease mechanism has yet to be studied thoroughly in IgG4-RD. This may be particularly relevant for those patients with hypocomplementemia and the associated manifestation of tubulointerstitial nephritis (TIN) [12]. The phenomenon of immune complex formation in IgG4-related disease remains incompletely understood, yet seems to be operative in some organ manifestations—particularly IgG4-related TIN. IgG4 does not bind complement well under most circumstances but other IgG subclasses that are often elevated to a lesser extent in IgG4-RD, i. e., IgG1 and IgG3, might account for this observation. In addition, the mannose binding lectin pathway of complement activation is one possible mechanism through which IgG4 could trigger this phenomenon.
The treatment effects of rituximab are generally observed rapidly, with symptomatic improvement within one month, a swift decline in serum IgG4 concentrations, and the ability to discontinue glucocorticoids entirely within a few weeks in most patients [14]. Independent of serum IgG4 levels, blood plasmablasts have also been shown to be a useful marker for disease monitoring while on rituximab [23]. Clinical benefit following rituximab treatment has been reported specifically in autoimmune pancreatitis [8], orbital pseudotumor [24], ascending cholangitis [22], and aortitis [19].
Plasmablasts as a target of treatment
Although rituximab indirectly depletes plasmablasts by leading to the depopulation (as measureable in peripheral blood) of their progenitor CD20 + B cells, plasmablast-targeted therapy may offer a more specific approach in treating IgG4-RD. XmAb5871, a monoclonal antibody (homodimer) with a high-affinity variable region binding to CD19 and enhanced Fc domain that binds to the FcγRIIb inhibitory receptor of B cells, is currently in phase II development for IgG4-RD treatment.
This nondepleting anti-CD19 therapy has been studied in phase 1 trials and mechanistic studies in both rheumatoid arthritis [4] and systemic lupus erythematosus [9]. Xmab5871 is currently administered via infusion every 14 days, but a subcutaneous formulation is under development and will likely be employed in advanced phase trials if the early experience is promising. The rapid on/off effect of Xmab5871, its fully-humanized structure, and its status as a non-depleting antibody may pose potential advantages over rituximab.
Future therapeutic directions
CD4 + CTL-directed treatments
Oligoclonally-expanded CD4 + effector-memory T cells with a cytotolytic phenotype (CD4 + CTLs) have been identified and characterized recently in IgG4-RD [17]. Through gene expression analysis, flow cytometry, and multicolor tissue imaging, these cells were found to express SLAMF7, IL-1β, TGF-β1, granzyme B, and perforin. Despite their lack of CD20 expression, the concentrations and percentages of the overall T cell pool of these novel CD4 + CTLs decreased substantially following rituximab administration, albeit more slowly than the concentrations of B cells and plasmablasts. The responsiveness to CD20-targeted B cell depletion is theoretically related to the interference of T and B cell collaboration as plasmablasts present antigen and activate effector/memory CD4 CTLs at the site of inflammation (Fig. 1). The clonal expansion, profibrotic phenotype and correlation to disease activity all point toward a central role for these cells in the disease mechanism of IgG4-RD (Fig. 1).
Antifibrosis therapies
Most therapies conceived and employed to date have focused on the “inflammatory” aspects of the disease rather than the “fibrotic” ones. Of course, fibrosis itself is an inflammatory response, so in this sense the dichotomy is artificial. Nevertheless, some patients with particular clinical presentations are plagued even at the time of diagnosis by disease features that result from such fibrosis. The fibrotic features are unlikely to respond to the currently available therapies. These disease manifestations are in great need of therapies designed specifically to address the problem of fibrosis.
Decreased fibrosis and myofibroblast activation markers following rituximab therapy
Some data indicate that the fibrosis of IgG4-RD may in many cases be at least partially reversible. Evidence for this statement comes from both studies of posttreatment tissue samples in the laboratory [5] and from serial clinical evaluations—particularly chest imaging—of patients following the institution of treatment. Della Torre et al. [5] reported a decrease in both circulating markers of fibrosis and myofibroblast activation in the effected tissue following rituximab therapy. The impact on fibrosis of Xmab5871 and potential future therapies such as those directed against the CD4 + CTL remain uncertain at the moment, but such effects will be a key aspect of the evaluation of any new treatment agent.
Conclusion
IgG4-RD is both an inflammatory and fibrotic condition that is exquisitely responsive to medical therapy. Until more effective and safer therapies are tested head-to-head with glucocorticoids in clinical trials, time-limited courses of glucocorticoids should remain the first-line therapy for many IgG4-RD patients. Unfortunately, a large percentage of patients with this condition have relative contraindications to prolonged glucocorticoid treatment, even in moderate to low doses. We often begin patients at 40 mg/day of prednisone and attempt to taper the medication to discontinuation over a period of 2–3 months. Exacerbations of disease during or shortly after this glucocorticoid course generally signal the need to consider alternative therapeutic approaches.
The rapid progress in understanding the pathophysiology of IgG4-RD has led to several exciting mechanism-based therapies, including B cell depletion, a first-in-class homodimer targeting both CD19 and FcγRIIb, and the possibility of directing therapy against a novel CD4 + CTL that may be at the heart of this condition.
References
Carruthers M, Topazian M, Khosroshahi A et al (2015) Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 74:1171–1177
Carruthers M, Stone JH, Desphande V, Khosroshahi A (2012) Development of an IgG4-RD Responder Index. Int J Rheumatol 2012:259408
Chari S, Smyrk T, Levy M (2006) Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 4:1010–1016
Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung S, Chen H et al (2014) Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcγ receptor IIb inhibitory receptor. Arthritis Rheum 66(5):1153–1164
Della-Torre E, Feeney E, Deshpande V, Mattoo H, Mahajan V et al (2015) B‑cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann Rheum Dis 74(12):2236–2243
Ebbo M, Daniel L, Pavic M et al (2012) IgG4-related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine (Baltimore) 91:49–56
Ghazale A, Chari S, Zhang L et al (2008) Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology 134:706–715
Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR et al (2013) Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut 62:1607–1615
Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IW, Jacob N et al (2011) Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol 186(7):4223–4233
Kamisawa T, Shimosegawa T, Okazaki K (2009) Standard steroid treatment for autoimmune pancreatitis. Gut 58:1504–1507
Kamisawa T, Okazaki K, Kawa S et al (2014) Amendment of the japanese consensus guidelines for autoimmune pancreatitis, 2013 III. treatment and prognosis of autoimmune pancreatitis. J Gastroenterol 49:961–970
Kawano M, Mizushima I, Yamaguchi Y et al (2012) Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: detailed analysis of 20 japanese cases. Int J Rheum 2012:1–9
Khosroshahi A, Carruthers MN, Deshpande V et al (2012) Rituximab for the treatment of IgG4-related disease lessons from 10 consecutive patients. Medicine (Baltimore) 91:57–66
Khosroshahi A, Bloch D, Deshpande V et al (2010) Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG-related systemic disease. Arthritis Rheum 62:1755–1762
Khosroshahi A, Wallace ZA, Crowe JL, Akamizu T, Azumi A, Carruthers MN et al (2015) International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheum 67:1688–1699
Mattoo H, Mahajan V, Della-Torre E et al (2014) De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol 134:679–687
Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS et al (2016) Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol (in Press)
Mattoo H, Della-Torre E, Mahajan VS, Stone JH, Pillai S (2014) Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy 69:399–402
Perugino CA, Wallace Z, Meyersohn N, Oliveira G, Stone JR, and Stone JH. Large Vessel Involvement by IgG4-Related Disease (accepted, Medicine). (in press)
Raina A, Yadav D, Krasinskas A et al (2009) Evaluation and management of autoimmune pancreatitis: experience at a large US center. Am J Gastroenterol 104:2295–2306
Stone JH, Zen Y, Deshpande V (2012) IgG4-related disease. N Engl J Med 366(6):539–551
Topazian M, Witzig TE, Smyrk TC, Pulido JS, Levy MJ, Kamath PS et al (2008) Rituximab therapy for refractory biliary strictures in immunoglobulin G4-associated cholangitis. Clin Gastroenterol Hepatol 6:364–366
Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, Kulikova M, Deshpande V, Pillai S, Stone JH (2015) Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis 74:190–195
Witzig TE, Inwards DJ, Habermann TM, Dogan A, Kurtin PJ, Gross JB Jr. et al (2007) Treatment of benign orbital pseudolymphomas with the monoclonal anti-CD20 antibody rituximab. Mayo Clin Proc 82:692–699
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C.A. Perugino state that there are no conflicts of interest. Dr. Stone has received research funding and performed consulting work in the area of IgG4-related disease for both Roche and Xencor.
The accompanying manuscript does not include studies on humans or animals.
Additional information
Redaktion
P. Lamprecht, Lübeck
F. Moosig, Neumünster
U. Müller-Ladner, Bad Nauheim
U. Lange, Bad Nauheim
Rights and permissions
About this article

Cite this article
Perugino, C.A., Stone, J.H. Treatment of IgG4-related disease. Z Rheumatol 75, 681–686 (2016). https://doi.org/10.1007/s00393-016-0142-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00393-016-0142-y